Abstract
Precursors of 3-pyridylnitrene and 2- and 4-pyrimidinylcarbenes all afford mixtures of 2- and 3-cyanopyrroles on flash vacuum thermolysis, but 3-cyanopyrroles are the first-formed products. 3-Quinolylnitrenes and 4-quinazolinylcarbenes similarly afford 3-cyanoindoles. 2-Pyrimidinylcarbenes rearrange to 3-pyridylnitrenes, but 4-pyrimidinylcarbenes and 4-quinazolinylcarbenes do not necessarily rearrange to the corresponding 3-pyridylnitrenes or 3-quinolylnitrenes. The ring contraction reactions are interpreted in terms of ring opening of either the nitrenes or the diazacycloheptatetraenes to nitrile ylides.

Graphical Abstract
Introduction
A multitude of rearrangements of heterocyclic nitrenes have been described in a recent review [1] and in several books [2-5]. This is illustrated by the ring expansion of 4-pyridylnitrene (1) and 2-pyrazinylcarbene (3) to 1,5-diazacyclohepta-1,2,4,6-tetraene (2, Scheme 1) [6]. Similarly, 2-pyridylnitrene (4) interconverts with 1,3-diazacyclohepta-1,2,4,6-tetraene (5) under conditions of photolysis in solution or in matrices as well as under flash vacuum thermolysis (FVT) (Scheme 2) [1,7-10].
![[1860-5397-9-84-i1]](/bjoc/content/inline/1860-5397-9-84-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: 4-Pyridylnitrene–2-pyrazinylcarbene interconversion.
Scheme 1: 4-Pyridylnitrene–2-pyrazinylcarbene interconversion.
![[1860-5397-9-84-i2]](/bjoc/content/inline/1860-5397-9-84-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Ring expansion and ring contraction in 2-pyridylnitrene (4).
Scheme 2: Ring expansion and ring contraction in 2-pyridylnitrene (4).
The end products under FVT conditions are the 2- and 3-cyanopyrroles 7 and 8 [10,11]. A competing ring-opening of 2-pyridylnitrenes to cyanobutadienylnitrenes can lead to the formation of glutacononitriles in low yields [12].
In contrast, 3-pyridylnitrene (10) undergoes a different type of ring opening to the observable nitrile ylide 11 and subsequently the ketenimine 12 (Scheme 3) [13]. Nitrile imines [14] and nitrile ylides [15,16] may have either allenic or propargylic structures, and for this reason their cumulene-type IR absorptions can occur over a wide frequency range, 1900–2300 cm−1, depending on substituents. The IR absorption of 11 was observed at 1961 cm−1, indicating an allenic structure. Nitrene 10 can also undergo ring expansion to two diazacycloheptatetraenes 15 and 16 via the azirenes 13 and 14 (Scheme 3) [13]. The diazacycloheptatetraenes were not observed directly in this study, but aza- and diazacycloheptatetraenes have been observed in several other cases [1,17,18] and the ring-expansion reactions have been the subject of detailed theoretical investigation [19].
![[1860-5397-9-84-i3]](/bjoc/content/inline/1860-5397-9-84-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Ring opening and ring expansion in 3-pyridylnitrene (10).
Scheme 3: Ring opening and ring expansion in 3-pyridylnitrene (10).
Results and Discussion
3-Pyridylnitrene
Flash vacuum thermolysis (FVT) of 3-azidopyridine (9) yields predominantly 3-cyanopyrrole (8, Scheme 4). Under the mildest conditions, FVT of 9 at 370 °C/10−3 mbar generates 8 and 7 in a 3:1 ratio. A temperature increase to 500 °C causes the ratio to drop to 1.2:1 due to the thermal interconversion of 7 and 8 [10,20]. The reaction mechanism was probed by calculations at the B3LYP/6-31G* level, which has been found to be adequate for other, similar reactions [11,13,17,21]. The transition state for the ring opening of the cyclic ketenimine 16 to the (s,Z)-nitrile ylide 11 lies 5 kcal/mol above the open shell singlet nitrene S1 10, i.e., the barrier is only 11 kcal/mol above the cyclic ketenimine 16. This is lower than the barrier for direct ring opening of the nitrene 10 itself (16 kcal/mol) (Scheme 4 and Figure 1) [13]. Thus, while both the nitrene 10 and the ketenimine 16 may undergo ring opening with rather low activation barriers, the ring opening of the ketenimine has the lowest barrier. Both of these reactions can take place with ease under conditions of FVT.
![[1860-5397-9-84-i4]](/bjoc/content/inline/1860-5397-9-84-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Ring opening and ring contraction in 3-pyridylnitrene (10) and diazacycloheptatetraene 16.
Scheme 4: Ring opening and ring contraction in 3-pyridylnitrene (10) and diazacycloheptatetraene 16.
![[1860-5397-9-84-1]](/bjoc/content/figures/1860-5397-9-84-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Energy profile for the ring opening and ring contraction in 3-pyridylnitrene 10 and 1,6-diazacyclohepta-1,2,4,6-tetraene (16, energies in kcal/mol at the B3LYP/6-31G* level).
Figure 1: Energy profile for the ring opening and ring contraction in 3-pyridylnitrene 10 and 1,6-diazacycloh...
With a low barrier for ring opening, recyclization of the nitrile ylide 11 now becomes the energetically most favourable mechanism of formation of 3-cyanopyrrole (8) via the 3H tautomer 18 with a barrier of only 10 kcal/mol above the S1 nitrene or 16 kcal/mol above the nitrile ylide (Scheme 4 and Figure 1). This explains why 3-cyanopyrrole (8) is the predominant isomer, in contrast to the reaction of 2-pyridylnitrene, where 2-cyanopyrrole is formed preferentially [11]. In the analogous case of 3-quinolylnitrene, 3-cyanoindole is formed exclusively under mild FVT conditions [21]. A direct, concerted ring contraction in 3-pyridylnitrene would be possible (via 17 and 18, Scheme 5), but such reactions have considerably higher activation barriers, ca. 30 kcal/mol in the case of phenylnitrene [22]. A transition state for the concerted ring contraction of 3-pyridylnitrene (10) to 3H-3-cyanopyrrole (18) was not found, but we calculate a barrier of 23 kcal/mol for the concerted ring contraction to 2-cyano-2H-pyrrole (17, Figure 1). Furthermore, a higher proportion of 2-cyanopyrrole would be expected if Scheme 5 was operating. Concerted ring contraction in the 7-membered ring ketenimine 16 is also possible [22], but this type of reaction has an even higher activation energy. The lowest-energy path for (di)azacycloheptatetraenes to undergo ring contraction is by ring opening.
![[1860-5397-9-84-i5]](/bjoc/content/inline/1860-5397-9-84-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Potential direct ring contraction in 3-pyridylnitrene (10).
Scheme 5: Potential direct ring contraction in 3-pyridylnitrene (10).
The nitrile ylide 11 is not directly observable under FVT conditions, because the barriers for its reactions are very low. The calculated barrier for cyclization to 3-cyano-3H-pyrrole (18) is 16 kcal/mol (Scheme 6 and Figure 1).
![[1860-5397-9-84-i6]](/bjoc/content/inline/1860-5397-9-84-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Ring contraction by ring opening to nitrile ylide 11.
