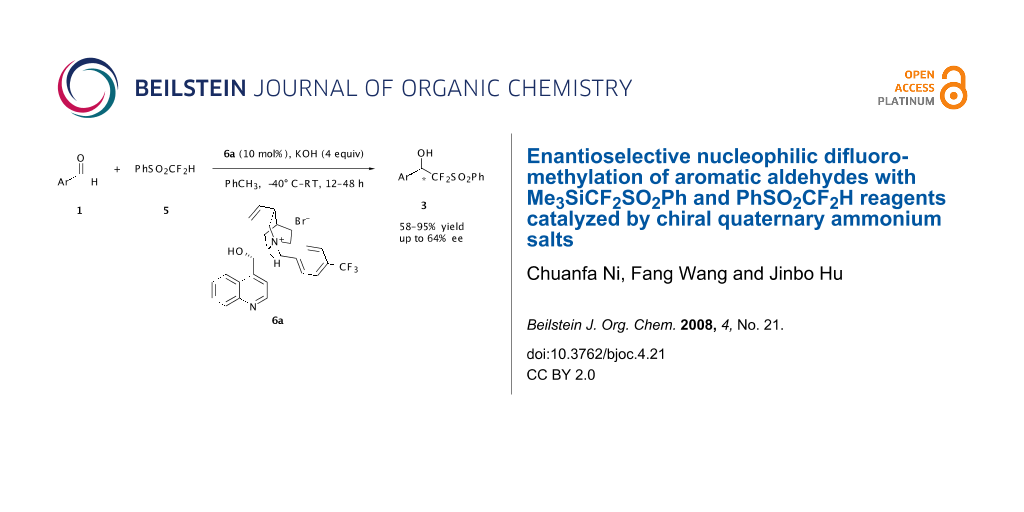
Abstract
Background
Although the nucleophilic difluoromethylation of aldehydes, ketones, and imines has been realized with PhSO2CF2H and related reagents, there are still no reports on the enantioselective nucleophilic reactions.
Results
With a chiral quaternary ammonium salt as the catalyst and KOH as the base, we describe the first enantioselective difluoromethylation of aromatic aldehydes with PhSO2CF2H or Me3SiCF2SO2Ph. The enantioselectivity is substrate-dependent and for 2-chlorinated benzaldehyde an ee up to 64% was obtained.
Conclusion
These results provide some insights into the enantioselective nucleophilic difluoromethylation chemistry, which will stimulate further progress in this field.

Graphical Abstract
Background
Because of the unique properties of fluorine, selective introduction of fluorine atom(s) or fluorine-containing moieties into organic molecules often dramatically alter their stability, lipophilicity, bioavailability, and biopotency. It is estimated that as many as 30–40% of agrochemicals and 20% of pharmaceuticals on the market contain fluorine [1,2]; as a result, fluorine is highlighted as the second most utilized hetero-element (after nitrogen) in life science-oriented research [3]. Nucleophilic fluoroalkylation, typically involving the transfer of a fluorinated carbanion (Rf−, the fluorine substitution being commonly on the carbanion center) to an electrophile, represents one of the major methods for the synthesis of organofluorine compounds [4-18]. In recent years, a few methods have been reported for the enantioselective introduction of a trifluoromethyl group into aldehydes and ketones with Ruppert-Prakash reagent, (trifluoromethyl)trimethylsilane, Me3SiCF3 and other reagents [19-29]. Although one-step nucleophilic difluoromethylation with R3SiCF2H is challenging regarding generality and efficiency [30], we found that both PhSO2CF2H and Me3SiCF2SO2Ph can be used as general and efficient nucleophilic difluoromethylation reagents [11,12,31]. We have successfully synthesized both α-difluoromethyl alcohols and β-difluoromethyl alcohols by using these two reagents [12,16,31]. Moreover, we have described a highly diastereoselective nucleophilic difluoromethylation method with N-(tert-butylsulfinyl)imines using PhSO2CF2H as a difluoromethylation reagent [13]. However, enantioselective introduction of the difluoromethyl group has not been previously reported.
