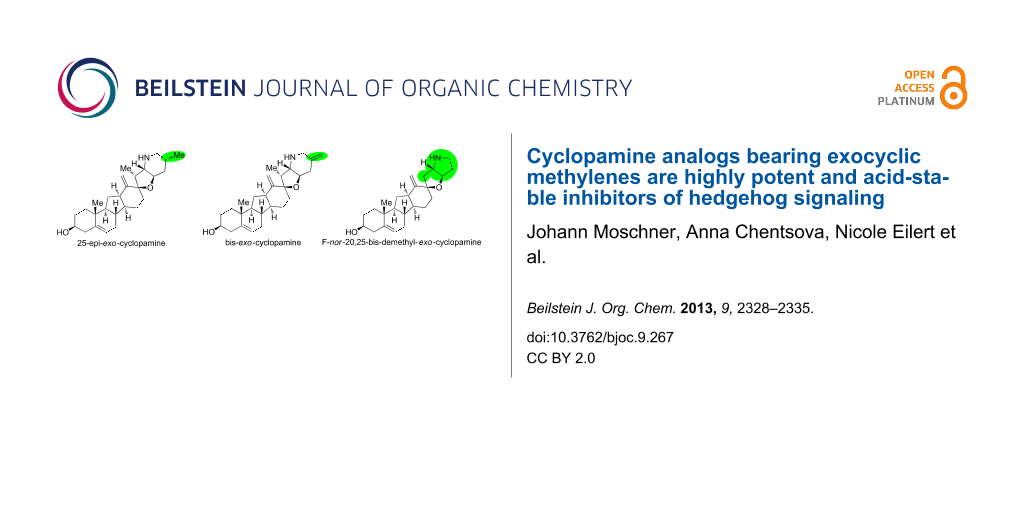
Abstract
The chemical synthesis and biological evaluation of new cyclopamine analogs bearing exocyclic methylenes in different positions is described. Bis-exo-cyclopamine 6 was identified as a potent inhibitor of the Gli1-dependent luciferase expression in Shh-LIGHTII cells. An extension of this study to F-ring-modified structures shows the necessity of a rigidly positioned nitrogen atom for bioactivity as well as the presence of the C21 methyl group for acid stability and bioactivity.

Graphical Abstract
Introduction
Hedgehog signaling is involved in embryonic development and plays an important role in the maintenance of stem cells, tissue repair and regeneration in adult organisms [1-4]. The erroneous activation of hedgehog signaling is tightly associated with the occurrence of basal cell carcinoma and medulloblastoma [5]. In addition, several other tumors are co-dependent on hedgehog signaling, examples for this type are cancers of the skin [6,7], brain [8,9], lung [10], pancreas [11], breast [12], prostate [13,14], colon [15], rhabdomyosarcoma [16], lymphoma [17-19], multiple myeloma [17,20], and chronic myeloic leukemia [21-23]. More recently, other diseases like diabetes [24,25], neurodegenerative disorders [26], and trisomy [27,28], have been linked with hedgehog signaling.
Cyclopamine (1, see Figure 1) was the first inhibitor of the hedgehog signaling pathway to be identified. As a highly selective inhibitor of the transmembrane protein Smoothened (Smo), an integral component of hedgehog signaling, it presents an attractive target for medicinal and pharmaceutical research [29,30]. Unfortunately, its direct development into a drug is hampered by its low metabolic stability (decomposition at pH < 3) [31] and rather moderate potency (IC50 ~ 5 µM).
![[1860-5397-9-267-1]](/bjoc/content/figures/1860-5397-9-267-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of cyclopamine and exo-cyclopamine.
Figure 1: Structures of cyclopamine and exo-cyclopamine.
We previously reported the first chemical synthesis of cyclopamine (1) starting from dehydroepiandrosterone and utilizing the C–H-functionalization logic and a biomimetic skeleton rearrangement [32,33]. Furthermore, quantum mechanical calculations guided our design and synthesis of exo-cyclopamine (2, see Figure 1), a ten-fold more potent and acid-stable analog with an exo-methylene unit at C13–C18 [34]. Herein, we describe a comprehensive study of cyclopamine analogs bearing exo-methylene units in different positions and extent this rational to F-ring-modified structures.
Results and Discussion
Our synthetic approach to the analogs described here started from previously reported azide 3 which was converted in six steps [32] to the protected bis-exo-methylene compound 4. Carefully chosen conditions for the deprotection of the benzyl ether (DDQ, DCE/pH 7 phosphate buffer, 40 °C, 86%) and the benzenesulfonylamine (sodium naphthalenide, DME, −78 °C, 79%) allowed for the isolation of bis-exo-cyclopamine 6 in 18% overall yield from 3. The hydrogenation of intermediate 4 by using Wilkinson’s catalyst in benzene yielded previously described exo-cyclopamine 2 and its C25 epimer 5. Deprotection with Raney-nickel (EtOH, 78 °C) and then sodium naphthalenide (DME, −78 °C, 41% over two steps) furnished 25-epi-exo-cyclopamine 5 in 3% overall yield from 3.