Scheme 6: Ring contraction by ring opening to nitrile ylide 11.
2- and 4-pyrimidinylcarbenes
FVT of tetrazolylpyrimidines of type 19 causes elimination of N2 to generate 4- and 2-diazomethylpyrimidines 20, which can ring-close to the corresponding 1,2,3-triazolopyrimidines 21 (Scheme 7) [23]. Endothermic ring-chain valence isomerization of the type 21 → 20, with free energies of activation of 18–22 kcal/mol in solution, has been demonstrated for 1,2,3-triazolo[1,5-a]pyrimidines [24] but not for 1,2,3-triazolo[1,5-c]pyrimidines [25,26]. However, 7-benzyl-3-ethoxycarbonyl-1,2,3-triazolo[1,5-c]pyrimidin-5-ol and its diazo valence tautomer, 6-(2-diazoethoxycarbonylmethylene)-2-(α-hydroxybenzyl)pyrimidin-4-(2H)-one, have been reported [27].
![[1860-5397-9-84-i7]](/bjoc/content/inline/1860-5397-9-84-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Generation of pyrimidinyldiazomethanes and pyrimidinylcarbenes.
Scheme 7: Generation of pyrimidinyldiazomethanes and pyrimidinylcarbenes.
We find that FVT of the 4- and 2-(5-tetrazolyl)pyrimidines 22–24 also affords cyanopyrroles (Scheme 8). FVT of 2-(5-tetrazolyl)pyrimidine (22) affords a ca. 1:1 ratio of 2- and 3-cyanopyrroles (Scheme 8). The results of FVT of tetrazoles 23 and 24 are collected in Table 1. The formation of different mixtures of the three cyanodimethylpyrroles 25–27 depending on the conditions can be explained in terms of chemical activation. The formation of (hetero)arylcarbenes and their rearrangement to (hetero)arylnitrenes and cyanopyrroles are strongly exothermic reactions. Consequently, when the reaction is performed in the low-pressure gas phase, the reaction products carry excess thermal (rovibrational) energy, which facilitates the sigmatropic shifts of H, CN, and CH3, which will cause interconversion of the cyanopyrroles [28]. In many cases, this cannot be completely avoided, even by using the mildest possible FVT temperatures, but an increase in pressure (1 hPa N2) will help to remove excess thermal energy and so preserve the initial reaction products. Therefore, as seen in Table 1, it can be concluded that the dimethylcyanopyrrole 25 is the primary reaction product of 23, and 27 is the predominant product from 24. That chemical activation is the cause is seen in the fact that FVT of the individual dimethylcyanopyrroles 25 and 27 afford mixtures very similar to those obtained from 23 and 24, respectively, but a temperature of 800 °C is required for this, whereas 400 °C suffices in the FVT of the tetrazoles (Table 1).
![[1860-5397-9-84-i8]](/bjoc/content/inline/1860-5397-9-84-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Formation of cyanopyrroles by FVT of tetrazolylpyrimidines.
Scheme 8: Formation of cyanopyrroles by FVT of tetrazolylpyrimidines.
Table 1: Dimethylcyanopyrroles from tetrazolylpyrimidines.
| Starting material | Conditions | Products, rel. yields (%) | ||
|---|---|---|---|---|
| T (°C)/p (hPa) | 25 | 26 | 27 | |
| 23 | 400/10−3 | 63 | 29 | 7 |
| 400/1 N2 | 98 | 2 | <0.5 | |
| 600/10−1 | 79 | 19 | 2 | |
| 24 | 400/10−3 | 34 | 23 | 43 |
| 400/1 N2 | 21 | 10 | 69 | |
| 600/10−1 | 25 | 23 | 44a | |
| 25 | 800/10−3 | 49 | 32 | 14a |
| 27 | 800/10−3 | 22 | 22 | 50a |
a6–8% yields of other, isomeric dimethylcyanopyrroles were also formed.
Following the analysis of the ring contraction in 3-pyridylnitrene (10) by ring expansion and ring opening to a nitrile ylide (Scheme 4 and Figure 1), we can interpret the reactions in terms of ring expansion of the pyrimidinylcarbenes 28 and 33 to diazacycloheptatetraenes 29 and 34, ring contraction to 3-pyridylnitrenes 30 and 35 and/or ring opening to nitrile ylides 31 and 37, and ring closure to cyanopyrroles (Scheme 9).
![[1860-5397-9-84-i9]](/bjoc/content/inline/1860-5397-9-84-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Rearrangements of pyrimidinylcarbenes to cyanopyrroles via nitrile ylides 31 and 37.
Scheme 9: Rearrangements of pyrimidinylcarbenes to cyanopyrroles via nitrile ylides 31 and 37.
In the case of the 4-pyrimidinylcarbene 28 a direct ring opening of the diazacycloheptatetraene 29 to the nitrile ylide 31 is possible. However, in the case of the 2-pyrimidinylcarbene 33, the first-formed diazacycloheptatetraene 34 cannot open directly to a nitrile ylide but must first rearrange to the 3-pyridylnitrene 35. Either the 3-pyridylnitrene 35 or the second diazacycloheptatetraene 36 may then undergo ring opening to the nitrile ylide 37. Sigmatropic shifts of H, CN, or CH3 in the 3H-pyrroles 32 and 38 lead to the final products.
We have previously reported strong evidence for the ring expansion of a 2-pyrimidinylcarbene 39 to a diazacycloheptatetraene 40 and subsequent ring contraction to a 3-pyridylnitrene 41, which undergoes cyclization to afford the pyridoindole (4-azacarbazole) 42 (Scheme 10) [29].
![[1860-5397-9-84-i10]](/bjoc/content/inline/1860-5397-9-84-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Rearrangements of phenyl(dimethylpyrimidinyl)carbene.
Scheme 10: Rearrangements of phenyl(dimethylpyrimidinyl)carbene.
The alternative ring expansion/ring contraction/recyclization to the pyrimidoisoindole 43 also takes place [29]. The formation of 42 is important, as it demonstrates that 2-pyrimidinylcarbenes can rearrange to 3-pyridylnitrenes, whereas 4-pyrimidinylcarbenes do not necessarily rearrange to 3-pyridylnitrenes (see 55 below).
2-Phenyl-3-quinolylnitrene
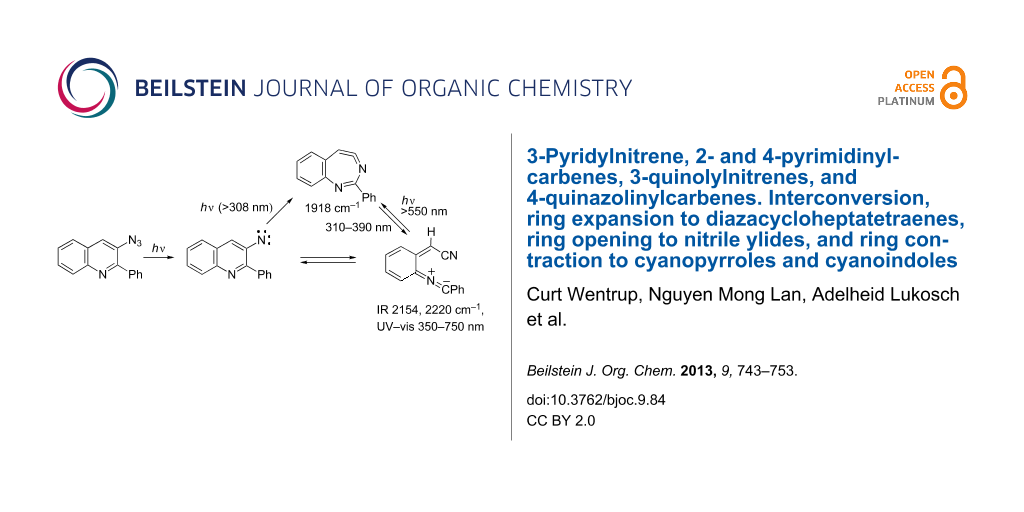
Matrix photolysis of 3-azido-2-phenylquinoline (44) at λ = 308 nm or 310–390 nm affords a blue nitrile ylide 47 (observed IR 2220, 2154 cm−1; calcd (B3LYL/6-31G*) 2233, 2171 cm−1; UV–vis λmax 400 and 680 nm) (Scheme 11, Figure 2 and Figure 3).