As a class of versatile catalysts for asymmetric synthesis, cinchona alkaloids and their derivatives can catalyze an amazing array of synthetically important reactions, providing access to chiral products of high enantiopurity [32,33]. Several examples have been reported on the enantioselective incorporation of trifluoromethyl [23-28] or monofluoromethyl group [34] into organic molecules catalyzed by chiral quaternary ammonium salts. In the course of our research on the fluoride ion-induced difluoromethylation of carbonyl compounds with Me3SiCF2SO2Ph, we found that the counterion of the fluoride source played an important role in tuning the fluoroalkylation reactivity towards aldehydes and ketones, i.e. aldehydes showed higher reactivity than ketones [12]. In connection with our studies on selective fluoroalkylation reactions, herein we disclose the first example of catalytic enantioselective difluoromethylation reaction of aromatic aldehydes with Me3SiCF2SO2Ph or PhSO2CF2H in the presence of a cinchona alkaloid-based chiral quaternary ammonium salt.
Results and Discussion
Enantioselective nucleophilic difluoromethylation of aromatic aldehydes with Me3SiCF2SO2Ph
We started our investigation with the reaction between benzaldehyde and Me3SiCF2SO2Ph, choosing the fluoride salt 4 as the initiator with 5 mol% loading (Table 1, entry 1). Catalyst 4 was reported to be a relatively effective catalyst in the reaction between Me3SiCF3 and aromatic aldehyde [23]. When the reaction was carried out in THF solvent at −78 °C, the reaction proceeded smoothly to afford the desired product in 91% yield with poor enantioselectivity (Table 1, entry 1). After a quick survey of the reaction solvent, the enantioselectivity was improved when toluene was used as solvent (Table 1, entries 1–4). It turned out that the least polar solvent (toluene) was an appropriate solvent for the reaction, while the use of polar solvents such as THF, Et2O and CH2Cl2 gave the product with relatively lower enantioselectivity. The increase of the catalyst loading was able to improve the enantioselectivity further. When 10 mol% of 4 was used, the ee was improved to 47% (Table 1, entry 5). However, when 2-naphthaldehyde or 4-methoxybenzaldehyde was used as the substrate, in each case lower ee was observed (Table 1, entries 6 and 7). Although catalyst 4 was ineffective in the catalysis of the nucleophilic difluoromethylation of ketones in toluene, the reaction could proceed smoothly when CH2Cl2 was added as a co-solvent. The tetrasubstituted difluoromethylated aryl alcohol was formed in 97% yield with 10% ee (Table 1, entry 8).
Table 1: Asymmetric nucleophilic difluoromethylation of aromatic aldehyde with PhSO2CF2SiMe3.
![[Graphic 1]](/bjoc/content/inline/1860-5397-4-21-i3.svg?max-width=637&scale=1.0)
|
|||||
| entry[a] | carbonyl compound | Initiator [mol%] | solvent | yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|
| 1 | PhCHO | 4 (5) | THF | 91 | 14 |
| 2 | PhCHO | 4 (5) | CH2Cl2 | 64 | 9 |
| 3 | PhCHO | 4 (5) | Et2O | 67 | 27 |
| 4 | PhCHO | 4 (5) | PhCH3 | 65 | 36 |
| 5 | PhCHO | 4 (10) | PhCH3 | 60 | 47 |
| 6 | 2-NapCHO | 4 (10) | PhCH3 | 64 | 26 |
| 7 | p-MeOC6H4CHO | 4 (10) | PhCH3 | 50 | 25 |
| 8[d] | PhCOCH3 | 4 (10) | PhCH3-CH2Cl2 (2:1, v/v) | 97 | 10 |
[a] Unless noted, reactions were carried out at 1.0 mmol scale. To a mixture of 1 (1.0 equiv) and 4 (x mol%) in 2 mL of toluene at −78 °C, 2 (1.2 equiv, dissolved in 2 mL of toluene) was added dropwise over 2 hours. The reaction mixture was then stirred at the same temperature for additional 3 hours.
[b] Isolated yield of the pure product.
[c] Enantiomeric excess was determined by HPLC analysis using a chiral column.
[d] The reaction was carried out at 0.25 mmol scale. 2 (4.0 equiv) in 0.5 mL PhCH3-CH2Cl2 was added in 5 minutes to the reaction mixture of 1 and 4 at −78 °C. The mixture was then stirred at the same temperature for another 12 hours.
