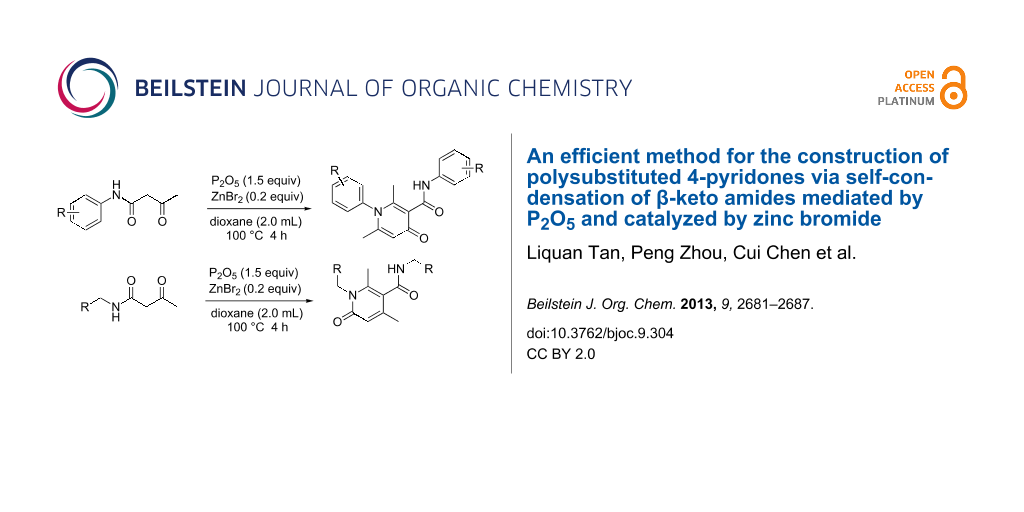
Abstract
A self-condensation cyclization reaction mediated by phosphorus pentoxide (P2O5) and catalyzed by zinc bromide (ZnBr2) is presented for the synthesis of polysubstituted 4-pyridones and 2-pyridones from β-keto amides. A variety of β-keto amides are used in this approach, and a wide range of functionalized 4-pyridones and 2-pyridones were obtained in good to excellent yields. When employing the N-aryl β-keto amides as the substrates in this protocol, 4-pyridones are resulted, however, when using N-aliphatic-substituted β-keto amides as the partners of N-aryl β-keto amides under the same conditions, 2-pyridones are afforded.

Graphical Abstract
Introduction
β-Keto amide and their derivatives are desired classes of intermediates for the synthesis of nitrogen- and oxygen-containing heterocyclic compounds since they possess six reactive sites in the same molecule (Scheme 1) [1-7]. A lot of reports can be found in the literature concerning the construction of different heterocyclic compounds from β-keto amides by modification of the six different reactive positions [8-13]. Our interest in the fields of cleavage or construction of C–C and C–hetero bonds using β-keto amides and their derivatives as the substrates prompted us to exploit the reactivity of the six different reactive positions of β-keto amides [14-18].
![[1860-5397-9-304-i1]](/bjoc/content/inline/1860-5397-9-304-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The six different reactive positions of β-keto amides.
Scheme 1: The six different reactive positions of β-keto amides.
Two groups reported the self-condensation of N-aryl β-keto amides. The group of Zhang used Na2S2O8 as the reagent to induce this condensation [19] and the group of Ovenden used tosic acid to catalyze this transformation [20]. In this article, we report an improved efficient method for the construction of polysubstituted 4-pyridones and 2-pyridones via phosphorus pentoxide-mediated self-condensation of β-ketobutylanilides catalyzed by zinc bromide (Scheme 2). It is well known that 4-pyridones are one of the most important classes of heterocyclic compounds as they are a key structural attribute of many bioactive natural products [21-27]. Besides, many of the 4-pyridones have shown interesting biological activities, such as antibacterial [24,28], antitumor [29], antiviral [30], and several other potentially useful activities [27,31].
![[1860-5397-9-304-i2]](/bjoc/content/inline/1860-5397-9-304-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of polysubstituted 4-pyridones from β-keto amides.
Scheme 2: Synthesis of polysubstituted 4-pyridones from β-keto amides.