![[1860-5397-9-84-i11]](/bjoc/content/inline/1860-5397-9-84-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Photolysis of 3-azido-2-phenylquinoline.
Scheme 11: Photolysis of 3-azido-2-phenylquinoline.
![[1860-5397-9-84-2]](/bjoc/content/figures/1860-5397-9-84-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: IR difference spectra from the photolysis of 3-azido-2-phenylquinoline (44) in Ar matrix. (a) Calculated spectrum of 46. (b) Initial difference spectrum after 240 s; the negative peaks are due to the azide. (c)–(e) Further irradiation as indicated for 900, 400 and 600 s, respectively. (f) Calculated spectrum of 47. Ordinate in arbitrary absorbance units. See also Figures S1–S3 in Supporting Information File 1.
Figure 2: IR difference spectra from the photolysis of 3-azido-2-phenylquinoline (44) in Ar matrix. (a) Calcu...
![[1860-5397-9-84-3]](/bjoc/content/figures/1860-5397-9-84-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra from the sequential photolysis of 3-azido-2-phenylquinoline (44) in Ar matrix at 310–390 nm and 550 nm. Abscissa in nanometres and ordinate in arbitrary absorbance units. These spectra correspond to the IR spectra in Figure 2. See Supporting Information File 1 for the calculated electronic transitions.
Figure 3: UV–vis spectra from the sequential photolysis of 3-azido-2-phenylquinoline (44) in Ar matrix at 310...
It could be converted to the seven-membered ring ketenimine 46 (IR: 1920; calcd: 1921 cm−1) on photolysis above 550 nm. The ketenimine 46 was again converted to ylide 47 on photolysis at 310–390 nm. We assume the nitrene 45 is formed initially, but it is converted to 46 and 47 at the same wavelength. As in the examples described above, the ring opening to the ylide 47 may take place either from the nitrene or from the cyclic ketenimine, although we only observed the latter reaction directly. It was possible to cycle many times between these two intermediates (Figure 2 and Figure 3), but eventually signal intensity was lost, probably because another reaction, the cyclization to the isocarbazole 49, took place. This reaction is analogous to the photocyclization of o-biphenylylnitrene to isocarbazole [30]. As shown below, the indoloquinoline 50 is indeed a major product under FVT conditions. It is possible that 49 could contribute to the observed visible spectrum, but it obviously cannot explain the IR spectrum. The same visible spectrum was obtained by photolysis of 44 embedded in a PVC film at 11 K, and the carrier of the spectrum was stable up to 90 K, as can be expected of a highly reactive nitrile ylide 47. The calculated IR and UV–vis spectra of 45, 46, 47 and 49 are listed in Supporting Information File 1.
Preparative FVT of azide 44 at 400–800 °C affords the indoloquinoline 50 in 52–60% yield (Scheme 12). Importantly, smaller amounts of amine 51 and the ring-contraction product 2-phenyl-3-cyanoindole (48, 21–15%) were also isolated.
![[1860-5397-9-84-i12]](/bjoc/content/inline/1860-5397-9-84-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Preparative FVT of 3-azido-2-phenylquinoline.
Scheme 12: Preparative FVT of 3-azido-2-phenylquinoline.
Formation of amines is diagnostic for triplet nitrenes, even in low-pressure gas-phase reactions [10,22]. The 3-cyanoindole 48 is expected to be formed by cyclization of the nitrile ylide 47 in the same way that the 3-cyanoindole is obtained from the unsubstituted 3-quinolylnitrene [21]. The three products, 48, 50 and 51 were formed even on thermolysis of 44 in solution.
4-Quinazolinylcarbenes
The tetrazolylquinazoline 52 and the triazoloquinazoline 53 undergo pyrolysis via the diazomethylquinazoline 54 (Scheme 13) [23]. Both afforded the 3-cyanoindole 48 in nearly quantitative yield (up to 98%) on FVT at 400–600 °C, but no traces of either indoloquinoline 50 or the 3-aminoquinoline 51 were detectable. The same was true when the thermolysis was performed in solution. In other words, nitrene 45 was not formed (Scheme 13).
![[1860-5397-9-84-i13]](/bjoc/content/inline/1860-5397-9-84-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: FVT of 2-phenyl-4-quinazolinylcarbene precursors.
Scheme 13: FVT of 2-phenyl-4-quinazolinylcarbene precursors.
This is in agreement with the conclusion reached for 4-quinazolinylcarbene itself [21], namely that ring contraction takes place via the pathway 2-phenyl-4-quinazolinylcarbene (55) → cyclic ketenimine 46 → nitrile ylide 47 → nitrile 48. The data demonstrate that carbene 55 does not convert thermally to nitrene 45. In other words, although carbene–nitrene rearrangements are known to occur [1,19] (see, e.g., Scheme 1 and Scheme 10 above), they may be bypassed when ring opening to nitrile ylides becomes more favourable.
Conclusion
The rearrangement of 2- and 4-pyrimidinylcarbenes 28 and 33 to mixtures of 2- and 3-cyanopyrroles on FVT implies prior isomerization to 3-pyridylnitrenes and/or diazacycloheptatetraenes and nitrile ylides. 3-Quinolylnitrenes and 4-quinazolinylcarbenes afford 3-cyanoindoles 48 on FVT. In addition, 2-phenyl-3-quinolylnitrene (45) cyclizes to indoloquinoline 50, but this compound is not formed at all from the isomeric 2-phenyl-4-quinazolinylcarbene (55). This demonstrates that carbene 55 does not isomerize to nitrene 45. Instead, the ring contraction to 48 takes place via the nitrile ylide 47. Matrix photolysis of 2-phenyl-3-azidoquinoline (44) revealed a reversible photochemical interconversion of the benzodiazacycloheptatetraene 46 and the nitrile ylide 47.