Enantioselective nucleophilic difluoromethylation of aromatic aldehydes with PhSO2CF2H
In 1989, Stahly reported the nucleophilic difluoromethylation of aromatic aldehydes with PhSO2CF2H under the phase transfer condition using Aliquat 336 (a commercially available quaternary ammonium salt) as the phase transfer catalyst [35]. With most aldehydes, the reaction affords moderate to excellent yields of products. Moreover, unlike the Me3SiCF2SO2Ph-based reactions, the reaction with PhSO2CF2H is not water-sensitive [34]. The use of PhSO2CF2H as a robust fluoroalkylating agent [11] has aroused our interest in developing its application in enantioselective difluoromethylation reactions. Thus, we decided to evaluate the ability of known chiral ammonium salts to promote the enantioselective difluoromethylation of aldehydes with PhSO2CF2H. In a preliminary study, we examined the reaction with the chiral quaternary ammonium salt 6a (see Scheme 1) as the phase transfer catalyst at room temperature, with 30% NaOH as the base and toluene as the solvent. After 12 h, the reaction afforded the desired product in 93% yield with a modest but significant enantioselectivity of 22% (Table 2, entry 1). When solid powdered KOH was used as the base, the ee could be slightly improved (Table 2, entry 2). Better enantioselectivity (47% ee) was observed on lowering the reaction temperature to −40 °C (Table 2, entry 3). However, at −78 °C, the reaction did not proceed.
![[1860-5397-4-21-i1]](/bjoc/content/inline/1860-5397-4-21-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Structure of chiral quaternary ammonium salts.
Scheme 1: Structure of chiral quaternary ammonium salts.
Table 2: Enantioselective difluoromethylation of benzaldehyde with PhSO2CF2H under various conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-4-21-i4.svg?max-width=637&scale=1.0)
|
|||||||
| Entry[a] | PTC | base | solvent | temperature [°C] | time [h] | yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|---|
| 1 | 6a | NaOH (30%) | PhCH3 | RT | 12 | 93 | 22 |
| 2 | 6a | solid KOH | PhCH3 | RT | 2 | 93 | 32 |
| 3 | 6a | solid KOH | PhCH3 | −40 | 48 | 67 | 47 |
| 4 | 7a | solid KOH | PhCH3 | −40 | 48 | 87 | 29 |
| 5 | 8a | solid KOH | PhCH3 | RT | 2 | 94 | 46 |
| 6 | 8a | solid KOH | PhCH3 | −40 | 48 | 55 | 11 |
| 7 | 9a | solid KOH | PhCH3 | RT | 2 | 91 | 41 |
| 8 | 8a | CsOH(H2O) | PhCH3 | RT | 12 | 81 | 20 |
| 9 | 8a | RbOH (50%) | PhCH3 | RT | 12 | 76 | 2 |
| 10 | 8a | solid KOH | THF | RT | 12 | 82 | 0 |
| 11 | 8a | solid KOH | CH2Cl2 | RT | 12 | 80 | 0 |
| 12 | 6b | solid KOH | PhCH3 | −40 | 48 | 40 | 9 |
| 13 | 6c | solid KOH | PhCH3 | −40 | 48 | 62 | 31 |
| 14 | 8b | solid KOH | PhCH3 | RT | 12 | 90 | 23 |
| 15 | 8c | RbOH (50%) | PhCH3 | RT | 12 | 0 | – |
| 16 | 8c | RbOH(H2O) | PhCH3 | RT | 12 | 80 | 5 |
[a] All reactions were carried out at 0.25 mmol scale with 1a (1.2 equiv) and 5 (1.0 equiv) in 1.5 mL solvent.
[b] Isolated yield of the pure product.
[c] Enantiomeric excess was determined by HPLC analysis using a chiral column (Chiralcel AD-H eluting with 20% i-PrOH in hexane).
Using toluene as the solvent and solid KOH as the base, we scanned four 4-trifluoromethylphenyl ammonium salts derived from quinine (QN), quinidine (QD), cinchonine (CN), and cinchonidine (CD) at different reaction temperatures. We found the structure of the cinchona alkaloid had some influence on the enantioselectivity. When a cinchonine or quinidine derivative was used, the main isomer was obtained as (+)-3a, and CN 6a was superior to QD 7a. The optimized reaction temperature was −40 °C (Table 2, entries 2 and 3). For quinine derivative 8a, it is interesting that a high temperature was beneficial for the enantioselectivity and (−)-3a was obtained as the main isomer. When the reaction was conducted at −40 °C, the reaction proceeded with moderate yield and poor enantioselectivity due to the low solubility of the catalyst in toluene (Table 2, entries 5 and 6). For CD derivative 9a, the enantioselectivity was slightly lower than 8a at rt (Table 2, entry 7). As reported, cinchonine derivatives and quinine derivatives yield products with the opposite configuration [32]. From the above screening, the quaternary salts 6a and 8a derived from the CN and QN were selected as the catalysts for further study.
