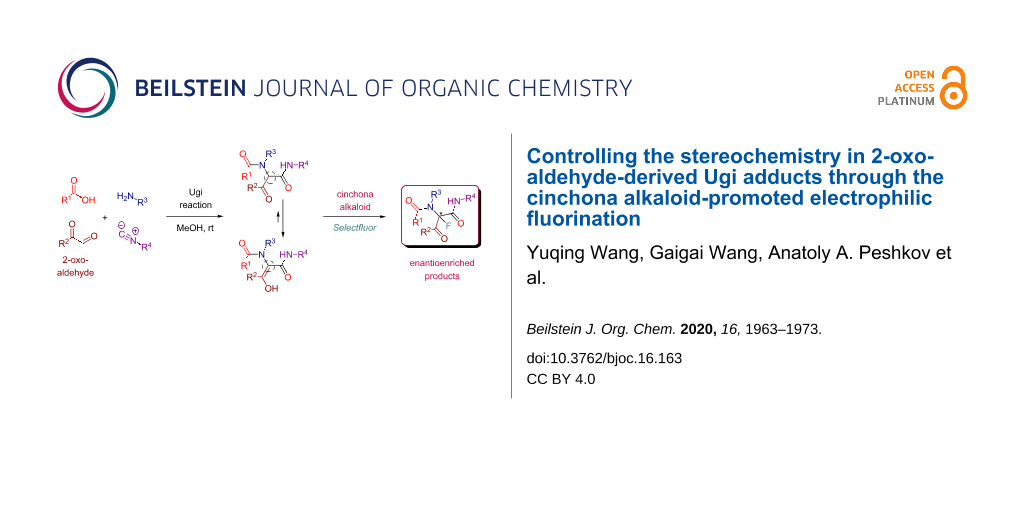
Abstract
In this report, we introduce a new strategy for controlling the stereochemistry in Ugi adducts. Instead of controlling stereochemistry directly during the Ugi reaction we have attempted to stereodefine the chiral center at the peptidyl position through the post-Ugi functionalization. In order to achieve this, we chose to study 2-oxo-aldehyde-derived Ugi adducts many of which partially or fully exist in the enol form that lacks the aforementioned chiral center. This in turn led to their increased nucleophilicity as compared to the standard Ugi adducts. As such, the stereocenter at the peptidyl position could be installed and stereodefined through the reaction with a suitable electrophile. Towards this end, we were able to deploy an asymmetric cinchona alkaloid-promoted electrophilic fluorination producing enantioenriched post-Ugi adducts fluorinated at the peptidyl position.

Graphical Abstract
Introduction
Multicomponent reactions (MCRs) [1-13] in general and isocyanide-based MCRs [14-18] in particular represent powerful tools of modern synthetic chemistry that allow to generate structurally diverse and complex products in a step and atom-economic manner from simple and accessible precursors. Three-component Passerini [19-22] and four-component Ugi [23,24] reactions that rely on the ability of organic isocyanides to participate in the nucleophilic attack onto the carbonyl or imine group are among the most studied MCRs. Accordingly, a wide range of post-MCR transformations have been elaborated allowing to upgrade the Passerini and Ugi adducts into potentially bioactive heterocycles [25-32]. While both Passerini and Ugi reactions proved to be quite robust towards different classes of substrates and possess broad functional group tolerance, the difficulties associated with controlling their stereochemical outcome [33-35] severely restrict their applicability in medicinal chemistry [36].
The typical Passerini three-component reaction (P-3CR) of a carboxylic acid 1, an aldehyde 2, and an isocyanide 3 being conducted in a polar-aprotic solvent such as THF or dichloromethane results in the assembly of an α-acyloxyamide adduct 4. Just a few protocols for the asymmetric P-3CR [37-40] and its modifications [41-47] have been reported over the course of the last two decades (Table 1). In the majority of those protocols [37-39], the stereocontrol was achieved with the aid of different Lewis acid promoters bearing chiral ligands, while the most recent and at the same time the most general procedure developed by Liu, Tan and co-workers relied on the stereoinduction by a chiral phosphoric acid (Table 1) [40].
Table 1: Asymmetric protocols for Passerini three-component reaction (P-3CR).