Experimental
General
The apparatus and procedures for preparative FVT [31] and for Ar matrix isolation [11,32,33] were as previously described. KBr and CsI windows were used for IR spectroscopy. FVT products were isolated in liquid nitrogen (77 K) in the preparative thermolysis, and at 22–25 K in Ar matrices for IR experiments. IR spectra of the Ar matrices were measured at 7–10 K with a resolution of 1 cm−1. Photolyses were done through quartz by using a 75 W low pressure Hg lamp (254 nm) or a 1000 W high pressure Hg/Xe lamp equipped with a monochromator and appropriate filters. A water filter was used to remove infrared radiation, and a 7.5% NiSO4 or a NiSO4/CuSO4 solution (7.5 and 2.5%, respectively) to remove visible light where required. Analytical gas chromatography used a 10% OV 17 column programmed at 120–180 °C at 2°/min with N2 as carrier gas at 0.6 bar. Preparative gas chromatography used a 20% Carbowax column at 200 °C with H2 as carrier gas at 60 mL/min.
Materials
3-Azidopyridine (9) [34,35], 2-(5-tetrazolyl)pyrimidine (22) [36,37], 2-phenyl-4-(5-tetrazolyl)quinazoline (52) [23], and 5-phenyl-1,2,3-triazolo[1,5-a]quinazoline (53) [23] were prepared according to literature procedures. 4,6-Dimethyl-2-(5-tetrazolyl)pyrimidine (24) and 2,4-dimethyl-6-(5-tetrazolylpyrimidine) (23) were prepared from the dimethylpyrimidine-carbonitriles [38,39] by adaptation of the literature method [36,37].
4,6-Dimethyl-2-(5-tetrazolyl)pyrimidine (24): From 3.0 g (22.5 mmol) of 4.6-dimethylpyrimidine-2-carbonitrile was obtained 2.8 g (71%); mp 209–210 °C (dec); 1H NMR (CDCl3) 12.0 (very broad), 7.37 (s, 1H), 2.50 (s, 6H); MS m/z: 176 (M+, 21), 148 (33), 134 (46), 120 (100), 119 (71), 107 (36), 105 (12), 93 (13), 92 (16 ), 80 (15), 67 (76), 66 (56), 53 (26); anal. calcd for C7H8N6: C, 47.72; H. 4.58; N, 47.70; found: C, 47.89; H, 4.40; N, 47.52.
2,4-Dimethyl-6-(5-tetrazolyl)pyrimidine (23): Prepared as the preceding entry in 41% yield; mp 208–209 °C (dec); 1H NMR (CDCl3) 7.43 (s, 1H), 2.66 (s, 3H), 2.56 (s, 3H); MS m/z: 176 (M+, 49), 148 (12), 135 (10), 134 (100), 120 (19), 119 (65), 107 (42), 93 (17), 97 (15), 78 (17), 66 (66), 52 (54); anal. calcd for C7H8N6: C, 47.72; H. 4.58; N, 47.70; found: C, 47.90; H, 4.80; N, 47.75.
3-Azido-2-phenylquinoline (44): This compound was prepared according to a standard literature procedure [40]. A sample of 2.54 g (11.5 mmol) of 3-amino-2-phenylquinoline [41] was dissolved in a mixture of 2 mL of conc. sulfuric acid (d = 1.84) and 13 mL water at 40 °C and then cooled to 0 °C. A solution of 900 mg (13 mmol) of NaNO2 in 8 mL water was added dropwise at 0 °C, causing the formation of a yellow, crystalline diazonium sulfate, which was not isolated. After 30 min at 0 °C a solution of 11 g (16.9 mmol) of NaN3 in 7 mL water was added. An amorphous, yellow precipitate formed immediately with simultaneous evolution of N2. After stirring for 3 h at rt, the yellow solid was filtered and recrystallized from petroleum ether, yielding 2.33 g (82%) of the azide, mp 93–94 °C; 1H NMR (DMSO-d6) 8.41 (s, 1H), 8.05–8.01 (m, 2H), 7.87–7.83 (m, 2H), 7.73 (dd, J = 8.2 Hz, J = 1.2 Hz, 1H), 7.64 (dd, J = 8.2 Hz, J = 1.2 Hz, 1H), 7.53–7.47 (m, 3H); 13C NMR (DMSO-d6) 151.4, 144.0, 137.3, 132.2, 129.6, 129.1, 128.9, 128.7, 127.9, 127.5, 127.4, 126.7, 124.9; IR (KBr): 3030 (w), 2100 (s), 1590 (w), 1485 (m), 1415 (s), 1340 (s), 1288 (m), 1270 (s), 1255 (m), 1245 (m), 1230 (m), 955 (m), 895 (s), 860 (m), 770 (s), 750 (s), 710 (s), 695 (s) cm−1; UV (CH3CN) λmax: 257, 260, 337 nm; MS m/z: 246 (M+, 5), 220 (9), 219 (26), 218 (100), 190 (8), 115 (20), 88 (9), 78 (3); anal. calcd for C15H10N4: C, 73.16; H, 4.09; N, 22.75; found: C, 73.10; H, 4.18; N, 22.68.
FVT of 3-azidopyridine (9): The azide (0.50 g) was distilled into the FVT apparatus from a sample flask held at −30 °C and thermolysed at 370–500 °C/10−3–10−4 mbar in the course of 2 h. The pyrolysate was examined by GC/MS and 1H NMR spectroscopy. The 2- and 3-cyanopyrroles 7 and 8 were separated by flash chromatography on silica gel 60, eluted with hexane/ethyl acetate 3:7 and identified by comparison with authentic materials reported previously [10,20]. Yields were determined by GC as previously described [10]. The following ratios of 3- to 2-cyanopyrroles at different temperatures were obtained: 370 °C: 2.29:1 (81% 3-azidopyridine remained unreacted); 400 °C: 1.63:1 (36% 3-azidopyridine remained unreacted); 500 °C: 1.48:1 (6% 3-azidopyridine remained unreacted). GC/MS retention times and molecular masses: 2-cyanopyrrole 3.60 min (m/z 92); 3-cyanpyrrole 5.45 min (m/z 92); 3-azidopyridine 2.19 min (m/z 120); 1H NMR (CDCl3) 2-cyanopyrrole: 9.01 (br, 1H), 6.98 (m, 1H), 6.88 (m, 1H), 6.28 (m, 1H); 3-cyanopyrrole: 8.80 (br, 1H), 7.36 (m, 1H), 6.84 (m, 1H), 6.53 (m, 1H); IR (Ar, 20 K): 2-cyanopyrrole: 2236 cm−1; 3-cyanopyrrole: 2234 and 2247 cm−1.
FVT of 2-(5-tetrazolyl)pyrimidine (22): A portion of 22 (300 mg) was sublimed into the pyrolysis tube at 170 °C and pyrolysed at 600 °C/10−3 mbar in the course of 12 h. The resulting pyrolysate (120 mg) was identified as a 1:1 mixture of 2- and 3-cyanopyrroles 7 and 8 by comparison of the 1H NMR, IR and mass spectra with those of authentic materials. Yields were determined by GC as above [10].
FVT of 2,4-dimethyl-6-(5-tetrazolyl)pyrimidine (23) and 4,6-dimethyl-2-(5-tetrazolyl)pyrimidine (24): A portion of 23 or 24 (500 mg) was sublimed at 150 °C and pyrolysed at 10−3 mbar in the course of 12–18 h. The pyrolyzates were extracted with diethyl ether, and the products were analysed and separated by GC on the Carbowax column. The yields of 2,6-dimethyl-3-cyanopyrrole (25), 2,4-dimethyl-3-cyanopyrrole (26) and 3,5-dimethyl-2-cyanopyrrole (27) at various temperatures are collected in Table 1. Yields were determined by GC, and the pyrroles were characterized as follows:
2,6-Dimethyl-3-cyanopyrrole (25): Retention time 63 min. 1H NMR (CDCl3) 8.4 (br, 1H), 5.95 (m, 1H), 2.38 (s, 3H), 2.20 (s, 3H); IR (KBr): 3470 (br), 2210 (s), 1600 (w), 1430 (w), 1050 (m), 780 (m); UV (EtOH) λmax: 245 nm; MS m/z: 120 (M+, 67), 119 (100), 106 (28), 105 (71), 78 (10), 65 (3). The spectra data were in agreement with literature values [42].