Subsequently, the solvent effect was examined with catalyst 8a in the presence of solid KOH as the base at room temperature. It was shown that the use of toluene as a reaction medium remarkably improved the enantioselectivity. When THF or CH2Cl2 was used, the complete loss of enantioselectivity was observed (Table 2, entries 10 and 11). Moreover, KOH gave the best results in enantioselectivity in comparison with the other metal hydroxides, such as CsOH and RbOH (Table 2, entries 8 and 9). Encouraged by these results, we further examined the influence of substituents on the chiral phase transfer catalysts. As shown in Table 2, the different substituents showed some influence on the enantioselectivity. The electron-withdrawing group CF3 at C-4 position of the benzyl ring afforded the product with good ee, though the unsubstituted one gave a significantly low ee (Table 2, entries 12 and 14). Although the 9-anthracenylmethyl and 2,3,4-trifluorobenzyl functionality were found to be quite useful for asymmetric alkylation of tert-butylglycinate Schiff base [36] or asymmetric synthesis of α,β-epoxysulfones [37], they were not as effective as 4-trifluoromethylphenyl functionality in the current asymmetric reaction (Table 2, entries 13, 15 and 16).
In the light of these results, we next examined the substrate scope of this enantioselective difluoromethylation reaction with catalyst 6a or 8a, and the results are shown in Table 3. Although the two types of cinchona alkaloids 6a and 8a are almost equally effective when benzaldehyde was tested, 6a was chosen due to its generality towards other aldehydes such as 4-chlorobenzaldehyde 1b. In general, the chemical yields were good to excellent, except in the case of 4-tert-butylbenzaldehyde 1k, where a moderate yield was obtained (although the reaction was performed at rt) (Table 3, entry 13). It is obvious that the enantioselectivity was dependent on the substrate structure. It is interesting that the aldehydes with halogen substitution (Table 3, entries 3, 5–9) showed better enantioselectivity than those with methyl and methoxy substituents (Table 3, entries 11,12). Among the halogenated benzaldehydes that were tested, the reaction with 2-chlorobenzaldehyde showed an enantiomeric excess up to 64% (Table 3, entry 7). The enantiomeric excess obtained from 2-naphthaldehyde was also modest (23% ee) (Table 3, entry 14).
Table 3: Asymmetric nucleophilic difluoromethylation of aromatic aldehydes with PhSO2CF2H.
![[Graphic 3]](/bjoc/content/inline/1860-5397-4-21-i5.svg?max-width=637&scale=1.0)
|
||||||
| entry[a] | aromatic aldehyde | PTC | T [°C] | time [h] | yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1 | 1a:Ar=C6H5- | 6a | −40 | 48 | 3a: 67 | 47 (S)[d] |
| 2 | 1a:Ar=C6H5- | 8a | 25 | 2 | 3a: 94 | 46 (R)[e] |
| 3 | 1b:Ar=4-Cl-C6H4- | 6a | −40 | 48 | 3b: 74 | 52 (S)[f] |
| 4 | 1b:Ar=4-Cl-C6H4- | 8a | 25 | 1 | 3b: 91 | 23 (R)[e] |
| 5 | 1c:Ar=2,4-Cl-C6H3- | 6a | −20 | 48 | 3c: 95 | 54 (S)[f] |
| 6 | 1d: Ar=3-Cl-C6H4- | 6a | −20 | 48 | 3d: 83 | 46 (S)[f] |
| 7 | 1e: Ar=2-Cl-C6H4- | 6a | −20 | 48 | 3e: 92 | 64 (S)[f] |
| 8 | 1f: Ar=4-F-C6H4- | 6a | −20 | 48 | 3f: 93 | 41 (S)[f] |
| 9 | 1g: Ar=4-Br-C6H4- | 6a | −20 | 48 | 3g: 95 | 36 (S)[f] |
| 10 | 1h:Ar=4-CF3-C6H4- | 6a | −20 | 48 | 3h: 68 | 36 (S)[f] |
| 11 | 1i: Ar=2-Me-C6H4- | 6a | −20 | 48 | 3i: 77 | 11 |
| 12 | 1j:Ar=4-MeO-C6H4- | 6a | −20 | 48 | 3j: 80 | 12 |
| 13 | 1k:Ar=4-tBu-C6H4- | 6a | 25 | 12 | 3k: 58 | 4 |
| 14 | 1l: Ar=2-naphthyl- | 6a | −20 | 48 | 3l: 72 | 23 (S)[f] |
[a] All reactions were carried out at 0.25 mmol scale with 1 (1.2 equiv) and 5 (1.0 equiv) in 1.5 mL toluene.
[b] Isolated yield of the pure product.