A first set of exo-cyclopamine analogs with a modified F-ring was obtained starting again from azide 3 (see Scheme 1). Reduction of the lactone moiety to give a tetrahydrofuran was accomplished in three steps including (1) partial reduction to the lactol (DIBAl-H, THF, −78 °C to −60 °C, 95%), (2) acetylation (Ac2O, pyridine, cat. DMAP, quant.) and (3) reductive removal of the so-obtained acetate (Et3SiH, BF3·Et2O, −78 °C to −20 °C, 79%). Removal of the benzyl ether, reduction of the azide moiety and concomitant alkylation was then all effected in one pot by using Raney-nickel in EtOH to give analog 8, an F-ring-opened exo-cyclopamine derivative in 27% overall yield from 3. Alternatively, the use of previously devised conditions for benzyl deprotection (DDQ, DCE/pH 7 phosphate buffer, 45 °C, 78%) and then sodium borohydride-mediated reduction of the azide (EtOH, 65 °C, 62%) cleanly furnished primary amine 9, an exo-cyclopamine derivative with no F-ring (35% overall yield from 3).
![[1860-5397-9-267-i1]](/bjoc/content/inline/1860-5397-9-267-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 25-epi-exo-cyclopamine 5, bis-exo-cyclopamine 6, and derivatives 8 and 9. Reaction conditions: (a) DIBAl-H, THF, −78 °C to −65 °C, 4 h, 95%; (b) Ac2O, pyridine, DMAP (cat.), 25 °C, 20 h, quant; (c) BF3·Et2O, Et3SiH, CH2Cl2, −78 °C to −20 °C, 7 h, 79%; (d) Rh(PPh3)3Cl, benzene, H2, 25 °C, 24 h, quant. (3:1 dr); (e) Raney-nickel (W2), EtOH, 78 °C, 5 min; (f) sodium naphthalenide, DME, −78 °C, 30 min, 41% over two steps; (g) DDQ, DCE/phosphate buffer (pH 7), 40 °C, 95 min, 86%; (h) sodium naphthalenide, DME, −78 °C, 1 h, 79%; (i) Raney-nickel (W2), EtOH, 37%; (j) DDQ, DCE/phosphate buffer (pH 7), 45 °C, 1.5 h, 78%, (k) NaBH4, EtOH, 65 °C, 5 d, 62%.
Scheme 1: Synthesis of 25-epi-exo-cyclopamine 5, bis-exo-cyclopamine 6, and derivatives 8 and 9. Reaction con...
A second set of F-ring-modified exo-cyclopamine derivatives was obtained starting from previously described 12-ß-triethylsilyloxy compound 10 (see Scheme 2) [32]. To construct the spirolactone 12, a three-step process consisting of (1) allylation (allylcerium chloride, THF, 0 °C, 93%), (2) hydroboration/oxidation (9-BBN, THF, 70 °C; then NaBO3, H2O, 50 °C, 91%) and finally, (3) oxidative cyclization (BAIB, cat. TEMPO, CH2Cl2, 25 °C, 73%) was employed. Deprotection of the silyl ether (HF, MeCN, 25 °C, 87%) and base-induced rearrangement of the steroid skeleton via the corresponding triflate (Tf2O, pyridine, 0 °C→50 °C, 46%) gave C-nor-D-homo-derivative 13 which, in turn, was transformed into lactone azide 14 by using Evans’ conditions (LDA, THF, −78 °C to −30 °C, 10 min; then trisylazide, THF, −78 °C, 1.5 h, 43%) [35]. From 14 two additional exo-cyclopamine derivatives became accessible. Partial reduction to the lactol (DIBAl-H, THF, −78 °C to −65 °C, 88%) and Horner–Wadsworth–Emmons reaction by using previously reported dimethyl {3-[(4-methoxybenzyl)oxy]-2-oxopropyl}phosphonate [32], (Ba(OH)2, THF/H2O, 80 °C, 50%) led to 15 which, in turn, was methylenated by employing the Peterson protocol (TMSCH2CeCl2, THF, −78 °C; then TMEDA; then HF, MeCN, 25 °C, 64%) [36]. Towards this end the azide moiety was reduced and monoprotection of the so-obtained amine as a sulfonamide (LiAlH4, THF, 0 °C→25 °C; then benzenesulfonyl chloride, Et3N, DMF, 0 °C, 69%) gave 17. Deprotetion of the 4-methoxybenzyl ether (DDQ, DCE/pH 7 phosphate buffer, 25 °C, 59%) and cyclization under Mitsunobu conditions (n-Bu3P, DEAD, toluene, 0 °C→25 °C, 93%) yielded piperidine 18. Deprotection of the benzyl ether by using previously devised conditions (DDQ, DCE/pH 7 phosphate buffer, 44 °C, 70%) and the benzenesulfonylamine (sodium naphthalenide, DME, −78 °C, 95%) gave 20-demethyl-bis-exo-cyclopamine 19 in 7% overall yield starting from 14.