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-163-i3.svg?max-width=637&scale=1.0)
|
||||
| Asymmetric protocol | Catalytic system | Conditions | Yield range, % | ee range, % |
| Dömling and co-workers, 2003 [37] |
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-163-i4.svg?max-width=637&scale=1.0)
(1 equiv) Ti(OiPr)4 (1.35 equiv) |
THF (0.5 M), overnight | 12–48 | 32–42 |
| Schreiber and co-workers, 2004 [38] |
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-163-i5.svg?max-width=637&scale=1.0)
(20 mol %) Cu(OTf)2 (20 mol %) |
DCM (0.04 M), molecular sieves, 18–48 h | 75–98 | 62–98 |
| Wang, Zhu and co-workers, 2008 [39] |
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-163-i6.svg?max-width=637&scale=1.0)
(10 mol %) |
toluene (0.33 M), argon, −40 °C, 48 h | 51–70 | 63 to >99 |
| Liu, Tan and co-workers, 2015 [40] |
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-163-i7.svg?max-width=637&scale=1.0)
(10–20 mol %) |
CHCl3 (0.033 M or 0.05 M), argon, MgSO4, rt or −20 °C or −35 °C, 36 h | 41–99 | 84–99 |
In the standard Ugi four-component reaction (U-4CR) a carboxylic acid 1, an aldehyde 2, and an isocyanide 3 are complemented by a primary amine 5 that altogether undergo a condensation into a peptide-like adduct 6. These reactions are typically conducted in polar protic solvents such as methanol or water. Several examples of diastereoselective U-4CR have been described [48-54]. Yet, the Ugi reaction turned out to be a rather challenging chemical transformation in terms of establishing the asymmetric version [33-35]. Apart from the two asymmetric three-component modifications lacking the acid component [55,56], no general asymmetric protocol for the four-component mode has been developed until very recently. In 2018, Houk, Tan and co-workers described an efficient enantioselective procedure operating in the presence of catalytic amounts of chiral phosphoric acids (Table 2) [57]. Two synthetic protocols have been elaborated allowing the engagement of different classes of substrates (Table 2). In addition, the group of Tan adapted their findings to the chiral phosphoric acid-catalyzed three-component Ugi reaction of an aldehyde 2, an isocyanide 3, and a primary amine 5 [58].
Table 2: Asymmetric protocols for Ugi four-component reaction (U-4CR) by Houk, Tan and co-workers.
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-163-i8.svg?max-width=637&scale=1.0)
|
||||
| Substrate scope | Catalytic system | Conditions | Yield range, % | ee range, % |
| R2 = Alk; R3 = Ar |
![[Graphic 7]](/bjoc/content/inline/1860-5397-16-163-i9.svg?max-width=637&scale=1.0)
(5 mol %) Cy = cyclohexyl |
DCM (0.05 M), argon, 5 Å MS, −20 °C, 12 h | 43–96 | 75–97 |
| R2 = Ar; R3 = Alk |
![[Graphic 8]](/bjoc/content/inline/1860-5397-16-163-i10.svg?max-width=637&scale=1.0)
(10 mol %) |
cyclohexane (0.025 M), argon, 5 Å MS, 20 °C, 36 h–7 d | 60–96 | 80–94 |
As a part of our ongoing research, we have been involved in the synthesis and utilization of 2-oxo-aldehyde-derived Ugi adducts 8 (Scheme 1). These products possess an increased nucleophilicity of the peptidyl position compared to the standard Ugi adducts owing to an additional electron-withdrawing group introduced with the 2-oxo-aldehyde 7. For example, we have explored the potential of the peptidyl reactive site in a number of enolization-driven post-Ugi cyclizations leading to the assembly of pyrrol-2-ones 9 and 10 [59,60]. We have also taken advantage of a 1,3-dicarbonyl moiety present in 8 by engaging them in an enolization-triggered complexation with boron trifluoride diethyl etherate. The resulting O,O-chelated boron complexes 11 turned out to be strong solid-state emitters featuring clear aggregation-induced emission (AIE) characteristics [61].
![[1860-5397-16-163-i1]](/bjoc/content/inline/1860-5397-16-163-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Post-transformations of 2-oxo-aldehyde-derived Ugi adducts 8.