Results and Discussion
In our initial study, 3-oxo-N-phenylbutanamide (1a) was selected as a model substrate to optimize the reaction conditions (Table 1). The preliminary results showed that this self-condensation cyclization reaction did not occur in the absence of ZnBr2 (Table 1, entry 1) or P2O5 (Table 1, entry 3) at room temperature. Notable efficacy was achieved when increasing the reaction temperature to 100 °C for 4 h and afforded the desired product 1,4-dihydro-2,6-dimethyl-4-oxo-N,1-diphenylpyridine-3-carboxamide (2a) in 94% GC yield (Table 1, entries 2, 4–6). Results of the screening study of the amount of ZnBr2 and P2O5 (Table 1, entries 6–8 and 12) indicated that 0.2 equiv of ZnBr2 and 1.5 equiv of P2O5 was sufficient for the completion of this transformation. In order to improve the efficiency of the reaction, we tested several zinc salts for this reaction (Table 1, entries 7, 9–11). The use of ZnBr2 and ZnCl2 greatly facilitated the reaction, and both gave 2a in excellent GC yield (Table 1, entries 7 and 9). Among the various solvents examined (Table 1, entries 7, 13–17), dioxane proved to be the most suitable solvent for this transformation (Table 1, entry 7). After screening, the optimal reaction conditions were obtained; these are, the mixture of 3-oxo-N-phenylbutanamide 1a with 0.2 equiv of ZnBr2 and 1.5 equiv of P2O5 reacted in dioxane (2.0 mL) at 100 °C for 4 h (Table 1, entry 7).
Table 1: Optimization of reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-9-304-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Zn(II) (equiv) | Temp. ( °C) | Yield (%)b |
|---|---|---|---|---|
| 1c | Dioxane | 0 | rt | 0 |
| 2c | Dioxane | ZnBr2 (1.0) | rt | 13 |
| 3c,d | Dioxane | ZnBr2 (1.0) | rt | 0 |
| 4e | Dioxane | ZnBr2 (1.0) | 100 | 72 |
| 5 | Dioxane | ZnBr2 (1.0) | 100 | 94 |
| 6c | Dioxane | ZnBr2 (1.0) | 100 | 94 |
| 7 | Dioxane | ZnBr2 (0.2) | 100 | 94 |
| 8 | Dioxane | ZnBr2 (0.5) | 100 | 94 |
| 9 | Dioxane | ZnCl2 (0.2) | 100 | 92 |
| 10 | Dioxane | Zn(OAc)2 (0.2) | 100 | 45 |
| 11 | Dioxane | ZnO (0.2) | 100 | 0 |
| 12f | Dioxane | ZnBr2 (0.2) | 100 | 84 |
| 13 | Cyclohexane | ZnBr2 (0.2) | reflux | 56 |
| 14 | DCE | ZnBr2 (0.2) | reflux | 81 |
| 15 | DMF | ZnBr2 (0.2) | 100 | 32 |
| 16 | AcOH | ZnBr2 (0.2) | 100 | 14 |
| 17 | MeOH | ZnBr2 (0.2) | reflux | 30 |
aAll reactions were carried out with 1a in 0.25 mmol scale and 2 mL solvent; bGC yield; creaction time: 6 h; dwithout P2O5; ereaction time: 2 h; fP2O5: 1.0 equiv.
To explore the substrate scope and limitations of this self-condensation cyclization reaction, a range of N-aryl β-keto amides were then examined under the optimal reaction conditions. The results are shown in Scheme 3.
![[1860-5397-9-304-i3]](/bjoc/content/inline/1860-5397-9-304-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The scope of the substrates. (Note: All the listed yields are isolated yields.)
Scheme 3: The scope of the substrates. (Note: All the listed yields are isolated yields.)