![[1860-5397-9-267-i2]](/bjoc/content/inline/1860-5397-9-267-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 20-demethyl-bis-exo-cyclopamine 19 and F-nor-20,25-bis-demethyl-exo-cyclopamine 23. Reaction conditions: (a) allylcerium chloride, THF, 0 °C, 30 min, 93%; (b) 9-BBN, THF, 70 °C, 6 h; then NaBO3, 50 °C, 12 h, 91%; (c) BAIB, TEMPO, CH2Cl2, 25 °C, 3 h, 73%; (d) HF 50 wt % in H2O, MeCN, 20 min, 25 °C, 87%; (e) Tf2O, pyridine, 0 °C→50 °C, 1.5 h; then additional Tf2O, 0 °C→50 °C, 2 h, 46%; (f) LDA, THF, −78 °C to −30 °C, 10 min; then trisylazide, THF, −78 °C, 1.5 h, 43%; g) DIBAl-H, THF, −78 °C to −65 °C, 2 h, 88%; (h) dimethyl (3-((4-methoxybenzyl)oxy)-2-oxopropyl)phosphonate, Ba(OH)2, THF/H2O, 80 °C, 13 h, 50%; (i) (trimethylsilyl)methylcerium chloride, THF, −78 °C, 30 min; then TMEDA, −78 °C, 15 min; then HF 50 wt % in H2O, MeCN, 25 °C, 10 min, 64%; (j) LiAlH4, THF, 0 °C→25 °C, 13 h; then benzenesulfonyl chloride, Et3N, DMF, 0 °C, 25 min, 69%; (k) DDQ, CH2Cl2/phosphate buffer (pH 7), 25 °C, 2 h, 59%; (l) n-Bu3P, DEAD, toluene, 0 °C→25 °C, 12 h, 93%; (m) DDQ, DCE/phosphate buffer (pH 7), 44 °C, 50 min, 70%; (n) sodium naphthalenide, DME, −78 °C, 30 min, 95%; (o) triethyl phosphonoacetate, Ba(OH)2, THF, 80 °C, 12 h, 55%; (p) PPh3, THF/H2O, 50 °C, 24 h; then benzenesulfonyl chloride, Et3N, CH2Cl2, 40 °C, 5 h, 97%; (q) DIBAl-H, THF, −78 °C to −40 °C, 3 h, 97%; (r) n-Bu3P, DEAD, toluene, 0 °C→25 °C, 24 h, 81%; (s) DDQ, DCE/phosphate buffer (pH 7), 40 °C, 30 min, 62%; (t) sodium naphthalenide, DME, −78 °C, 40 min, 74%.
Scheme 2: Synthesis of 20-demethyl-bis-exo-cyclopamine 19 and F-nor-20,25-bis-demethyl-exo-cyclopamine 23. Re...
Starting from azido lactone 14 (see Scheme 2) a partial reduction to the lactol (DIBAl-H, THF, −78 °C to −65 °C, 88%) and the Horner–Wadsworth–Emmons reaction by using triethyl phosphonoacetate (Ba(OH)2, THF/H2O, 80 °C, 55%) led to ethyl ester 20. Reduction of the azide moiety in 20 was carried out by using Staudinger’s protocol (Ph3P, THF/H2O, 50 °C). Immediate protection of the obtained amine (benzenesulfonyl chloride, Et3N, CH2Cl2, 40 °C, 97% over 2 steps) gave sulfonylamide 21. Reduction of the ester in 21 to the primary alcohol (DIBAl-H, THF, −78 °C to −40 °C, 97%) and cyclization by employing Mitsunobu conditions (n-Bu3P, DEAD, toluene, 0 °C→25 °C, 81%) yielded pyrrolidine 22. Previously devised conditions for the deprotection (1. DDQ, DCE/pH 7 phosphate buffer, 40 °C; 2. sodium naphthalenide, DME, −78 °C, 46%) finally afforded F-nor-20,25-bis-demethyl-exo-cyclopamine 23 in 9% overall yield starting from 14.