Scheme 1: Post-transformations of 2-oxo-aldehyde-derived Ugi adducts 8.
Encouraged by these results, we decided to attempt the reaction of 2-oxo-aldehyde-derived Ugi adducts 8 with suitable electrophiles in the presence of chiral catalyst or promotor. In this way, the peptidyl position of 8 could be simultaneously functionalized and stereodefined leading to the formation of enantioenriched products 12 (Scheme 1). Towards this goal, we have previously tested the reactivity of 8 in the intermolecular aldol addition to ethyl glyoxalate [62]. As these attempts met with failure, we have turned our attention to enantioselective fluorination reactions [63-65]. Specifically, we decided to explore the enantioselective electrophilic fluorination of carbonyl compounds promoted by cinchona alkaloid derivatives developed independently by Shibata, Takeuchi and co-workers [65,66] and by the group of Cahard [67-70]. The method proved to be applicable to a broad range of substrates under a variety of conditions [71-78] and was successfully utilized for the synthesis of several bioactive molecules [79-81]. Herein we present its adaptation for the enantioselective fluorination of 2-oxo-aldehyde-derived Ugi adducts 8 leading to the installment of a fluorine-bound quaternary stereocenter at the peptidyl position. It should be stressed that the exploration of methods for the enantioselective fluorination continues to be an important topic considering the ever-growing role of fluorine derivatives in drug design and development [82,83]. An alternative strategy to prepare related quaternary carbon-containing adducts bearing fluorine and nitrogen atoms involves the asymmetric addition of α-fluoro-β-ketoesters or α-fluoro-α-nitro esters to appropriate electrophiles [84-89].
Results and Discussion
Initially, we prepared a series of Ugi adducts 8 by varying acid, 2-oxo-aldehyde, amine, and isocyanide components (Scheme 2). The product 8a obtained from benzoic acid, phenylglyoxal, benzylamine, and tert-butyl isocyanide was selected for the screening of the reaction parameters (Table 3). The overall process was conducted in a two-step one-pot fashion as was originally designed by Shibata. In the first step, a cinchona alkaloid derivative was allowed to react with an electrophilic fluorinating agent to afford an N-fluoroammonium salt of the cinchona alkaloid via transfer fluorination. In the second step, the Ugi adduct 8a was added to this in situ generated N-fluorocinchona intermediate to afford the desired fluorinated product 12a. Thus, according to this synthetic and mechanistic path, the N-fluorocinchona intermediate acted as an asymmetric electrophilic fluorinating agent.
![[1860-5397-16-163-i2]](/bjoc/content/inline/1860-5397-16-163-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2-oxo-aldehyde-derived Ugi adducts.
Scheme 2: Synthesis of 2-oxo-aldehyde-derived Ugi adducts.
Table 3: Screening of the conditions for enantioselective fluorination of Ugi adduct 8a.a
![[Graphic 9]](/bjoc/content/inline/1860-5397-16-163-i11.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Cinchona alkaloid derivative | Fluorinating agent | Solvent | Time, h | Yield,b % | ee (major enantiomer),c % |
| 1 | Q | Selectfluor | MeCN | 23 | 88 | 19 (F) |
| 2 | Q | Selectfluor | THF | 19 | 94 | 21 (F) |
| 3 | Q | Selectfluor | DCM | 21 | 74 | 26 (F) |
| 4d | Q | Selectfluor | DCM | 70 | 42 | 20 (F) |
| 5 | Q | Selectfluor | CHCl3 | 21 | 71–79 | 31–44 (F) |
| 6 | CPN | Selectfluor | CHCl3 | 21 | –e | –e |
| 7 | DHQ | Selectfluor | CHCl3 | 16-21 | 85–90 | 46–57 (F) |
| 8 | DHQ | Selectfluor | THF | 12 | 76 | 43 (F) |
| 9 | Q-Bn | Selectfluor | CHCl3 | 21 | 78–83 | 39–41 (F) |
| 10 | Q-Ac | Selectfluor | CHCl3 | 21–24 | 35–59 | 42–51 (F) |
| 11 | Q-Bz | Selectfluor | CHCl3 | 20 | 62 | 32 (F) |
| 12 | DHQ-Bn | Selectfluor or NFSI | CHCl3 | 16–22 | 60–81 | 57–73 (F) |
| 13 | QD | Selectfluor or NFSI | CHCl3 | 21–23 | 83–94 | 51–69 (S) |
| 14 | DHQD | Selectfluor | CHCl3 | 21 | 86–92 | 15–21 (S) |
| 15 | QD-Me | Selectfluor | CHCl3 | 23 | 75 | 14 (S) |
| 16 | QD-Ac | Selectfluor | CHCl3 | 20 | 29 | 27 (S) |
| 17 | QD-Bn | Selectfluor | CHCl3 | 21 | 80 | 29 (S) |
| 18 | DHQD-Bn | Selectfluor | CHCl3 | 21 | 87 | 23 (S) |
aThe reactions were run on a 0.15–0.2 mmol scale; bisolated yield; сdetermined by chiral HPLC analysis, S: slower enantiomer, F: faster enantiomer; dthe reaction was conducted with 0.2 equiv of Q; eno product 12a was formed.