2,4-Dimethyl-3-cyanopyrrole (26): Retention time 57 min; 1H NMR (CDCl3) 8.60 (br, 1H), 6.38 (m, 1H), 2.38 (s 3H), 2.12 (s, 3H); IR (KBr): 3460 (br), 2210 (s), 1580 (w), 1400 (m), 1110 (m), 800 (m); MS m/z: 120 (M+, 55), 119 (100), 106 (25), 105 (60), 78 (12), 65 (3); UV (EtOH) λmax: 240 nm. The spectra data were in agreement with literature values [42].
3,5-Dimethyl-2-cyanopyrrole (27): Residence time 31 min; 1H NMR (CDCl3) 9.0 (br, 1H), 5.76 (m, 1H), 2.23 (s, 3H), 2.17 (s, 3H); IR (KBr): 3460 (br), 2210 (s), 1580 (w), 1460 (w), 1390 (w), 1300 (w), 1270 (m), 800 (m); MS m/z: 120 (M+, 73), 119 (100), 106 (48), 105 (86), 92 (8), 78 (14), 65 (10); UV (EtOH) λmax: 257, 234 nm. The spectra data were in agreement with literature values [42]. Anal. calcd for C7H8N2: C, 69.97; H, 6.71; N, 23.31; found: C, 69.61; H, 6.31; N, 23.21
Matrix photolysis of 3-azido-2-phenyl-3-quinoline (44): The azide was sublimed at 80 °C and deposited with Ar at 22 K to form a matrix. Principal absorptions of the azide at 11 K: 2100, 1590, 1420, 1340, 1270, 1100 cm−1. Irradiation of the azide 44 at 308 nm or at 310–390 nm for 30 s afforded the nitrile ylide 47 together with a smaller amount of the cyclic ketenimine 46 (2220, 2154, 1920, 1610, 1516, 1500, 1450, 1390, 1088 cm−1). Further photolysis for 210 s caused additional formation of the ylide 47 (IR, Ar, 10 K: 2220, 2154, 1609, 1516, 1453, 1088, 740 cm−1 (Figure 2); UV–vis λmax ca. 350 and 585, 635, 700, 775 nm (Figure 3)). Subsequent irradiation at λ > 550 nm bleached the long-wavelength band in the visible spectrum and the IR bands at 2154 and 2220 cm−1 in the IR. The intensity of the band at 1918 cm−1 due to 46 increased substantially at the same time (Figure 2). Renewed irradiation at 310–390 nm caused diminution of the IR bands ascribed to 46 and reformation of the IR and UV–vis bands ascribed to the nitrile ylide 47. It was possible to cycle several times between these two species by using λ > 550 nm and λ = 310–390 nm, respectively (Figure 2 and Figure 3).
FVT of 3-azido-2-phenyl-3-quinoline (44): (a) A sample of the azide (0.2 g, 0.8 mmol) was sublimed at 60–80 °C and pyrolysed at 500 °C/10−4 mbar in the course of 3 h. The products were separated by flash chromatography on silica gel, eluting with chloroform. Indolo[3,2-b]quinoline (50) was obtained in 56–65% yield in different experiments: mp 250–251 °C (lit. [43] 248 °C; lit. [44] 251–252 °C); 1H NMR (DMSO-d6) 11.40 (s, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.27 (s, 1H), 8.18 (d, J = 8.3 Hz, 1H), 8.09 (d, J = 8.3 Hz, 1H), 7.66–7.52 (m, 4H), 7.28 (dd, J = 7.4 Hz, J = 1.0 Hz, 1H); 13C NMR 145.6, 144.0, 143.3, 132.4, 129.6, 128.5, 127.4, 126.6, 125.9, 124.7, 121.3, 120.9, 119.2, 113.0, 111.4; IR (KBr): 3300–3000 (br), 1610 (s), 1490 (s), 1460 (m), 1400 (s), 1340 (s), 1220 (s), 1120 (m), 750 (m), 730 (s) cm−1; UV (EtOH) λmax: 347, 276 nm; MS m/z: 219 (M+ + 1, 16), 218 (M+, 100), 217 (16), 190 (15), 109 (M++, 11), 108 (10), 96 (17), 95 (10), 89 (20), 77 (10). 3-Cyano-2-phenylindole (48) was obtained in 18–20% yield in different experiments: mp 241–243 °C (lit. [45] 246–248 °C, lit. [46] 240 °C); 1H NMR (CDCl3) 8.91 (br s, 1H), 7.89–7.96 (m, 2H), 7.76 (dd, J = 7.2 Hz, J = 0.6 Hz, 1H), 7.56–7.42 (m, 4H), 7.35–7.27 (m, 2H); 13C NMR (CDCl3) 144.7, 135.5, 129.9, 129.4, 129.2, 128.3, 126.9, 123.8, 122.0, 118.3, 117.0, 112.6, 81.4; IR (KBr): 3240–3180 (br s), 2220 (s), 1490 (m), 1450 (s), 1420 (m), 1370 (w), 1320 (br w), 1240 (m), 770 (m), 730 (s), 710 (w), 680 (m); MS m/z: 219 (M+ + 1, 17), 218 (M+, 100), 190 (9), 115 (5), 109 (8), 96 (6). (b) In analogous experiments at 450–700 °C the products of FVT were condensed on a KBr window at 77 K for IR spectroscopy. In each case the product was a mixture of 3-cyano-2-phenylindole and indolo[3,2-b]quinoline according to IR spectroscopy of the crude and TLC of the isolated material.
Thermolysis of 3-azido-2-phenylquinoline (44) in solution: A solution of 200 mg (0.81 mmol) of the azide in xylene was heated under reflux for 3 days. After distillation of the solvent, the residue was chromatographed on silica, eluting with petroleum ether/chloroform to yield 3-cyano-2-phenylindole (48, 3 mg; 2%), 3-amino-2-phenylquinoline (51, 80 mg; 45%), and indolo[3,2-b]quinoline (52 mg; 29%).
FVT of 2-phenyl-4-(5-tetrazolyl)quinazoline (52): (a) A sample of 300 mg (1.10 mmol) was sublimed at 150 °C and pyrolysed at 600 °C/10−3–10−4 mbar in the course of 12 h. Chromatography yielded 178 mg (75%) of 3-cyano-2-phenylindole (48). (b) In similar experiments with FVT at 600–800 °C the product was isolated on a KBr window at 77 K for IR spectroscopy. In each case 3-cyano-2-phenylindole (48) was the exclusive product.