Next, we examined all synthesized compounds for their ability to inhibit the Gli1-dependent luciferase expression in Shh-LIGHTII cells, a clonal mouse fibroblast cell line which stably incorporates a Gli-dependent firefly luciferase reporter and a constitutive Renilla luciferase reporter [37]. The compounds were tested in a concentration range from 0.01 μM to 10 μM. While analogs 8, 9, and 19 showed no activity (data not shown) in this concentration range, 5, 6, and 23 were active with 25-epi-exo-cyclopamine 5 having an IC50 of 3.29 ± 0.31 μM, F-nor-20,25-bis-demethyl-exo-cyclopamine 23 of 6.4 ± 0.9 μM, and bis-exo-cyclopamine 6 being the most active with an IC50 of 0.20 ± 0.01 μM (see Figure 2).
![[1860-5397-9-267-2]](/bjoc/content/figures/1860-5397-9-267-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: IC50 values of Shh inhibition by compounds 5, 6 and 23 in a Gli1-reporter gene assay. Data were obtained from three independent experiments and represent the mean ± standard deviation.
Figure 2: IC50 values of Shh inhibition by compounds 5, 6 and 23 in a Gli1-reporter gene assay. Data were obt...
Given the negative test results of compounds 8 and 9 it becomes evident that the F-ring is necessary for bioactivity. The piperidine moiety provides a rather rigidly placed nitrogen atom. Nevertheless, a pyrrolidine as in compound 23 still provides the correct orientation of the nitrogen atom. Despite compounds 8 and 9 being inactive in the assay, both derivatives induced cytotoxicity in the concentration range tested. Very subtle changes of the conformation of the piperidine ring significantly change bioactivity: While 25-epi-exo-cyclopamine 5 shows reduced activity in comparison to exo-cyclopamine 2, bis-exo-cyclopamine 6 is the most active compound tested in this study. Furthermore, the methyl group at C-20 seems to have a pronounced effect on the bioactivity, with 20-demethyl-bis-exo-cyclopamine 19 being completely inactive in the tested concentration range.
Finally, we studied the stability of all newly synthesized compounds towards acidic conditions. Therefore, they were exposed to a pH of approximately 1 (MeOH, 1 M HCl) for 24 h. After evaporation of all volatiles, 1H NMR spectra were acquired and compared to the initially obtained spectra of the pure compounds. While compounds 5, 6, 8, and 9 remained unchanged, compounds 19 and 23 showed decomposition. This experiment emphasizes the importance of the C-21 methyl group for the stability of exo-cyclopamine derivatives. All synthesized compounds, the number of steps required, and the respective overall yield starting from 3 or 14, as well as their biological activity and stability under acidic conditions are summarized in Table 1.
Table 1: Synthesized exo-cyclopamine derivatives, number of steps, yields, results of their biological testing, and stability towards acid.
| Compound | Number of steps from 3 or 14 | Overall yield | Potency in Gli-assay | Stability towards acid (pH ~1, 24 h) |
|---|---|---|---|---|
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-267-i3.svg?max-width=637&scale=1.0)
5: 25-epi-exo-cyclopamine |
9 steps from 3 | 3% | 3.29 ± 0.31 µM | stable |
![[Graphic 2]](/bjoc/content/inline/1860-5397-9-267-i4.svg?max-width=637&scale=1.0)
6: bis-exo-cyclopamine |
8 steps from 3 | 18% | 0.2 ± 0.01 µM | stable |
![[Graphic 3]](/bjoc/content/inline/1860-5397-9-267-i5.svg?max-width=637&scale=1.0)
8 |
4 steps from 3 | 27% | > 10 µM | stable |
![[Graphic 4]](/bjoc/content/inline/1860-5397-9-267-i6.svg?max-width=637&scale=1.0)
9 |
5 steps from 3 | 35% | > 10 µM | stable |
![[Graphic 5]](/bjoc/content/inline/1860-5397-9-267-i7.svg?max-width=637&scale=1.0)
19: 20-demethyl-bis-exo-cyclopamine |
8 steps from 14 | 7% | > 10 µM | decomp. |
![[Graphic 6]](/bjoc/content/inline/1860-5397-9-267-i8.svg?max-width=637&scale=1.0)
23: F-nor-20,25-bis-demethyl-exo-cyclopamine |
7 steps from 14 | 9% | 6.40 ± 0.90 µM | decomp. |
| Previously synthesized and biologically evaluated: | ||||
![[Graphic 7]](/bjoc/content/inline/1860-5397-9-267-i9.svg?max-width=637&scale=1.0)
2: exo-cyclopamine |
9 steps from 3 | 7% | 0.5 µM [34] | stable |
![[Graphic 8]](/bjoc/content/inline/1860-5397-9-267-i10.svg?max-width=637&scale=1.0)
1: cyclopamine |
10 steps from 3 | 5% | 5 µM [34] | decomp. |
Conclusion
In conclusion, we succeeded in identifying the new cyclopamine derivative bis-exo-cyclopamine 6 which surpasses the biological potency of the parent compound by the 25-fold and is stable at pH 1. Further insights were gained into the structure–activity relationship of F-ring-modified analogs of cyclopamine and the necessity of the C-21 methyl group for bioactivity and acid stability was revealed. Our designed analogs of cyclopamine are accessible in noticeably shorter and higher yielding synthetic routes than the parent compound, a fact that will further contribute to their usefulness in biological and medicinal studies.