[c] Enantiomeric excess was determined by HPLC analysis using a chiral column (Chiralcel AD-H, OD or IC).
[d] The absolute configuration was determined to be S after chemical derivatization.
[e] The absolute configuration was determined to be R by comparing the retention time on chiral HPLC.
[f] The absolute stereochemistry was tentatively determined by 19F NMR analysis of the corresponding Mosher's esters comparing with (S)-3a.
The absolute configuration of the alcohol (+)-3a (Table 3, entry 1) was determined to be S by comparing the optical rotation with that of the corresponding difluoromethyl alcohol (after desulfonylation) with the known data (Scheme 2) [38]. For other alcohols, the stereochemistry was tentatively determined by 19F NMR analysis of the corresponding Mosher's esters comparing with (+)-3a [39].
![[1860-5397-4-21-i2]](/bjoc/content/inline/1860-5397-4-21-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Determination of the absolute configuration of (+)-3a.
Scheme 2: Determination of the absolute configuration of (+)-3a.
Conclusion
In conclusion, we have described the first chiral quaternary ammonium salts catalyzed enantioselective difluoromethylation of aromatic aldehydes with Me3SiCF2SO2Ph and PhSO2CF2H. The enantioselectivity is substrate-dependent and for 2-chlorinated benzaldehydes, an ee up to 64% was obtained. The easy preparation of the chiral cinchonium salts and the convenient experimental procedure make the reaction operationally simple. These results provide some insights into enantioselective nucleophilic difluoromethylation chemistry, which will stimulate further progress in this field.
Supporting Information
| Supporting Information File 1: Full experimental details and compound characterization data for all new compounds described. | ||
| Format: DOC | Size: 71.0 KB | Download |
References
-
Thayer, A. M. Chem. Eng. News 2006, 84 (23), 15–24.
Return to citation in text: [1] -
Thayer, A. M. Chem. Eng. News 2006, 84 (23), 27–32.
Return to citation in text: [1] -
Cottet, F.; Marull, M.; Lefebvre, O.; Schlosser, M. Eur. J. Org. Chem. 2003, 1559–1568. doi:10.1002/ejoc.200390215
Return to citation in text: [1] -
Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991
Return to citation in text: [1] -
Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8
Return to citation in text: [1] -
Singh, R. P.; Shreeve, J. M. Tetrahedron 2000, 56, 7613–7632. doi:10.1016/S0040-4020(00)00550-0
Return to citation in text: [1] -
Langlois, B. R.; Billard, T. Synthesis 2003, 185–194. doi:10.1055/s-2003-36812
Return to citation in text: [1] -
Aït-Mohand, S.; Takechi, N.; Médebielle, M.; Dolbier, W. R., Jr. Org. Lett. 2001, 3, 4271–4273. doi:10.1021/ol016933x
Return to citation in text: [1] -
McClinton, M. A.; McClinton, D. A. Tetrahedron 1992, 48, 6555–6666. doi:10.1016/S0040-4020(01)80011-9
Return to citation in text: [1] -
Burton, D. J.; Yang, Z.-Y. Tetrahedron 1992, 48, 189–275. doi:10.1016/S0040-4020(01)88139-4
Return to citation in text: [1] -
Prakash, G. K. S.; Hu, J. Acc. Chem. Res. 2007, 40, 921–930. doi:10.1021/ar700149s
Return to citation in text: [1] [2] [3] -
Ni, C.; Hu, J. Tetrahedron Lett. 2005, 46, 8273–8277. doi:10.1016/j.tetlet.2005.09.162