Thermolysis of 2-phenyl-4-(5-tetrazolyl)quinazoline (52) in solution: (a) A solution of 250 mg (0.91 mmol) in 50 mL of xylene was heated under reflux for 6 d. Chromatography after distillation of the solvent yielded 166 mg (74%) 2-phenyl-1,2,3-triazolo[1,5-c]quinazoline 53, mp 192–193 °C (dec) (lit. [23] 192–193 °C (dec)). (b) A solution of 50 mg (0.18 mmol) in 10 mL of diphenylmethane was heated at 180 °C for 1 h. After distilling the solvent vacuum and chromatography of the residue, 6 mg (15%) of 3-cyano-2-phenylindole (48) was obtained.
FVT of 2-phenyl-1,2,3-triazolo[1,5-c]quinazoline (53): (a) A sample of 50 mg (0.20 mmol) was sublimed at a temperature increasing gradually to 190 °C and pyrolysed at 660 °C/10−3 mbar. The product (43 mg; 98%) consisted exclusively of 3-cyano-2-phenylindole (48). (b) In similar experiments with FVT at 300–600 °C the product was isolated on a KBr window at 77 K for IR spectroscopy. No reaction was observable below 400 °C. The 4-diazomethyl-2-phenylquinazoline (54) was detectable by absorption at 2095 cm−1 in the 400 °C experiment. In each case, at FVT temperatures of 400–600 °C, 3-cyano-2-phenylindole (48) was the only other identified product, characterized by its absorption at 2220 cm−1.
Thermolysis of 2-phenyl-1,2,3-triazolo[1,5-c]quinazoline (53) in solution: A solution of 20 mg (0.08 mmol) in 5 mL of diphenylmethane was heated at 180 °C for 1 h. Chromatography of the resulting mixture yielded 2 mg (10%) of 3-cyano-2-phenylindole (48).
Supporting Information
| Supporting Information File 1: Computational details, calculated and experimental IR spectra, and calculated electronic transitions. | ||
| Format: PDF | Size: 645.0 KB | Download |
References
-
Wentrup, C. Acc. Chem. Res. 2011, 44, 393. doi:10.1021/ar700198z
Return to citation in text: [1] [2] [3] [4] -
Lwowski, W. Nitrenes; Wiley-VCH: New York, NY, USA, 1970.
Return to citation in text: [1] -
Scriven, E. F. V., Ed. Azides and Nitrenes; Academic Press: Orlando, FL, USA, 1984.
Return to citation in text: [1] -
Bräse, S.; Banert, K. Organic Azides – Synthesis and Applications; Wiley-VCH: Chichester, UK, 2010.
Return to citation in text: [1] -
Falvey, D. E.; Gudmundsdottir, A. D., Eds. Nitrenes and Nitrenium Ions; Wiley-VCH: Hoboken, NJ, USA, 2013.
Return to citation in text: [1] -
Wentrup, C.; Reisinger, A.; Kvaskoff, D. Beilstein J. Org. Chem. 2013, 9, 754–760. doi:10.3762/bjoc.9.85
Return to citation in text: [1] -
Reisinger, A.; Bernhardt, P. V.; Wentrup, C. Org. Biomol. Chem. 2004, 2, 246. doi:10.1039/b311247k
Return to citation in text: [1] -
Reisinger, A.; Koch, R.; Bernhardt, P. V.; Wentrup, C. Org. Biomol. Chem. 2004, 2, 1227. doi:10.1039/b317099c
Return to citation in text: [1] -
Addicott, C.; Wentrup, C. Aust. J. Chem. 2008, 61, 592. doi:10.1071/CH08252
Return to citation in text: [1] -
McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Kvaskoff, D.; Bednarek, P.; Wentrup, C. J. Org. Chem. 2010, 75, 1600. doi:10.1021/jo902570d
Return to citation in text: [1] [2] [3] [4] -
Wentrup, C.; Kvaskoff, D. Aust. J. Chem. 2013, 66, 286–296. doi:10.1071/CH12502.
Return to citation in text: [1] -
Bednarek, P.; Wentrup, C. J. Am. Chem. Soc. 2003, 125, 9083. doi:10.1021/ja035632a
Return to citation in text: [1] [2] [3] [4] -
Bégué, D.; Qiao, G. G.; Wentrup, C. J. Am. Chem. Soc. 2012, 134, 5339. doi:10.1021/ja2118442
Return to citation in text: [1] -
Orton, E.; Collins, S. T.; Pimentel, G. C. J. Phys. Chem. 1986, 90, 6139. doi:10.1021/j100281a018
Return to citation in text: [1] -
Rajam, S.; Murthy, R. S.; Jadhav, A. V.; Li, Q.; Keller, C.; Carra, C.; Pace, T. C. S.; Bohne, C.; Ault, B. S.; Gudmundsdottir, A. D. J. Org. Chem. 2011, 76, 9934. doi:10.1021/jo200877k
Return to citation in text: [1] -
Kvaskoff, D.; Vosswinkel, M.; Wentrup, C. J. Am. Chem. Soc. 2011, 133, 5413. doi:10.1021/ja111155r
Return to citation in text: [1] [2] -
Crabtree, K. N.; Hostetler, K. J.; Munsch, T. E.; Neuhaus, P.; Lahti, P. M.; Sander, W.; Poole, J. S. J. Org. Chem. 2008, 73, 3441. doi:10.1021/jo8001936
Return to citation in text: [1] -
Karney, W. L.; Borden, W. T. Differences between Phenylcarbene and Phenylnitrene and the Ring Expansion Reactions they Undergo. In Advances in Carbene Chemistry; Brinker, U. H., Ed.; Elsevier: Amsterdam, 2001; Vol. 3, p 205.
Return to citation in text: [1] [2] -
Wentrup, C.; Crow, W. D. Tetrahedron 1970, 26, 3965. doi:10.1016/S0040-4020(01)93037-6
Return to citation in text: [1] [2] -
Kvaskoff, D.; Mitschke, U.; Addicott, C.; Finnerty, J.; Bednarek, P.; Wentrup, C. Aust. J. Chem. 2009, 62, 275. doi:10.1071/CH08523
Return to citation in text: [1] [2] [3] [4] -
Kvaskoff, D.; Bednarek, P.; George, L.; Pankajakshan, S.; Wentrup, C. J. Org. Chem. 2005, 70, 7947. doi:10.1021/jo050898g
Return to citation in text: [1] [2] [3] -
Wentrup, C. Helv. Chim. Acta 1978, 61, 1755. doi:10.1002/hlca.19780610522
Return to citation in text: [1] [2] [3] [4] [5] -
Tennant, G.; Vevers, R. J. S. J. Chem. Soc., Chem. Commun. 1974, 671. doi:10.1039/C3974000671B
Return to citation in text: [1] -
Maury, G.; Paugam, J.-P.; Pougam, R. J. Heterocycl. Chem. 1978, 15, 1041. doi:10.1002/jhet.5570150627
Return to citation in text: [1] -
Abarca, B.; Ballesteros, R.; Chadlaoui, M.; Miralles, J.; Murillo, J. V.; Colonna, D. Tetrahedron 2001, 57, 10111. doi:10.1016/S0040-4020(01)01053-5
Return to citation in text: [1] -
Ezema, B. E.; Akpanisi, L. E. S.; Ezema, C. G.; Onoabedje, A. E. Chem. Mater. Res. 2012, 2, 12.