Supporting Information
| Supporting Information File 1: Experimental details and analytical data of all synthesized compounds are provided. | ||
| Format: PDF | Size: 9.3 MB | Download |
References
-
Keeler, R. F. Phytochemistry 1968, 7, 303. doi:10.1016/S0031-9422(00)86328-1
Return to citation in text: [1] -
Keeler, R. F. Teratology 1970, 3, 169. doi:10.1002/tera.1420030209
Return to citation in text: [1] -
Beachy, P. A.; Karhadkar, S. S.; Berman, D. M. Nature 2004, 432, 324. doi:10.1038/nature03100
Return to citation in text: [1] -
Taipale, J.; Beachy, P. A. Nature 2001, 411, 349. doi:10.1038/35077219
Return to citation in text: [1] -
Hidalgo, M.; Maitra, A. N. Engl. J. Med. 2009, 361, 2094. doi:10.1056/NEJMcibr0905857
Return to citation in text: [1] -
Gailani, M. R.; Ståhle-Bäckdahl, M.; Leffell, D. J.; Glyn, M.; Zaphiropoulos, G.; Undén, A. B.; Dean, M.; Brash, D. E.; Bale, A. E.; Toftgård, R. Nat. Genet. 1996, 14, 78. doi:10.1038/ng0996-78
Return to citation in text: [1] -
Hahn, H.; Wicking, C.; Zaphiropoulos, P. G.; Gailani, M. R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Undén, A. B.; Gillies, S.; Negus, K.; Smyth, I.; Pressman, C.; Leffell, D. J.; Gerrard, B.; Goldstein, A. M.; Dean, D.; Toftgård, R.; Chenevix-Trench, G.; Wainwright, B.; Bale, A. E. Cell 1996, 85, 841. doi:10.1016/S0092-8674(00)81268-4
Return to citation in text: [1] -
Zurawel, R. H.; Allen, C.; Chiappa, S.; Cato, W.; Biegel, J.; Cogen, P.; de Sauvage, F.; Raffel, C. Genes, Chromosomes Cancer 2000, 27, 44. doi:10.1002/(SICI)1098-2264(200001)27:1<44::AID-GCC6>3.0.CO;2-V
Return to citation in text: [1] -
Bar, E. E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A. L.; DiMeco, F.; Olivi, A.; Eberhart, C. G. Stem Cells 2007, 25, 2524. doi:10.1634/stemcells.2007-0166
Return to citation in text: [1] -
Watkins, D. N.; Berman, D. M.; Baylin, S. B. Cell Cycle 2003, 2, 195. doi:10.4161/cc.2.3.378
Return to citation in text: [1] -
Dosch, J. S.; di Magliano, M. P.; Simeone, D. M. Pancreatology 2010, 10, 151. doi:10.1159/000225923
Return to citation in text: [1] -
Vořechovský, I.; Benediktsson, K. P.; Toftgård, R. Eur. J. Cancer 1999, 35, 711. doi:10.1016/S0959-8049(99)00017-9
Return to citation in text: [1] -
Fan, L.; Pepicelli, C. V.; Dibble, C. C.; Catbagan, W.; Zarycki, J. L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M. L. G.; Munoz, A.; Lipinski, R.; Thrasher, J. B.; Bushman, W. Endocrinology 2004, 145, 3961. doi:10.1210/en.2004-0079
Return to citation in text: [1] -
Karhadkar, S. S.; Bova, G. S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J. T.; Berman, D. M.; Beachy, P. A. Nature 2004, 431, 707. doi:10.1038/nature02962
Return to citation in text: [1] -
Berman, D. M.; Karhadkar, S. S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M. R.; Briggs, K.; Parker, A. R.; Shimada, Y.; Eshleman, J. R.; Watkins, D. N.; Beachy, P. A. Nature 2003, 425, 846. doi:10.1038/nature01972
Return to citation in text: [1] -
Teglund, S.; Toftgård, R. Biochim. Biophys. Acta, Rev. Cancer 2010, 1805, 181. doi:10.1016/j.bbcan.2010.01.003
Return to citation in text: [1] -
Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944. doi:10.1038/nm1614
Return to citation in text: [1] [2] -
Hegde, G. V.; Munger, C. M.; Emanuel, K.; Joshi, A. D.; Greiner, T. C.; Weisenburger, D. D.; Vose, J. M.; Joshi, S. S. Mol. Cancer Ther. 2008, 7, 1450. doi:10.1158/1535-7163.MCT-07-2118
Return to citation in text: [1] -
Kawahara, T.; Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Anticancer Res. 2009, 29, 4629.