Conducting the fluorination of 8a with a quinine (Q)/Selectfluor combination in different solvents revealed chloroform as a preferred reaction medium for this process with acetonitrile, tetrahydrofuran, and dichloromethane being less efficient in terms of ee of the obtained product 12a (Table 3, entries 1–5). Using catalytic quantities of quinine led to a prolonged reaction time and diminished yield and ee of 12a compared to the analogous reaction with stoichiometric amounts of quinine (Table 3, entry 4 versus entry 3). Thus, we focused on the exploration of a stoichiometric version considering that most of cinchona alkaloid derivatives are readily accessible and recoverable. The original Shibata’s protocol also relied on the use of stoichiometric amounts of the chiral promotor that was in an agreement with a two-step reaction mode requiring the initial formation of the asymmetric fluorinating agent. An attempt to use cupreine, a member of the cinchona family featuring free phenolic hydroxy group, met with failure producing no desired product 12a (Table 3, entry 6). Using dihydroquinine (DHQ) led to an improved ee of 12a compared to the analogous reactions with quinine (Table 3, entries 7 and 8 versus entries 5 and 2, respectively). Testing ether and ester derivatives of Q and DHQ (Table 3, entries 9–12) led to further improvement allowing to obtain 12a with up to 73% ee through the reaction with dihydroquinine benzyl ether (DHQ-Bn, Table 3, entry 12). Switching to quinidine (QD), dihydroquinidine (DHQD), and their derivatives allowed to alter the stereoselectivity of the process leading to the preferred formation of another enantiomer of 12a (Table 3, entries 13–18) with the QD-promoted reaction affording the best ee value (Table 3, entry 13). Additionally, we tested N-fluorobenzenesulfonimide (NFSI) as an alternative fluorinating agent in a combination with DHQ-Bn and QD with the outcome being indistinguishable with the analogous reactions with Selectfluor.
Having these results in hand, we moved to investigating the scope and limitation of this procedure with the Ugi adducts 8 obtained previously (Table 4). In order to have a more balanced representation we decided to test most of these substrates 8 with eight different cinchona alkaloid derivatives including Q, DHQ, QD, DHQD, and their benzyl ether derivatives. To assure reproducibility, some of these reactions were repeated two to five times. In all such cases a range for the yield and ee value is reported in Table 4. Testing Ugi adducts 8b and 8c that similarly to 8a were obtained from benzylamine and tert-butyl isocyanide by varying either acid or 2-oxo-aldehyde components revealed that it was difficult to achieve high enantioselectivity for such substrates. Specifically, none of the studied chiral promoters could afford the desired products 12b and 12c with the ee value consistently higher than 50%. Turning to the substrates 8d–g derived from aromatic amines we were able to achieve improved enantioselectivity. The best results were obtained with Q-Bn, DHQ-Bn, DHQD, and QD-Bn that performed consistently well with each of the substrates 8d–g delivering the corresponding fluorinated products 12d–g with the ee values higher than 50% and in some cases close or even over 70%. Furthermore, it was possible to divert the selectivity of these reactions towards the preferred formation of either of the enantiomers by choosing an appropriate chiral promotor. Testing Ugi adduct 8h allowed us to highlight the possibility of simultaneous variation of acid, 2-oxo-aldehyde, and amine components. Overall, four cinchona derivatives were screened with 8h producing the fluorinated product 12h with moderate to good enantioselectivity, which was consistent with the performance of other arylamine-derived Ugi adducts 8d–g. The last investigated Ugi adduct 8i featured the variation of the isocyanide component. Conducting fluorinations with this n-butyl isocyanide-derived Ugi adduct 8i, the desired product 12i could be successfully obtained. However, the degree of enantioselective induction was lower than in the reactions with the analogous tert-butyl isocyanide-derived substrate 8d. This could probably be ascribed to a reduced rigidity and bulkiness of the n-butyl group as compared to tert-butyl.