Return to citation in text: [1] [2] [3] [4] -
Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2005, 44, 5882–5886. doi:10.1002/anie.200501769
Return to citation in text: [1] [2] -
Li, Y.; Ni, C.; Liu, J.; Zhang, L.; Zheng, J.; Zhu, L.; Hu, J. Org. Lett. 2006, 8, 1693–1696. doi:10.1021/ol060322t
Return to citation in text: [1] -
Ni, C.; Li, Y.; Hu, J. J. Org. Chem. 2006, 71, 6829–6833. doi:10.1021/jo060955l
Return to citation in text: [1] -
Ni, C.; Liu, J.; Zhang, L.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 786–789. doi:10.1002/anie.200603983
Return to citation in text: [1] [2] -
Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 2489–2492. doi:10.1002/anie.200604783
Return to citation in text: [1] -
Liu, J.; Ni, C.; Wang, F.; Hu, J. Tetrahedron Lett. 2008, 49, 1605–1608. doi:10.1016/j.tetlet.2008.01.034
Return to citation in text: [1] -
Ma, J.-A.; Cahard, D. Chem. Rev. 2004, 104, 6119–6146. doi:10.1021/cr030143e
Return to citation in text: [1] -
Ma, J.-A.; Cahard, D. J. Fluorine Chem. 2007, 128, 975–996. doi:10.1016/j.jfluchem.2007.04.026
Return to citation in text: [1] -
Roussel, S.; Billard, T.; Langlois, B. R.; Saint-James, L. Chem.–Eur. J. 2005, 11, 939–944. doi:10.1002/chem.200400777
Return to citation in text: [1] -
Kuroki, Y.; Iseki, K. Tetrahedron Lett. 1999, 40, 8231–8234. doi:10.1016/S0040-4039(99)01724-4
Return to citation in text: [1] -
Iseki, K.; Nagai, T.; Kobayashi, Y. Tetrahedron Lett. 1994, 35, 3137–3138. doi:10.1016/S0040-4039(00)76850-X
Return to citation in text: [1] [2] [3] -
Caron, S.; Do, N. M.; Arpin, P.; Larivée, A. Synthesis 2003, 11, 1693–1698. doi:10.1055/s-2003-40889
Return to citation in text: [1] [2] -
Nagao, H.; Yamane, Y.; Mukaiyama, T. Chem. Lett. 2007, 36, 666–667. doi:10.1246/cl.2007.666
Return to citation in text: [1] [2] -
Nagao, H.; Kawano, Y.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2007, 80, 2406–2412. doi:10.1246/bcsj.80.2406
Return to citation in text: [1] [2] -
Mizuta, S.; Shibata, N.; Hibino, M.; Nagano, S.; Nakamura, S.; Toru, T. Tetrahedron 2007, 63, 8521–8528. doi:10.1016/j.tet.2007.05.024
Return to citation in text: [1] [2] -
Zhao, H.; Qin, B.; Liu, X.; Feng, X. Tetrahedron 2007, 63, 6822–6826. doi:10.1016/j.tet.2007.04.068
Return to citation in text: [1] [2] -
Mizuta, S.; Shibata, N.; Akiti, S.; Fujimoto, H.; Nakamura, S.; Toru, T. Org. Lett. 2007, 9, 3707–3710. doi:10.1021/ol701791r
Return to citation in text: [1] -
Hagiwara, T.; Fuchikami, T. Synlett 1995, 717–718. doi:10.1055/s-1995-5070
Return to citation in text: [1] -
Prakash, G. K. S.; Hu, J.; Wang, Y.; Olah, G. A. Eur. J. Org. Chem. 2005, 2218–2223. doi:10.1002/ejoc.200500101
Return to citation in text: [1] [2] -
Kacprzak, K.; Gawroński, J. Synthesis 2001, 961–998. doi:10.1055/s-2001-14560
Return to citation in text: [1] [2] -
Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. doi:10.1002/anie.200601737
Return to citation in text: [1] -
Mizuta, S.; Shibata, N.; Goto, Y.; Furukawa, T.; Nakamura, S.; Toru, T. J. Am. Chem. Soc. 2007, 129, 6394–6395. doi:10.1021/ja071509y
Return to citation in text: [1] [2] -
Stahly, G. P. J. Fluorine Chem. 1989, 43, 53–66. doi:10.1016/S0022-1139(00)81636-X
Return to citation in text: [1] -
Okino, T.; Takemoto, Y. Org. Lett. 2001, 3, 1515–1517. doi:10.1021/ol015829c