The results indicated that a variety of substituted N-aryl acetoacetamides 1a–1k could be easily converted into their corresponding 4-pyridone 2a–2k. All the reactions proceeded smoothly and afforded the desired product in good to excellent isolated yields (72–90%) in 4 h. This transformation appears quite tolerant with respect to the positions of the substituents on the aryl group (para-, meta- and ortho-positions). For example, the reactions of N-(2-chlorophenyl)-3-oxobutanamide (1c), N-(3-chlorophenyl)-3-oxobutanamide (1d), N-(4-chlorophenyl)-3-oxobutanamide (1e), as well as 3-oxo-N-o-tolylbutanamide (1j), 3-oxo-N-m-tolylbutanamide (1k) and 3-oxo-N-p-tolylbutanamide (1l) all lead to their corresponding product (2c, 2d, 2e, 2j, 2k and 2l) in good isolated yield. Furthermore, various electron-withdrawing groups (EWG) (Cl and CO2Et) and electron-donating groups (EDG) (Me, OMe and OEt) on the benzene ring had no obvious influence on the reaction, including without a substituent or with both an EWG and EDG substituent on the benzene ring (Scheme 3), all could be easily converted into their corresponding product with good isolated yield. Besides, the scope of this protocol was further examined by the N-aliphatic-substituted β-keto amides (1n and 1o) (Scheme 4). We found that the reactions did not afford the 4-pyridones, but provided 2-pyridones 2n and 4o with 82% and 81% isolated yield, respectively.
![[1860-5397-9-304-i4]](/bjoc/content/inline/1860-5397-9-304-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of polysubstituted 4-pyridones from N-aliphatic-substituted β-keto amides.
Scheme 4: Synthesis of polysubstituted 4-pyridones from N-aliphatic-substituted β-keto amides.
What is the result when employing different β-keto amides? Is it possible to achieve the cross-condensation products? Therefore, we attempted the reactions under the same conditions (Scheme 5). We were astonished to find that the results only included the self-condensation products (2a, 2l, 2j) and achieved not the cross-condensation products.
![[1860-5397-9-304-i5]](/bjoc/content/inline/1860-5397-9-304-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Construct the cross-condensation products.
Scheme 5: Construct the cross-condensation products.
It is well known that Lewis acids can activate β-keto amides [32]. Under mild conditions, phosphorus pentoxide (P2O5) is often used as the dehydrating agent in organic synthesis for the condensation of amines with carbonyl compounds [33,34]. Based on these evidences, a plausible reaction pathway of this self-condensation cyclization reaction was hypothesized as shown in Scheme 6. The first step of this transformation involved the coordination of β-keto amides 1 with ZnBr2 to activate the carbonyl group and to form intermediates 4 and 5, which was followed by the attack of the lone-pair electrons of the amide nitrogen to the carbonyl carbon in the presence of P2O5 and intermediate 6 was formed by eliminating one equivalent of H2O. Next, when R is aryl, a 1,3-acyl migration occurred from N to C of intermediate 6 [35-39], and affords imine 7 and its equilibrium compound 8, the subsequent intramolecular nucleophilic cyclization of intermediate 8 provides the final product 4-pyridones [20,38]. Alternatively, when R is aliphatic, the 1,3-acyl migration is not observed – maybe the migration leads to an unstable imine intermediate. Therefore, under the acidic conditions, a cationic species 10 is generated from the intermediate 6 and the subsequent intramolecular dehydration process provides 2-pyridones 11 [19].
![[1860-5397-9-304-i6]](/bjoc/content/inline/1860-5397-9-304-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have established an improved efficient synthetic protocol for the synthesis of 4-pyridone derivatives by a sequence of intermolecular dehydration of β-keto amides in the presence of phosphorus pentoxide (P2O5), 1,3-acyl migration and intramolecular dehydration. Comparing to the similar findings, the simple operation with satisfactory yields, relatively cheap additive P2O5 and zinc catalyst, and the mild reaction conditions are the advantages of this protocol. Further investigations concerning the scope of this reaction, applications, and mechanistic details are currently on-going in our laboratory.
Supporting Information
| Supporting Information File 1: Full experimental details and copies of NMR spectral data. | ||
| Format: PDF | Size: 196.6 KB | Download |
References
-
Clemens, R. J. Chem. Rev. 1986, 86, 241–318. doi:10.1021/cr00072a001
Return to citation in text: [1] -
Nishiwaki, N.; Nakaike, Y.; Ariga, M. J. Oleo Sci. 2008, 57, 53–54. doi:10.5650/jos.57.53
Return to citation in text: [1] -
Jayalakshmi, R.; Neelakatan, P.; Rao, S. N.; Iyengar, D. S.; Bhalerao, U. T. Indian J. Chem., Sect. B 1979, 18B, 366–372.