Table 4: Scope and limitations of the asymmetric electrophilic fluorination of 2-oxo-aldehyde-derived Ugi adducts 8.a
![[Graphic 10]](/bjoc/content/inline/1860-5397-16-163-i12.svg?max-width=637&scale=1.0)
|
|||||||||
| Q | DHQ | Q-Bn | DHQ‑Bn | QD | DHQD | QD-Bn | DHQD-Bn | ||
![[Graphic 11]](/bjoc/content/inline/1860-5397-16-163-i13.svg?max-width=637&scale=1.0)
12a |
yield,b % | 71–79 | 85–90 | 78–83 | 60–81 | 83–94 | 86–92 | 80 | 87 |
| ee,c % |
31–44
(F) |
46–57
(F) |
39–41
(F) |
57–73
(F) |
51–69
(S) |
15–21
(S) |
29
(S) |
23
(S) |
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-16-163-i14.svg?max-width=637&scale=1.0)
12b |
yield,b % | 78–85 | 84–97 | 79 | 76–90 | 83–89 | 95–97 | 67–80 | 81 |
| ee,c % |
23–34
(S) |
29–39
(S) |
39
(S) |
38–51
(S) |
24–28
(F) |
9–15
(F) |
20–24
(F) |
21
(F) |
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-16-163-i15.svg?max-width=637&scale=1.0)
12c |
yield,b % | 59–81 | 60–81 | 83 | 74–89 | 68–85 | 72–78 | 81 | 78 |
| ee,c % |
39–46
(S) |
39–47
(S) |
35
(S) |
36
(S) |
30–43
(F) |
23–25
(F) |
25
(F) |
23
(F) |
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-16-163-i16.svg?max-width=637&scale=1.0)
12d |
yield,b % | 91 | 81 | 78 | 96 | 71–91 | 84 | 85 | 82 |
| ee,c % |
24
(F) |
22
(F) |
66
(S) |
71
(S) |
47–60
(S) |
57
(S) |
59
(F) |
38
(F) |
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-16-163-i17.svg?max-width=637&scale=1.0)
12e |
yield,b % | 68–78 | 91–97 | 78–82 | 86–97 | 89–98 | 84–91 | 95–98 | 85 |
| ee,c % |
18–30
(F) |
19–23
(F) |
65–69
(S) |
71–74
(S) |
49–50
(S) |
58–65
(S) |
61–67
(F) |
42
(F) |
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-16-163-i18.svg?max-width=637&scale=1.0)
12f |
yield,b % | 88–89 | 91–92 | 84–88 | 85–86 | 71–78 | 89–98 | 89 | 79 |
| ee,c % |
23–27
(F) |
18–19
(F) |
66–67
(S) |
69-72
(S) |
50–55
(S) |
53–64
(S) |
57–58
(F) |
40
(F) |
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-16-163-i19.svg?max-width=637&scale=1.0)
12g |
yield,b % | 74–85 | 90 | 82–84 | 86–87 | 82–88 | 75–76 | 75–84 | 78–81 |
| ee,c % |
25–37
(F) |
18–24
(F) |
65–66
(S) |
63–70
(S) |
48–59
(S) |
51–61
(S) |
59–60
(F) |
37–54
(F) |
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-16-163-i20.svg?max-width=637&scale=1.0)
12h |
yield,b % | –d | –d | 64 | 86 | 84 | –d | 75 | –d |
| ee,c % | –d | –d |
71
(F) |
73
(F) |
54
(F) |
–d |
55
(S) |
–d | |
![[Graphic 19]](/bjoc/content/inline/1860-5397-16-163-i21.svg?max-width=637&scale=1.0)
12i |
yield,b % | –d | –d | 51 | 54 | 67 | –d | 49 | –d |
| ee,c % | –d | –d |
44
(F) |
48
(F) |
55
(F) |
–d |
29
(S) |
–d | |
aThe reactions were run on a 0.15–0.2 mmol scale; bisolated yield; сdetermined by chiral HPLC analysis, S: slower enantiomer, F: faster enantiomer; dnot conducted.