Return to citation in text: [1] -
Wentrup, C. Tetrahedron 1974, 30, 1301. doi:10.1016/S0040-4020(01)97303-X
Return to citation in text: [1] -
Mayor, C.; Wentrup, C. J. Am. Chem. Soc. 1975, 97, 7467. doi:10.1021/ja00859a014
Return to citation in text: [1] [2] -
Tsao, M.-L.; Gritsan, N.; James, T. R.; Platz, M. S.; Hrovat, D. A.; Borden, W. T. J. Am. Chem. Soc. 2003, 125, 9343. doi:10.1021/ja0351591
Return to citation in text: [1] -
Wentrup, C.; Blanch, R.; Briehl, H.; Gross, G. J. Am. Chem. Soc. 1988, 110, 1874. doi:10.1021/ja00214a034
Return to citation in text: [1] -
Kuhn, A.; Plüg, C.; Wentrup, C. J. Am. Chem. Soc. 2000, 122, 1945. doi:10.1021/ja993859t
Return to citation in text: [1] -
Kappe, C. O.; Wong, M. W.; Wentrup, C. J. Org. Chem. 1995, 60, 1686. doi:10.1021/jo00111a029
Return to citation in text: [1] -
Dyall, L. K.; Moloney, D. W. J.; Harvey, J. J.; Fulloon, B. E. Aust. J. Chem. 1996, 49, 761. doi:10.1071/CH9960761
Return to citation in text: [1] -
Dyall, L. K.; Wong, M. W. Aust. J. Chem. 1985, 38, 1045. doi:10.1071/CH9851045
Return to citation in text: [1] -
Robba, M. Ann. Chim. (Cachan, Fr.) 1960, 5, 351.
Return to citation in text: [1] [2] -
Holland, G. F.; Pereira, J. N. J. Med. Chem. 1967, 10, 149. doi:10.1021/jm00314a004
Return to citation in text: [1] [2] -
Klötzer, W. Monatsh. Chem. 1956, 87, 131. doi:10.1007/BF00903597
Return to citation in text: [1] -
Klötzer, W. Monatsh. Chem. 1956, 87, 526. doi:10.1007/BF00917846
Return to citation in text: [1] -
Smith, P. A. S.; Brown, B. B. J. Am. Chem. Soc. 1951, 73, 2438. doi:10.1021/ja01150a009
Return to citation in text: [1] -
Baumgarten, H. E.; Saylor, J. L. J. Am. Chem. Soc. 1957, 79, 1502. doi:10.1021/ja01563a063
Return to citation in text: [1] -
Elsom, L. F.; Jones, R. A. J. Chem. Soc. B 1970, 79. doi:10.1039/j29700000079
Return to citation in text: [1] [2] [3] -
Long, L. M.; Troutman, H. D. J. Am. Chem. Soc. 1949, 71, 2469. doi:10.1021/ja01175a067
Return to citation in text: [1] -
Clemo, G. R.; Felton, D. G. I. J. Chem. Soc. 1952, 1658. doi:10.1039/jr9520001658
Return to citation in text: [1] -
Tamura, Y.; Adachi, M.; Kawasaki, T.; Yasuda, H.; Kita, Y. J. Chem. Soc., Perkin Trans. 1 1980, 1132. doi:10.1039/P19800001132
Return to citation in text: [1] -
Mehta, G. Synthesis 1978, 374. doi:10.1055/s-1978-24751
Return to citation in text: [1]
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 22. | Kvaskoff, D.; Bednarek, P.; George, L.; Pankajakshan, S.; Wentrup, C. J. Org. Chem. 2005, 70, 7947. doi:10.1021/jo050898g |
| 21. | Kvaskoff, D.; Mitschke, U.; Addicott, C.; Finnerty, J.; Bednarek, P.; Wentrup, C. Aust. J. Chem. 2009, 62, 275. doi:10.1071/CH08523 |
| 34. | Dyall, L. K.; Moloney, D. W. J.; Harvey, J. J.; Fulloon, B. E. Aust. J. Chem. 1996, 49, 761. doi:10.1071/CH9960761 |
| 35. | Dyall, L. K.; Wong, M. W. Aust. J. Chem. 1985, 38, 1045. doi:10.1071/CH9851045 |
| 36. | Robba, M. Ann. Chim. (Cachan, Fr.) 1960, 5, 351. |
| 37. | Holland, G. F.; Pereira, J. N. J. Med. Chem. 1967, 10, 149. doi:10.1021/jm00314a004 |
| 31. | Wentrup, C.; Blanch, R.; Briehl, H.; Gross, G. J. Am. Chem. Soc. 1988, 110, 1874. doi:10.1021/ja00214a034 |
| 11. | Kvaskoff, D.; Bednarek, P.; Wentrup, C. J. Org. Chem. 2010, 75, 1600. doi:10.1021/jo902570d |
| 32. | Kuhn, A.; Plüg, C.; Wentrup, C. J. Am. Chem. Soc. 2000, 122, 1945. doi:10.1021/ja993859t |
| 33. | Kappe, C. O.; Wong, M. W.; Wentrup, C. J. Org. Chem. 1995, 60, 1686. doi:10.1021/jo00111a029 |
| 21. | Kvaskoff, D.; Mitschke, U.; Addicott, C.; Finnerty, J.; Bednarek, P.; Wentrup, C. Aust. J. Chem. 2009, 62, 275. doi:10.1071/CH08523 |
| 1. | Wentrup, C. Acc. Chem. Res. 2011, 44, 393. doi:10.1021/ar700198z |
| 19. | Karney, W. L.; Borden, W. T. Differences between Phenylcarbene and Phenylnitrene and the Ring Expansion Reactions they Undergo. In Advances in Carbene Chemistry; Brinker, U. H., Ed.; Elsevier: Amsterdam, 2001; Vol. 3, p 205. |
| 38. | Klötzer, W. Monatsh. Chem. 1956, 87, 131. doi:10.1007/BF00903597 |
| 39. | Klötzer, W. Monatsh. Chem. 1956, 87, 526. doi:10.1007/BF00917846 |
| 36. | Robba, M. Ann. Chim. (Cachan, Fr.) 1960, 5, 351. |
| 37. | Holland, G. F.; Pereira, J. N. J. Med. Chem. 1967, 10, 149. doi:10.1021/jm00314a004 |
| 40. | Smith, P. A. S.; Brown, B. B. J. Am. Chem. Soc. 1951, 73, 2438. doi:10.1021/ja01150a009 |
| 42. | Elsom, L. F.; Jones, R. A. J. Chem. Soc. B 1970, 79. doi:10.1039/j29700000079 |
| 43. | Long, L. M.; Troutman, H. D. J. Am. Chem. Soc. 1949, 71, 2469. doi:10.1021/ja01175a067 |
| 42. | Elsom, L. F.; Jones, R. A. J. Chem. Soc. B 1970, 79. doi:10.1039/j29700000079 |
| 42. | Elsom, L. F.; Jones, R. A. J. Chem. Soc. B 1970, 79. doi:10.1039/j29700000079 |
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 41. | Baumgarten, H. E.; Saylor, J. L. J. Am. Chem. Soc. 1957, 79, 1502. doi:10.1021/ja01563a063 |