Return to citation in text: [1] -
Ehm, W. Justus Liebigs Ann. Chem. 1977, 1642–1660. doi:10.1002/jlac.197719771009
Return to citation in text: [1] -
Wang, Y.; Xin, X.; Liang, Y.; Lin, Y.; Duan, H.; Dong, D. Adv. Synth. Catal. 2009, 351, 2217–2223. doi:10.1002/adsc.200900310
Return to citation in text: [1] -
Han, M.; Nam, K.-D.; Hahn, H.-G.; Shin, D. Tetrahedron Lett. 2008, 49, 5217–5219. doi:10.1016/j.tetlet.2008.05.132
Return to citation in text: [1] -
Zaleska, B.; Lis, S. Synth. Commun. 2001, 31, 189–197. doi:10.1081/SCC-100000198
Return to citation in text: [1] -
Maiti, A.; Reddy, P. V. N.; Sturdy, M.; Marler, L.; Pegan, S. D.; Mesecar, A. D.; Pezzuto, J. M.; Cushman, M. J. Med. Chem. 2009, 52, 1873–1884. doi:10.1021/jm801335z
Return to citation in text: [1] -
Zhang, R.; Zhang, D.; Liang, Y.; Zhou, G.; Dong, D. J. Org. Chem. 2011, 76, 2880–2883. doi:10.1021/jo101949y
Return to citation in text: [1] -
Zhang, Z.; Zhang, Q.; Yan, Z.; Liu, Q. J. Org. Chem. 2007, 72, 9808–9810. doi:10.1021/jo701551f
Return to citation in text: [1] -
Zhang, R.; Zhang, D.; Guo, Y.; Zhou, G.; Jiang, Z.; Dong, D. J. Org. Chem. 2008, 73, 9504–9507. doi:10.1021/jo801959j
Return to citation in text: [1] -
Xiang, D.; Wang, K.; Liang, Y.; Zhou, G.; Dong, D. Org. Lett. 2008, 10, 345–348. doi:10.1021/ol702846t
Return to citation in text: [1] -
Knapp, J. M.; Zhu, J. S.; Wood, A. B.; Kurth, M. J. ACS Comb. Sci. 2012, 14, 85–88. doi:10.1021/co200199h
Return to citation in text: [1] -
Liu, W.-B.; Chen, C.; Zhang, Q.; Zhu, Z.-B. Beilstein J. Org. Chem. 2011, 7, 1436–1440. doi:10.3762/bjoc.7.167
Return to citation in text: [1] -
Liu, W.-B.; Chen, C.; Zhang, Q.; Zhu, Z.-B. Beilstein J. Org. Chem. 2012, 8, 344–348. doi:10.3762/bjoc.8.38
Return to citation in text: [1] -
Hu, W.; Deng, Z.; Chen, C.; Liu, W.-B. J. Chem. Res. 2012, 36, 571–572. doi:10.3184/174751912X13445852334047
Return to citation in text: [1] -
Liu, W.; Zhou, P.; Chen, C.; Zhang, Q.; Zhu, Z. Org. Biomol. Chem. 2013, 11, 542–544. doi:10.1039/c2ob27145a
Return to citation in text: [1] -
Zhang, Q.; Liu, W.; Chen, C.; Tan, L. Chin. J. Chem. 2013, 31, 453–455. doi:10.1002/cjoc.201300007
Return to citation in text: [1] -
Zhang, Z.; Fang, S.; Liu, Q.; Zhang, G. J. Org. Chem. 2012, 77, 7665–7670. doi:10.1021/jo3010217
Return to citation in text: [1] [2] -
Pierce, J. B.; Ariyan, Z. S.; Ovenden, G. S. J. Med. Chem. 1982, 25, 131–136. doi:10.1021/jm00344a008
Return to citation in text: [1] [2] -
Edwards, M. L.; Erickson, R. C. J. Med. Chem. 1979, 22, 1416–1418. doi:10.1021/jm00197a026
Return to citation in text: [1] -
Liu, J.-K. Chem. Rev. 2005, 105, 2723–2744. doi:10.1021/cr0400818
Return to citation in text: [1] -