For the substrates 8a–g tested with eight cinchona alkaloid derivatives fluorinations with QD and DHQD led to the reversed stereoselectivity as compared to the reactions with Q and DHQ. For the benzylamine-derived substrates 8a–c benzyl ether derivatives of cinchona alkaloids favored the formation of the same enantiomer of 12 as their free alcohol counterparts (e.g., both Q-Bn and Q favored the formation of the same enantiomer of 12). In contrast, for the substrates 8d–g derived from aromatic amines a reversed trend was observed. In case of 8d–g, benzyl ether derivatives of cinchona alkaloids favored the formation of the opposite enantiomer of 12 as compared to the one favored by the parent alkaloids featuring free alcohol moiety (e.g., Q-Bn favored the formation of the enantiomer of 12 with the absolute configuration opposite to the one favored by Q). Substrates 8h and 8i were tested with only four cinchona alkaloid derivatives, but the observed stereochemical trends appeared to be consistent with those established for the rest of arylamine-derived Ugi adducts 8d–g.
Finally, we attempted to determine the absolute configuration of a stereocenter for the major enantiomer of the representative fluorinated product. Using a sample of product 12e with the ee value of 74% we were able to obtain crystals suitable for the structure determination via X-ray crystallographic analysis. It was found that a part of the substance crystallized in a racemic form while the rest crystallized in an enantiopure form. By resolving both types of crystals [90], the configuration of the major slow enantiomer was successfully established as S (Figure 1).
![[1860-5397-16-163-1]](/bjoc/content/figures/1860-5397-16-163-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular representation of the X-ray crystal structure of (S)-12e (slow enantiomer).
Figure 1: Molecular representation of the X-ray crystal structure of (S)-12e (slow enantiomer).
Conclusion
In summary, we have explored a new strategy for controlling the stereochemistry in the Ugi adducts through the enantioselective post-Ugi functionalization. Specifically, we have adapted the cinchona alkaloid-promoted asymmetric electrophilic fluorination for derivatizing the 2-oxo-aldehyde-derived Ugi adducts. This allowed us to obtain the post-Ugi products fluorinated at the peptidyl position with the enantiomeric excess values in several instances reaching more than 70%.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of spectra. | ||
| Format: PDF | Size: 4.1 MB | Download |
| Supporting Information File 2: Crystallographic data (CIF) for compound (S)-12e. | ||
| Format: CIF | Size: 2.8 MB | Download |
| Supporting Information File 3: Crystallographic data (CIF) for compound (rac)-12e. | ||
| Format: CIF | Size: 985.4 KB | Download |
Acknowledgements
We thank Prof. Xingwang Wang for his advice to explore the electrophilic fluorination as a tool for the enantioselective post-Ugi functionalization.
Funding
This work was supported by the start-up fund from Soochow University (grant Q410900714), National Natural Science Foundation of China (grant 21650110445), Natural Science Foundation of Jiangsu Province of China (grant BK20160310), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the project of scientific and technologic infrastructure of Suzhou (grant SZS201708). M.H. and M.Z. are grateful to the Chinese Scholarship Council (CSC) for providing doctoral scholarships.