Return to citation in text: [1] -
Peacock, C. D.; Wang, Q.; Gesell, G. S.; Corcoran-Schwartz, I. M.; Jones, E.; Kim, J.; Devereux, W. L.; Rhodes, J. T.; Huff, C. A.; Beachy, P. A.; Watkins, D. N.; Matsui, W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4048. doi:10.1073/pnas.0611682104
Return to citation in text: [1] -
Zhao, C.; Chen, A.; Jamieson, C. H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H. Y.; Kim, J.; Chute, J. P.; Rizzieri, D.; Munchhof, M.; VanArsdale, T.; Beachy, P. A.; Reya, T. Nature 2009, 458, 776. doi:10.1038/nature07737
Return to citation in text: [1] -
Bai, L.-Y.; Chiu, C.-F.; Lin, C.-W.; Hsu, N.-Y.; Lin, C.-L.; Lo, W.-J.; Kao, M.-C. Leukemia 2008, 22, 226. doi:10.1038/sj.leu.2404978
Return to citation in text: [1] -
Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M. R.; Manley, P.; Trussel, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; Warmuth, M. Cancer Cell 2008, 14, 238. doi:10.1016/j.ccr.2008.08.003
Return to citation in text: [1] -
Suh, J. M.; Gao, X.; McKay, J.; McKay, R.; Salo, Z.; Graff, J. M. Cell Metab. 2006, 3, 25. doi:10.1016/j.cmet.2005.11.012
Return to citation in text: [1] -
Teperino, R.; Amann, S.; Bayer, M.; McGee, S. L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; Dalgaard, K.; Selvaraj, M.; Gaster, M.; Lee-Young, R. S.; Febbraio, M. A.; Knauf, C.; Cani, P. D.; Aberger, F.; Penninger, J. M.; Pospilik, J. A.; Esterbauer, H. Cell 2012, 151, 414. doi:10.1016/j.cell.2012.09.021
Return to citation in text: [1] -
Gonzalez-Reyes, L. E.; Verbitsky, M.; Blesa, J.; Jackson-Lewis, V.; Paredes, D.; Tillack, K.; Phani, S.; Kramer, E. R.; Przedborski, S.; Kottmann, A. H. Neuron 2012, 75, 306. doi:10.1016/j.neuron.2012.05.018
Return to citation in text: [1] -
Nowaczyk, M. J.; Huggins, M. J.; Tomkins, D. J.; Rossi, E.; Ramsay, J. A.; Woulfe, J.; Scherer, S. W.; Belloni, E. Clin. Genet. 2000, 57, 388. doi:10.1034/j.1399-0004.2000.570510.x
Return to citation in text: [1] -
Decker, S.; Zirlik, K.; Djebatchie, L.; Hartmann, D.; Ihorst, G.; Schmitt-Graeff, A.; Herchenbach, D.; Jumaa, H.; Warmuth, M.; Veelken, H.; Dierks, C. Blood 2012, 119, 997. doi:10.1182/blood-2011-06-359075
Return to citation in text: [1] -
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Bioorg. Med. Chem. 2010, 18, 6613. doi:10.1016/j.bmc.2010.07.038
Return to citation in text: [1] -
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Angew. Chem. 2010, 122, 3492. doi:10.1002/ange.200906967
Return to citation in text: [1] -
Wilson, S. R.; Strand, M. F.; Krapp, A.; Rise, F.; Petersen, D.; Krauss, S. J. Pharm. Biomed. Anal. 2010, 52, 707. doi:10.1016/j.jpba.2010.02.017
Return to citation in text: [1] -
Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem. 2009, 121, 8052. doi:10.1002/ange.200902520
Return to citation in text: [1] [2] [3] [4] -
Heretsch, P.; Rabe, S.; Giannis, A. J. Am. Chem. Soc. 2010, 132, 9968. doi:10.1021/ja103152k
Return to citation in text: [1] -
Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362. doi:10.1039/C1CC11782C
Return to citation in text: [1] [2] [3] -
Evans, D. A.; Britton, T. C. J. Am. Chem. Soc. 1987, 109, 6881. doi:10.1021/ja00256a069
Return to citation in text: [1] -
Johnson, C. R.; Tait, B. D. J. Org. Chem. 1987, 52, 281. doi:10.1021/jo00378a024
Return to citation in text: [1] -