Ye, Y. H.; Zhu, H. L.; Song, Y. C.; Liu, J. Y.; Tan, R. X. J. Nat. Prod. 2005, 68, 1106–1108. doi:10.1021/np050059p
Return to citation in text: [1] -
Kitagawa, H.; Ozawa, T.; Takahata, S.; Iida, M.; Saito, J.; Yamada, M. J. Med. Chem. 2007, 50, 4710–4720. doi:10.1021/jm0705354
Return to citation in text: [1] [2] -
Stierle, A. A.; Stierle, D. B.; Patacini, B. J. Nat. Prod. 2008, 71, 856–860. doi:10.1021/np0705054
Return to citation in text: [1] -
Maloney, K. N.; MacMillan, J. B.; Kauffman, C. A.; Jensen, P. R.; DiPasquale, A. G.; Rheingold, A. L.; Fenical, W. Org. Lett. 2009, 11, 5422–5424. doi:10.1021/ol901997k
Return to citation in text: [1] -
Jessen, H. J.; Schumacher, A.; Schmid, F.; Pfaltz, A.; Gademann, K. Org. Lett. 2011, 13, 4368–4370. doi:10.1021/ol201692h
Return to citation in text: [1] [2] -
Mitscher, L. A. Chem. Rev. 2005, 105, 559–592. doi:10.1021/cr030101q
Return to citation in text: [1] -
Mao, D. T.; Driscoll, J. S.; Marquez, V. E. J. Med. Chem. 1984, 27, 160–164. doi:10.1021/jm00368a010
Return to citation in text: [1] -
Woodford, W. J.; Swartz, B. A.; Pillar, C. J.; Kampf, A.; Mertes, M. P. J. Med. Chem. 1974, 17, 1027–1029. doi:10.1021/jm00255a030
Return to citation in text: [1] -
Kim, K. S.; Zhang, L.; Schmidt, R.; Cai, Z.-W.; Wei, D.; Williams, D. K.; Lombardo, L. J.; Trainor, G. L.; Xie, D.; Zhang, Y.; An, Y.; Sack, J. S.; Tokarski, J. S.; Darienzo, C.; Kamath, A.; Marathe, P.; Zhang, Y.; Lippy, J.; Jeyaseelan, R., Sr.; Wautlet, B.; Henley, B.; Gullo-Brown, J.; Manne, V.; Hunt, J. T.; Fargnoli, J.; Borzilleri, R. M. J. Med. Chem. 2008, 51, 5330–5341. doi:10.1021/jm800476q
Return to citation in text: [1] -
Christoffers, J. Chem. Commun. 1997, 943–944. doi:10.1039/a700838d
Return to citation in text: [1] -
Ohtsuka, Y. J. Org. Chem. 1976, 41, 629–633. doi:10.1021/jo00866a010
Return to citation in text: [1] -
So, Y.-H.; Heeschen, J. P. J. Org. Chem. 1997, 62, 3552–3561. doi:10.1021/jo960441u
Return to citation in text: [1] -
Shimada, T.; Nakamura, I.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 10546–10547. doi:10.1021/ja047542r
Return to citation in text: [1] -
Liso, G.; Trapani, G.; Reho, A.; Latrofa, A. Tetrahedron Lett. 1981, 22, 1641–1644. doi:10.1016/S0040-4039(01)90399-5
Return to citation in text: [1] -
Mészáros, Z.; Hennecz, I. Tetrahedron Lett. 1975, 16, 1019–1020. doi:10.1016/S0040-4039(00)72632-3
Return to citation in text: [1] -
Hermecz, I.; Mészáros, Z.; Vasvári-Debreczy, L.; Horváth, A.; Horváth, G.; Pongor-Csákvári, M. J. Chem. Soc., Perkin Trans. 1 1977, 789–795. doi:10.1039/P19770000789
Return to citation in text: [1] [2] -
Dimroth, P.; Radtke, V. Liebigs Ann. Chem. 1979, 769–775. doi:10.1002/jlac.197919790606
Return to citation in text: [1]