References
-
Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Chem. – Eur. J. 2000, 6, 3321–3329. doi:10.1002/1521-3765(20000915)6:18<3321::aid-chem3321>3.0.co;2-a
Return to citation in text: [1] -
Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions, 1st ed.; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527605118
Return to citation in text: [1] -
Sunderhaus, J. D.; Martin, S. F. Chem. – Eur. J. 2009, 15, 1300–1308. doi:10.1002/chem.200802140
Return to citation in text: [1] -
Ganem, B. Acc. Chem. Res. 2009, 42, 463–472. doi:10.1021/ar800214s
Return to citation in text: [1] -
Orru, R. V. A.; Ruijter, E., Eds. Synthesis of Heterocycles via Multicomponent Reactions I; Topics in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2010. doi:10.1007/978-3-642-12675-8
Return to citation in text: [1] -
Orru, R. V. A.; Ruijter, E., Eds. Synthesis of Heterocycles via Multicomponent Reactions II; Topics in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2010. doi:10.1007/978-3-642-15455-3
Return to citation in text: [1] -
Jiang, B.; Rajale, T.; Wever, W.; Tu, S.-J.; Li, G. Chem. – Asian J. 2010, 5, 2318–2335. doi:10.1002/asia.201000310
Return to citation in text: [1] -
Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r
Return to citation in text: [1] -
Zhu, J.; Wang, Q.; Wang, M.-X., Eds. Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527678174
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Moni, L.; Riva, R. Eur. J. Org. Chem. 2014, 2005–2015. doi:10.1002/ejoc.201301715
Return to citation in text: [1] -
Cioc, R. C.; Ruijter, E.; Orru, R. V. A. Green Chem. 2014, 16, 2958–2975. doi:10.1039/c4gc00013g
Return to citation in text: [1] -
Levi, L.; Müller, T. J. J. Chem. Soc. Rev. 2016, 45, 2825–2846. doi:10.1039/c5cs00805k
Return to citation in text: [1] -
Murlykina, M. V.; Morozova, A. D.; Zviagin, I. M.; Sakhno, Y. I.; Desenko, S. M.; Chebanov, V. A. Front. Chem. (Lausanne, Switz.) 2018, 6, 527. doi:10.3389/fchem.2018.00527
Return to citation in text: [1] -
Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u
Return to citation in text: [1] -
Akritopoulou-Zanze, I. Curr. Opin. Chem. Biol. 2008, 12, 324–331. doi:10.1016/j.cbpa.2008.02.004
Return to citation in text: [1] -
Koopmanschap, G.; Ruijter, E.; Orru, R. V. A. Beilstein J. Org. Chem. 2014, 10, 544–598. doi:10.3762/bjoc.10.50
Return to citation in text: [1] -
Váradi, A.; Palmer, T. C.; Notis Dardashti, R.; Majumdar, S. Molecules 2016, 21, 19. doi:10.3390/molecules21010019
Return to citation in text: [1] -
Bode, M. L.; Gravestock, D.; Rousseau, A. L. Org. Prep. Proced. Int. 2016, 48, 89–221. doi:10.1080/00304948.2016.1138072
Return to citation in text: [1] -
Passerini, M. Gazz. Chim. Ital. 1921, 51, 126–129.
Return to citation in text: [1] -
Banfi, L.; Riva, R. Org. React. 2005, 65, 1–140. doi:10.1002/0471264180.or065.01
Return to citation in text: [1] -
Kazemizadeh, A. R.; Ramazani, A. Curr. Org. Chem. 2012, 16, 418–450. doi:10.2174/138527212799499868
Return to citation in text: [1] -
Chandgude, A. Multicomponent Reactions: Development, Scope, and Applications. Ph.D. Thesis, University of Groningen, Groningen, Netherlands, 2017.