Return to citation in text: [1] -
Ku, J.-M.; Yoo, M.-S.; Park, H.; Jew, S.; Jeong, B.-S. Tetrahedron 2007, 63, 8099–8103. doi:10.1016/j.tet.2007.06.012
Return to citation in text: [1] -
Matsuda, T.; Harada, T.; Nakajima, N.; Itoh, T.; Nakamura, K. J. Org. Chem. 2000, 65, 157–163. doi:10.1021/jo991283k
Return to citation in text: [1] -
Gaspar, J.; Guerrero, A. Tetrahedron: Asymmetry 1995, 6, 231–238. doi:10.1016/0957-4166(94)00379-P
Return to citation in text: [1]
| 37. | Ku, J.-M.; Yoo, M.-S.; Park, H.; Jew, S.; Jeong, B.-S. Tetrahedron 2007, 63, 8099–8103. doi:10.1016/j.tet.2007.06.012 |
| 32. | Kacprzak, K.; Gawroński, J. Synthesis 2001, 961–998. doi:10.1055/s-2001-14560 |
| 1. | Thayer, A. M. Chem. Eng. News 2006, 84 (23), 15–24. |
| 2. | Thayer, A. M. Chem. Eng. News 2006, 84 (23), 27–32. |
| 34. | Mizuta, S.; Shibata, N.; Goto, Y.; Furukawa, T.; Nakamura, S.; Toru, T. J. Am. Chem. Soc. 2007, 129, 6394–6395. doi:10.1021/ja071509y |
| 19. | Ma, J.-A.; Cahard, D. Chem. Rev. 2004, 104, 6119–6146. doi:10.1021/cr030143e |
| 20. | Ma, J.-A.; Cahard, D. J. Fluorine Chem. 2007, 128, 975–996. doi:10.1016/j.jfluchem.2007.04.026 |
| 21. | Roussel, S.; Billard, T.; Langlois, B. R.; Saint-James, L. Chem.–Eur. J. 2005, 11, 939–944. doi:10.1002/chem.200400777 |
| 22. | Kuroki, Y.; Iseki, K. Tetrahedron Lett. 1999, 40, 8231–8234. doi:10.1016/S0040-4039(99)01724-4 |
| 23. | Iseki, K.; Nagai, T.; Kobayashi, Y. Tetrahedron Lett. 1994, 35, 3137–3138. doi:10.1016/S0040-4039(00)76850-X |
| 24. | Caron, S.; Do, N. M.; Arpin, P.; Larivée, A. Synthesis 2003, 11, 1693–1698. doi:10.1055/s-2003-40889 |
| 25. | Nagao, H.; Yamane, Y.; Mukaiyama, T. Chem. Lett. 2007, 36, 666–667. doi:10.1246/cl.2007.666 |
| 26. | Nagao, H.; Kawano, Y.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2007, 80, 2406–2412. doi:10.1246/bcsj.80.2406 |
| 27. | Mizuta, S.; Shibata, N.; Hibino, M.; Nagano, S.; Nakamura, S.; Toru, T. Tetrahedron 2007, 63, 8521–8528. doi:10.1016/j.tet.2007.05.024 |
| 28. | Zhao, H.; Qin, B.; Liu, X.; Feng, X. Tetrahedron 2007, 63, 6822–6826. doi:10.1016/j.tet.2007.04.068 |
| 29. | Mizuta, S.; Shibata, N.; Akiti, S.; Fujimoto, H.; Nakamura, S.; Toru, T. Org. Lett. 2007, 9, 3707–3710. doi:10.1021/ol701791r |
| 11. | Prakash, G. K. S.; Hu, J. Acc. Chem. Res. 2007, 40, 921–930. doi:10.1021/ar700149s |
| 4. | Prakash, G. K. S.; Yudin, A. K. Chem. Rev. 1997, 97, 757–786. doi:10.1021/cr9408991 |
| 5. | Prakash, G. K. S.; Mandal, M. J. Fluorine Chem. 2001, 112, 123–131. doi:10.1016/S0022-1139(01)00477-8 |
| 6. | Singh, R. P.; Shreeve, J. M. Tetrahedron 2000, 56, 7613–7632. doi:10.1016/S0040-4020(00)00550-0 |
| 7. | Langlois, B. R.; Billard, T. Synthesis 2003, 185–194. doi:10.1055/s-2003-36812 |
| 8. | Aït-Mohand, S.; Takechi, N.; Médebielle, M.; Dolbier, W. R., Jr. Org. Lett. 2001, 3, 4271–4273. doi:10.1021/ol016933x |
| 9. | McClinton, M. A.; McClinton, D. A. Tetrahedron 1992, 48, 6555–6666. doi:10.1016/S0040-4020(01)80011-9 |
| 10. | Burton, D. J.; Yang, Z.-Y. Tetrahedron 1992, 48, 189–275. doi:10.1016/S0040-4020(01)88139-4 |
| 11. | Prakash, G. K. S.; Hu, J. Acc. Chem. Res. 2007, 40, 921–930. doi:10.1021/ar700149s |
| 12. | Ni, C.; Hu, J. Tetrahedron Lett. 2005, 46, 8273–8277. doi:10.1016/j.tetlet.2005.09.162 |