| 1. | Clemens, R. J. Chem. Rev. 1986, 86, 241–318. doi:10.1021/cr00072a001 |
| 2. | Nishiwaki, N.; Nakaike, Y.; Ariga, M. J. Oleo Sci. 2008, 57, 53–54. doi:10.5650/jos.57.53 |
| 3. | Jayalakshmi, R.; Neelakatan, P.; Rao, S. N.; Iyengar, D. S.; Bhalerao, U. T. Indian J. Chem., Sect. B 1979, 18B, 366–372. |
| 4. | Ehm, W. Justus Liebigs Ann. Chem. 1977, 1642–1660. doi:10.1002/jlac.197719771009 |
| 5. | Wang, Y.; Xin, X.; Liang, Y.; Lin, Y.; Duan, H.; Dong, D. Adv. Synth. Catal. 2009, 351, 2217–2223. doi:10.1002/adsc.200900310 |
| 6. | Han, M.; Nam, K.-D.; Hahn, H.-G.; Shin, D. Tetrahedron Lett. 2008, 49, 5217–5219. doi:10.1016/j.tetlet.2008.05.132 |
| 7. | Zaleska, B.; Lis, S. Synth. Commun. 2001, 31, 189–197. doi:10.1081/SCC-100000198 |
| 20. | Pierce, J. B.; Ariyan, Z. S.; Ovenden, G. S. J. Med. Chem. 1982, 25, 131–136. doi:10.1021/jm00344a008 |
| 19. | Zhang, Z.; Fang, S.; Liu, Q.; Zhang, G. J. Org. Chem. 2012, 77, 7665–7670. doi:10.1021/jo3010217 |
| 19. | Zhang, Z.; Fang, S.; Liu, Q.; Zhang, G. J. Org. Chem. 2012, 77, 7665–7670. doi:10.1021/jo3010217 |
| 14. | Liu, W.-B.; Chen, C.; Zhang, Q.; Zhu, Z.-B. Beilstein J. Org. Chem. 2011, 7, 1436–1440. doi:10.3762/bjoc.7.167 |
| 15. | Liu, W.-B.; Chen, C.; Zhang, Q.; Zhu, Z.-B. Beilstein J. Org. Chem. 2012, 8, 344–348. doi:10.3762/bjoc.8.38 |
| 16. | Hu, W.; Deng, Z.; Chen, C.; Liu, W.-B. J. Chem. Res. 2012, 36, 571–572. doi:10.3184/174751912X13445852334047 |
| 17. | Liu, W.; Zhou, P.; Chen, C.; Zhang, Q.; Zhu, Z. Org. Biomol. Chem. 2013, 11, 542–544. doi:10.1039/c2ob27145a |
| 18. | Zhang, Q.; Liu, W.; Chen, C.; Tan, L. Chin. J. Chem. 2013, 31, 453–455. doi:10.1002/cjoc.201300007 |
| 35. | Shimada, T.; Nakamura, I.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 10546–10547. doi:10.1021/ja047542r |
| 36. | Liso, G.; Trapani, G.; Reho, A.; Latrofa, A. Tetrahedron Lett. 1981, 22, 1641–1644. doi:10.1016/S0040-4039(01)90399-5 |
| 37. | Mészáros, Z.; Hennecz, I. Tetrahedron Lett. 1975, 16, 1019–1020. doi:10.1016/S0040-4039(00)72632-3 |
| 38. | Hermecz, I.; Mészáros, Z.; Vasvári-Debreczy, L.; Horváth, A.; Horváth, G.; Pongor-Csákvári, M. J. Chem. Soc., Perkin Trans. 1 1977, 789–795. doi:10.1039/P19770000789 |
| 39. | Dimroth, P.; Radtke, V. Liebigs Ann. Chem. 1979, 769–775. doi:10.1002/jlac.197919790606 |
| 8. | Maiti, A.; Reddy, P. V. N.; Sturdy, M.; Marler, L.; Pegan, S. D.; Mesecar, A. D.; Pezzuto, J. M.; Cushman, M. J. Med. Chem. 2009, 52, 1873–1884. doi:10.1021/jm801335z |
| 9. | Zhang, R.; Zhang, D.; Liang, Y.; Zhou, G.; Dong, D. J. Org. Chem. 2011, 76, 2880–2883. doi:10.1021/jo101949y |
| 10. | Zhang, Z.; Zhang, Q.; Yan, Z.; Liu, Q. J. Org. Chem. 2007, 72, 9808–9810. doi:10.1021/jo701551f |