Taipale, J.; Chen, J. K.; Cooper, M. K.; Wang, B.; Mann, R. K.; Milenkovic, L.; Scott, M. P.; Beachy, P. A. Nature 2000, 406, 1005. doi:10.1038/35023008
Return to citation in text: [1]
| 34. | Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362. doi:10.1039/C1CC11782C |
| 1. | Keeler, R. F. Phytochemistry 1968, 7, 303. doi:10.1016/S0031-9422(00)86328-1 |
| 2. | Keeler, R. F. Teratology 1970, 3, 169. doi:10.1002/tera.1420030209 |
| 3. | Beachy, P. A.; Karhadkar, S. S.; Berman, D. M. Nature 2004, 432, 324. doi:10.1038/nature03100 |
| 4. | Taipale, J.; Beachy, P. A. Nature 2001, 411, 349. doi:10.1038/35077219 |
| 10. | Watkins, D. N.; Berman, D. M.; Baylin, S. B. Cell Cycle 2003, 2, 195. doi:10.4161/cc.2.3.378 |
| 26. | Gonzalez-Reyes, L. E.; Verbitsky, M.; Blesa, J.; Jackson-Lewis, V.; Paredes, D.; Tillack, K.; Phani, S.; Kramer, E. R.; Przedborski, S.; Kottmann, A. H. Neuron 2012, 75, 306. doi:10.1016/j.neuron.2012.05.018 |
| 8. | Zurawel, R. H.; Allen, C.; Chiappa, S.; Cato, W.; Biegel, J.; Cogen, P.; de Sauvage, F.; Raffel, C. Genes, Chromosomes Cancer 2000, 27, 44. doi:10.1002/(SICI)1098-2264(200001)27:1<44::AID-GCC6>3.0.CO;2-V |
| 9. | Bar, E. E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A. L.; DiMeco, F.; Olivi, A.; Eberhart, C. G. Stem Cells 2007, 25, 2524. doi:10.1634/stemcells.2007-0166 |
| 27. | Nowaczyk, M. J.; Huggins, M. J.; Tomkins, D. J.; Rossi, E.; Ramsay, J. A.; Woulfe, J.; Scherer, S. W.; Belloni, E. Clin. Genet. 2000, 57, 388. doi:10.1034/j.1399-0004.2000.570510.x |
| 28. | Decker, S.; Zirlik, K.; Djebatchie, L.; Hartmann, D.; Ihorst, G.; Schmitt-Graeff, A.; Herchenbach, D.; Jumaa, H.; Warmuth, M.; Veelken, H.; Dierks, C. Blood 2012, 119, 997. doi:10.1182/blood-2011-06-359075 |
| 6. | Gailani, M. R.; Ståhle-Bäckdahl, M.; Leffell, D. J.; Glyn, M.; Zaphiropoulos, G.; Undén, A. B.; Dean, M.; Brash, D. E.; Bale, A. E.; Toftgård, R. Nat. Genet. 1996, 14, 78. doi:10.1038/ng0996-78 |
| 7. | Hahn, H.; Wicking, C.; Zaphiropoulos, P. G.; Gailani, M. R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Undén, A. B.; Gillies, S.; Negus, K.; Smyth, I.; Pressman, C.; Leffell, D. J.; Gerrard, B.; Goldstein, A. M.; Dean, D.; Toftgård, R.; Chenevix-Trench, G.; Wainwright, B.; Bale, A. E. Cell 1996, 85, 841. doi:10.1016/S0092-8674(00)81268-4 |
| 21. | Zhao, C.; Chen, A.; Jamieson, C. H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H. Y.; Kim, J.; Chute, J. P.; Rizzieri, D.; Munchhof, M.; VanArsdale, T.; Beachy, P. A.; Reya, T. Nature 2009, 458, 776. doi:10.1038/nature07737 |
| 22. | Bai, L.-Y.; Chiu, C.-F.; Lin, C.-W.; Hsu, N.-Y.; Lin, C.-L.; Lo, W.-J.; Kao, M.-C. Leukemia 2008, 22, 226. doi:10.1038/sj.leu.2404978 |
| 23. | Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M. R.; Manley, P.; Trussel, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; Warmuth, M. Cancer Cell 2008, 14, 238. doi:10.1016/j.ccr.2008.08.003 |
| 5. | Hidalgo, M.; Maitra, A. N. Engl. J. Med. 2009, 361, 2094. doi:10.1056/NEJMcibr0905857 |
| 24. | Suh, J. M.; Gao, X.; McKay, J.; McKay, R.; Salo, Z.; Graff, J. M. Cell Metab. 2006, 3, 25. doi:10.1016/j.cmet.2005.11.012 |
| 25. | Teperino, R.; Amann, S.; Bayer, M.; McGee, S. L.; Loipetzberger, A.; Connor, T.; Jaeger, C.; Kammerer, B.; Winter, L.; Wiche, G.; Dalgaard, K.; Selvaraj, M.; Gaster, M.; Lee-Young, R. S.; Febbraio, M. A.; Knauf, C.; Cani, P. D.; Aberger, F.; Penninger, J. M.; Pospilik, J. A.; Esterbauer, H. Cell 2012, 151, 414. doi:10.1016/j.cell.2012.09.021 |