| 13. | Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2005, 44, 5882–5886. doi:10.1002/anie.200501769 |
| 14. | Li, Y.; Ni, C.; Liu, J.; Zhang, L.; Zheng, J.; Zhu, L.; Hu, J. Org. Lett. 2006, 8, 1693–1696. doi:10.1021/ol060322t |
| 15. | Ni, C.; Li, Y.; Hu, J. J. Org. Chem. 2006, 71, 6829–6833. doi:10.1021/jo060955l |
| 16. | Ni, C.; Liu, J.; Zhang, L.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 786–789. doi:10.1002/anie.200603983 |
| 17. | Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 2489–2492. doi:10.1002/anie.200604783 |
| 18. | Liu, J.; Ni, C.; Wang, F.; Hu, J. Tetrahedron Lett. 2008, 49, 1605–1608. doi:10.1016/j.tetlet.2008.01.034 |
| 23. | Iseki, K.; Nagai, T.; Kobayashi, Y. Tetrahedron Lett. 1994, 35, 3137–3138. doi:10.1016/S0040-4039(00)76850-X |
| 3. | Cottet, F.; Marull, M.; Lefebvre, O.; Schlosser, M. Eur. J. Org. Chem. 2003, 1559–1568. doi:10.1002/ejoc.200390215 |
| 35. | Stahly, G. P. J. Fluorine Chem. 1989, 43, 53–66. doi:10.1016/S0022-1139(00)81636-X |
| 32. | Kacprzak, K.; Gawroński, J. Synthesis 2001, 961–998. doi:10.1055/s-2001-14560 |
| 33. | Ooi, T.; Maruoka, K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. doi:10.1002/anie.200601737 |
| 34. | Mizuta, S.; Shibata, N.; Goto, Y.; Furukawa, T.; Nakamura, S.; Toru, T. J. Am. Chem. Soc. 2007, 129, 6394–6395. doi:10.1021/ja071509y |
| 13. | Li, Y.; Hu, J. Angew. Chem., Int. Ed. 2005, 44, 5882–5886. doi:10.1002/anie.200501769 |
| 12. | Ni, C.; Hu, J. Tetrahedron Lett. 2005, 46, 8273–8277. doi:10.1016/j.tetlet.2005.09.162 |
| 12. | Ni, C.; Hu, J. Tetrahedron Lett. 2005, 46, 8273–8277. doi:10.1016/j.tetlet.2005.09.162 |
| 16. | Ni, C.; Liu, J.; Zhang, L.; Hu, J. Angew. Chem., Int. Ed. 2007, 46, 786–789. doi:10.1002/anie.200603983 |
| 31. | Prakash, G. K. S.; Hu, J.; Wang, Y.; Olah, G. A. Eur. J. Org. Chem. 2005, 2218–2223. doi:10.1002/ejoc.200500101 |
| 38. | Matsuda, T.; Harada, T.; Nakajima, N.; Itoh, T.; Nakamura, K. J. Org. Chem. 2000, 65, 157–163. doi:10.1021/jo991283k |
| 11. | Prakash, G. K. S.; Hu, J. Acc. Chem. Res. 2007, 40, 921–930. doi:10.1021/ar700149s |
| 12. | Ni, C.; Hu, J. Tetrahedron Lett. 2005, 46, 8273–8277. doi:10.1016/j.tetlet.2005.09.162 |
| 31. | Prakash, G. K. S.; Hu, J.; Wang, Y.; Olah, G. A. Eur. J. Org. Chem. 2005, 2218–2223. doi:10.1002/ejoc.200500101 |
| 23. | Iseki, K.; Nagai, T.; Kobayashi, Y. Tetrahedron Lett. 1994, 35, 3137–3138. doi:10.1016/S0040-4039(00)76850-X |
| 24. | Caron, S.; Do, N. M.; Arpin, P.; Larivée, A. Synthesis 2003, 11, 1693–1698. doi:10.1055/s-2003-40889 |
| 25. | Nagao, H.; Yamane, Y.; Mukaiyama, T. Chem. Lett. 2007, 36, 666–667. doi:10.1246/cl.2007.666 |
| 26. | Nagao, H.; Kawano, Y.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2007, 80, 2406–2412. doi:10.1246/bcsj.80.2406 |
| 27. | Mizuta, S.; Shibata, N.; Hibino, M.; Nagano, S.; Nakamura, S.; Toru, T. Tetrahedron 2007, 63, 8521–8528. doi:10.1016/j.tet.2007.05.024 |
| 28. | Zhao, H.; Qin, B.; Liu, X.; Feng, X. Tetrahedron 2007, 63, 6822–6826. doi:10.1016/j.tet.2007.04.068 |
| 39. | Gaspar, J.; Guerrero, A. Tetrahedron: Asymmetry 1995, 6, 231–238. doi:10.1016/0957-4166(94)00379-P |
© 2008 Ni et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)