| 11. | Zhang, R.; Zhang, D.; Guo, Y.; Zhou, G.; Jiang, Z.; Dong, D. J. Org. Chem. 2008, 73, 9504–9507. doi:10.1021/jo801959j |
| 12. | Xiang, D.; Wang, K.; Liang, Y.; Zhou, G.; Dong, D. Org. Lett. 2008, 10, 345–348. doi:10.1021/ol702846t |
| 13. | Knapp, J. M.; Zhu, J. S.; Wood, A. B.; Kurth, M. J. ACS Comb. Sci. 2012, 14, 85–88. doi:10.1021/co200199h |
| 20. | Pierce, J. B.; Ariyan, Z. S.; Ovenden, G. S. J. Med. Chem. 1982, 25, 131–136. doi:10.1021/jm00344a008 |
| 38. | Hermecz, I.; Mészáros, Z.; Vasvári-Debreczy, L.; Horváth, A.; Horváth, G.; Pongor-Csákvári, M. J. Chem. Soc., Perkin Trans. 1 1977, 789–795. doi:10.1039/P19770000789 |
| 30. | Woodford, W. J.; Swartz, B. A.; Pillar, C. J.; Kampf, A.; Mertes, M. P. J. Med. Chem. 1974, 17, 1027–1029. doi:10.1021/jm00255a030 |
| 29. | Mao, D. T.; Driscoll, J. S.; Marquez, V. E. J. Med. Chem. 1984, 27, 160–164. doi:10.1021/jm00368a010 |
| 33. | Ohtsuka, Y. J. Org. Chem. 1976, 41, 629–633. doi:10.1021/jo00866a010 |
| 34. | So, Y.-H.; Heeschen, J. P. J. Org. Chem. 1997, 62, 3552–3561. doi:10.1021/jo960441u |
| 24. | Kitagawa, H.; Ozawa, T.; Takahata, S.; Iida, M.; Saito, J.; Yamada, M. J. Med. Chem. 2007, 50, 4710–4720. doi:10.1021/jm0705354 |
| 28. | Mitscher, L. A. Chem. Rev. 2005, 105, 559–592. doi:10.1021/cr030101q |
| 21. | Edwards, M. L.; Erickson, R. C. J. Med. Chem. 1979, 22, 1416–1418. doi:10.1021/jm00197a026 |
| 22. | Liu, J.-K. Chem. Rev. 2005, 105, 2723–2744. doi:10.1021/cr0400818 |
| 23. | Ye, Y. H.; Zhu, H. L.; Song, Y. C.; Liu, J. Y.; Tan, R. X. J. Nat. Prod. 2005, 68, 1106–1108. doi:10.1021/np050059p |
| 24. | Kitagawa, H.; Ozawa, T.; Takahata, S.; Iida, M.; Saito, J.; Yamada, M. J. Med. Chem. 2007, 50, 4710–4720. doi:10.1021/jm0705354 |
| 25. | Stierle, A. A.; Stierle, D. B.; Patacini, B. J. Nat. Prod. 2008, 71, 856–860. doi:10.1021/np0705054 |
| 26. | Maloney, K. N.; MacMillan, J. B.; Kauffman, C. A.; Jensen, P. R.; DiPasquale, A. G.; Rheingold, A. L.; Fenical, W. Org. Lett. 2009, 11, 5422–5424. doi:10.1021/ol901997k |
| 27. | Jessen, H. J.; Schumacher, A.; Schmid, F.; Pfaltz, A.; Gademann, K. Org. Lett. 2011, 13, 4368–4370. doi:10.1021/ol201692h |
| 27. | Jessen, H. J.; Schumacher, A.; Schmid, F.; Pfaltz, A.; Gademann, K. Org. Lett. 2011, 13, 4368–4370. doi:10.1021/ol201692h |
| 31. | Kim, K. S.; Zhang, L.; Schmidt, R.; Cai, Z.-W.; Wei, D.; Williams, D. K.; Lombardo, L. J.; Trainor, G. L.; Xie, D.; Zhang, Y.; An, Y.; Sack, J. S.; Tokarski, J. S.; Darienzo, C.; Kamath, A.; Marathe, P.; Zhang, Y.; Lippy, J.; Jeyaseelan, R., Sr.; Wautlet, B.; Henley, B.; Gullo-Brown, J.; Manne, V.; Hunt, J. T.; Fargnoli, J.; Borzilleri, R. M. J. Med. Chem. 2008, 51, 5330–5341. doi:10.1021/jm800476q |
© 2013 Tan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)