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 20. | Wentrup, C.; Crow, W. D. Tetrahedron 1970, 26, 3965. doi:10.1016/S0040-4020(01)93037-6 |
| 45. | Tamura, Y.; Adachi, M.; Kawasaki, T.; Yasuda, H.; Kita, Y. J. Chem. Soc., Perkin Trans. 1 1980, 1132. doi:10.1039/P19800001132 |
| 44. | Clemo, G. R.; Felton, D. G. I. J. Chem. Soc. 1952, 1658. doi:10.1039/jr9520001658 |
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 11. | Kvaskoff, D.; Bednarek, P.; Wentrup, C. J. Org. Chem. 2010, 75, 1600. doi:10.1021/jo902570d |
| 13. | Bednarek, P.; Wentrup, C. J. Am. Chem. Soc. 2003, 125, 9083. doi:10.1021/ja035632a |
| 1. | Wentrup, C. Acc. Chem. Res. 2011, 44, 393. doi:10.1021/ar700198z |
| 7. | Reisinger, A.; Bernhardt, P. V.; Wentrup, C. Org. Biomol. Chem. 2004, 2, 246. doi:10.1039/b311247k |
| 8. | Reisinger, A.; Koch, R.; Bernhardt, P. V.; Wentrup, C. Org. Biomol. Chem. 2004, 2, 1227. doi:10.1039/b317099c |
| 9. | Addicott, C.; Wentrup, C. Aust. J. Chem. 2008, 61, 592. doi:10.1071/CH08252 |
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 11. | Kvaskoff, D.; Bednarek, P.; Wentrup, C. J. Org. Chem. 2010, 75, 1600. doi:10.1021/jo902570d |
| 6. | Wentrup, C.; Reisinger, A.; Kvaskoff, D. Beilstein J. Org. Chem. 2013, 9, 754–760. doi:10.3762/bjoc.9.85 |
| 10. | McCluskey, A.; Wentrup, C. J. Org. Chem. 2008, 73, 6265. doi:10.1021/jo800899t |
| 20. | Wentrup, C.; Crow, W. D. Tetrahedron 1970, 26, 3965. doi:10.1016/S0040-4020(01)93037-6 |
| 2. | Lwowski, W. Nitrenes; Wiley-VCH: New York, NY, USA, 1970. |
| 3. | Scriven, E. F. V., Ed. Azides and Nitrenes; Academic Press: Orlando, FL, USA, 1984. |
| 4. | Bräse, S.; Banert, K. Organic Azides – Synthesis and Applications; Wiley-VCH: Chichester, UK, 2010. |
| 5. | Falvey, D. E.; Gudmundsdottir, A. D., Eds. Nitrenes and Nitrenium Ions; Wiley-VCH: Hoboken, NJ, USA, 2013. |
| 11. | Kvaskoff, D.; Bednarek, P.; Wentrup, C. J. Org. Chem. 2010, 75, 1600. doi:10.1021/jo902570d |
| 13. | Bednarek, P.; Wentrup, C. J. Am. Chem. Soc. 2003, 125, 9083. doi:10.1021/ja035632a |
| 17. | Kvaskoff, D.; Vosswinkel, M.; Wentrup, C. J. Am. Chem. Soc. 2011, 133, 5413. doi:10.1021/ja111155r |
| 21. | Kvaskoff, D.; Mitschke, U.; Addicott, C.; Finnerty, J.; Bednarek, P.; Wentrup, C. Aust. J. Chem. 2009, 62, 275. doi:10.1071/CH08523 |
| 15. | Orton, E.; Collins, S. T.; Pimentel, G. C. J. Phys. Chem. 1986, 90, 6139. doi:10.1021/j100281a018 |
| 16. | Rajam, S.; Murthy, R. S.; Jadhav, A. V.; Li, Q.; Keller, C.; Carra, C.; Pace, T. C. S.; Bohne, C.; Ault, B. S.; Gudmundsdottir, A. D. J. Org. Chem. 2011, 76, 9934. doi:10.1021/jo200877k |
| 1. | Wentrup, C. Acc. Chem. Res. 2011, 44, 393. doi:10.1021/ar700198z |
| 17. | Kvaskoff, D.; Vosswinkel, M.; Wentrup, C. J. Am. Chem. Soc. 2011, 133, 5413. doi:10.1021/ja111155r |
| 18. | Crabtree, K. N.; Hostetler, K. J.; Munsch, T. E.; Neuhaus, P.; Lahti, P. M.; Sander, W.; Poole, J. S. J. Org. Chem. 2008, 73, 3441. doi:10.1021/jo8001936 |
| 14. | Bégué, D.; Qiao, G. G.; Wentrup, C. J. Am. Chem. Soc. 2012, 134, 5339. doi:10.1021/ja2118442 |
| 19. | Karney, W. L.; Borden, W. T. Differences between Phenylcarbene and Phenylnitrene and the Ring Expansion Reactions they Undergo. In Advances in Carbene Chemistry; Brinker, U. H., Ed.; Elsevier: Amsterdam, 2001; Vol. 3, p 205. |
| 13. | Bednarek, P.; Wentrup, C. J. Am. Chem. Soc. 2003, 125, 9083. doi:10.1021/ja035632a |
| 12. | Wentrup, C.; Kvaskoff, D. Aust. J. Chem. 2013, 66, 286–296. doi:10.1071/CH12502. |
| 13. | Bednarek, P.; Wentrup, C. J. Am. Chem. Soc. 2003, 125, 9083. doi:10.1021/ja035632a |
| 22. | Kvaskoff, D.; Bednarek, P.; George, L.; Pankajakshan, S.; Wentrup, C. J. Org. Chem. 2005, 70, 7947. doi:10.1021/jo050898g |
| 21. | Kvaskoff, D.; Mitschke, U.; Addicott, C.; Finnerty, J.; Bednarek, P.; Wentrup, C. Aust. J. Chem. 2009, 62, 275. doi:10.1071/CH08523 |
| 22. | Kvaskoff, D.; Bednarek, P.; George, L.; Pankajakshan, S.; Wentrup, C. J. Org. Chem. 2005, 70, 7947. doi:10.1021/jo050898g |
| 29. | Mayor, C.; Wentrup, C. J. Am. Chem. Soc. 1975, 97, 7467. doi:10.1021/ja00859a014 |
| 30. | Tsao, M.-L.; Gritsan, N.; James, T. R.; Platz, M. S.; Hrovat, D. A.; Borden, W. T. J. Am. Chem. Soc. 2003, 125, 9343. doi:10.1021/ja0351591 |
| 29. | Mayor, C.; Wentrup, C. J. Am. Chem. Soc. 1975, 97, 7467. doi:10.1021/ja00859a014 |
| 25. | Maury, G.; Paugam, J.-P.; Pougam, R. J. Heterocycl. Chem. 1978, 15, 1041. doi:10.1002/jhet.5570150627 |
| 26. | Abarca, B.; Ballesteros, R.; Chadlaoui, M.; Miralles, J.; Murillo, J. V.; Colonna, D. Tetrahedron 2001, 57, 10111. doi:10.1016/S0040-4020(01)01053-5 |
| 27. | Ezema, B. E.; Akpanisi, L. E. S.; Ezema, C. G.; Onoabedje, A. E. Chem. Mater. Res. 2012, 2, 12. |
| 24. | Tennant, G.; Vevers, R. J. S. J. Chem. Soc., Chem. Commun. 1974, 671. doi:10.1039/C3974000671B |
© 2013 Wentrup et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)