| 15. | Berman, D. M.; Karhadkar, S. S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M. R.; Briggs, K.; Parker, A. R.; Shimada, Y.; Eshleman, J. R.; Watkins, D. N.; Beachy, P. A. Nature 2003, 425, 846. doi:10.1038/nature01972 |
| 17. | Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944. doi:10.1038/nm1614 |
| 18. | Hegde, G. V.; Munger, C. M.; Emanuel, K.; Joshi, A. D.; Greiner, T. C.; Weisenburger, D. D.; Vose, J. M.; Joshi, S. S. Mol. Cancer Ther. 2008, 7, 1450. doi:10.1158/1535-7163.MCT-07-2118 |
| 19. | Kawahara, T.; Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Anticancer Res. 2009, 29, 4629. |
| 13. | Fan, L.; Pepicelli, C. V.; Dibble, C. C.; Catbagan, W.; Zarycki, J. L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M. L. G.; Munoz, A.; Lipinski, R.; Thrasher, J. B.; Bushman, W. Endocrinology 2004, 145, 3961. doi:10.1210/en.2004-0079 |
| 14. | Karhadkar, S. S.; Bova, G. S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J. T.; Berman, D. M.; Beachy, P. A. Nature 2004, 431, 707. doi:10.1038/nature02962 |
| 17. | Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944. doi:10.1038/nm1614 |
| 20. | Peacock, C. D.; Wang, Q.; Gesell, G. S.; Corcoran-Schwartz, I. M.; Jones, E.; Kim, J.; Devereux, W. L.; Rhodes, J. T.; Huff, C. A.; Beachy, P. A.; Watkins, D. N.; Matsui, W. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 4048. doi:10.1073/pnas.0611682104 |
| 12. | Vořechovský, I.; Benediktsson, K. P.; Toftgård, R. Eur. J. Cancer 1999, 35, 711. doi:10.1016/S0959-8049(99)00017-9 |
| 11. | Dosch, J. S.; di Magliano, M. P.; Simeone, D. M. Pancreatology 2010, 10, 151. doi:10.1159/000225923 |
| 16. | Teglund, S.; Toftgård, R. Biochim. Biophys. Acta, Rev. Cancer 2010, 1805, 181. doi:10.1016/j.bbcan.2010.01.003 |
| 32. | Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem. 2009, 121, 8052. doi:10.1002/ange.200902520 |
| 33. | Heretsch, P.; Rabe, S.; Giannis, A. J. Am. Chem. Soc. 2010, 132, 9968. doi:10.1021/ja103152k |
| 29. | Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Bioorg. Med. Chem. 2010, 18, 6613. doi:10.1016/j.bmc.2010.07.038 |
| 30. | Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Angew. Chem. 2010, 122, 3492. doi:10.1002/ange.200906967 |
| 31. | Wilson, S. R.; Strand, M. F.; Krapp, A.; Rise, F.; Petersen, D.; Krauss, S. J. Pharm. Biomed. Anal. 2010, 52, 707. doi:10.1016/j.jpba.2010.02.017 |
| 37. | Taipale, J.; Chen, J. K.; Cooper, M. K.; Wang, B.; Mann, R. K.; Milenkovic, L.; Scott, M. P.; Beachy, P. A. Nature 2000, 406, 1005. doi:10.1038/35023008 |
| 34. | Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362. doi:10.1039/C1CC11782C |
| 32. | Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem. 2009, 121, 8052. doi:10.1002/ange.200902520 |
| 36. | Johnson, C. R.; Tait, B. D. J. Org. Chem. 1987, 52, 281. doi:10.1021/jo00378a024 |
| 32. | Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem. 2009, 121, 8052. doi:10.1002/ange.200902520 |
| 35. | Evans, D. A.; Britton, T. C. J. Am. Chem. Soc. 1987, 109, 6881. doi:10.1021/ja00256a069 |
| 34. | Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362. doi:10.1039/C1CC11782C |
| 32. | Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem. 2009, 121, 8052. doi:10.1002/ange.200902520 |
© 2013 Moschner et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)