Return to citation in text: [1] -
Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C. Angew. Chem. 1959, 71, 386. doi:10.1002/ange.19590711110
Return to citation in text: [1] -
Ugi, I.; Steinbrückner, C. Angew. Chem. 1960, 72, 267–268. doi:10.1002/ange.19600720709
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Riva, R. Synthesis of Heterocycles Through Classical Ugi and Passerini Reactions Followed by Secondary Transformations Involving One or Two Additional Functional Groups. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A.; Ruijter, E., Eds.; Topics in Heterocyclic Chemistry, Vol. 23; Springer: Berlin, Germany, 2010; pp 1–39. doi:10.1007/7081_2009_23
Return to citation in text: [1] -
Sharma, U. K.; Sharma, N.; Vachhani, D. D.; Van der Eycken, E. V. Chem. Soc. Rev. 2015, 44, 1836–1860. doi:10.1039/c4cs00253a
Return to citation in text: [1] -
Zhang, Z.; Zheng, X.; Guo, C. Chin. J. Org. Chem. 2016, 36, 1241–1265. doi:10.6023/cjoc201511004
Return to citation in text: [1] -
Li, X.; Jia, X.; Yin, L. Chin. J. Org. Chem. 2017, 37, 2237–2249. doi:10.6023/cjoc201704026
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Lambruschini, C.; Moni, L.; Riva, R. Chem. Heterocycl. Compd. 2017, 53, 382–408. doi:10.1007/s10593-017-2065-1
Return to citation in text: [1] -
Heravi, M. M.; Mohammadkhani, L. Synthesis of various N-heterocycles using the four-component Ugi reaction. Advances in Heterocyclic Chemistry; Elsevier: Amsterdam, Netherlands, 2020; Vol. 131, pp 351–403. doi:10.1016/bs.aihch.2019.04.001
Return to citation in text: [1] -
Bariwal, J.; Kaur, R.; Voskressensky, L. G.; Van der Eycken, E. V. Front. Chem. (Lausanne, Switz.) 2018, 6, 557. doi:10.3389/fchem.2018.00557
Return to citation in text: [1] -
Mohammadi-Khanaposhtani, M.; Jalalimanesh, N.; Saeedi, M.; Larijani, B.; Mahdavi, M. Mol. Diversity 2020, 24, 855–887. doi:10.1007/s11030-019-09975-y
Return to citation in text: [1] -
Wang, Q.; Wang, D.-X.; Wang, M.-X.; Zhu, J. Acc. Chem. Res. 2018, 51, 1290–1300. doi:10.1021/acs.accounts.8b00105
Return to citation in text: [1] [2] -
Shaabani, S.; Dömling, A. Angew. Chem., Int. Ed. 2018, 57, 16266–16268. doi:10.1002/anie.201811129
Return to citation in text: [1] [2] -
Riva, R. Science 2018, 361, 1072–1073. doi:10.1126/science.aau7497
Return to citation in text: [1] [2] -
Huang, Y.; Wolf, S.; Koes, D.; Popowicz, G. M.; Camacho, C. J.; Holak, T. A.; Dömling, A. ChemMedChem 2012, 7, 49–52. doi:10.1002/cmdc.201100428
Return to citation in text: [1] -
Kusebauch, U.; Beck, B.; Messer, K.; Herdtweck, E.; Dömling, A. Org. Lett. 2003, 5, 4021–4024. doi:10.1021/ol035010u
Return to citation in text: [1] [2] [3] -
Andreana, P. R.; Liu, C. C.; Schreiber, S. L. Org. Lett. 2004, 6, 4231–4233. doi:10.1021/ol0482893
Return to citation in text: [1] [2] [3] -
Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Angew. Chem., Int. Ed. 2008, 47, 388–391. doi:10.1002/anie.200704315
Return to citation in text: [1] [2] [3] -
Zhang, J.; Lin, S.-X.; Cheng, D.-J.; Liu, X.-Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 14039–14042. doi:10.1021/jacs.5b09117
Return to citation in text: [1] [2] [3] -
Denmark, S. E.; Fan, Y. J. Org. Chem. 2005, 70, 9667–9676. doi:10.1021/jo050549m
Return to citation in text: [1] -
Wang, S.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Eur. J. Org. Chem. 2007, 4076–4080. doi:10.1002/ejoc.200700340
Return to citation in text: [1] -
Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Org. Lett. 2007, 9, 3615–3618. doi:10.1021/ol7014658
Return to citation in text: [1] -
Yue, T.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Angew. Chem., Int. Ed. 2008, 47, 9454–9457. doi:10.1002/anie.200804213
Return to citation in text: [1] -
Yue, T.; Wang, M.-X.; Wang, D.-X.; Masson, G.; Zhu, J. J. Org. Chem. 2009, 74, 8396–8399. doi:10.1021/jo9017765
Return to citation in text: [1] -
Mihara, H.; Xu, Y.; Shepherd, N. E.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 8384–8385. doi:10.1021/ja903158x
Return to citation in text: [1] -
Zeng, X.; Ye, K.; Lu, M.; Chua, P. J.; Tan, B.; Zhong, G. Org. Lett. 2010, 12, 2414–2417. doi:10.1021/ol1007789
Return to citation in text: [1] -
Gulevich, A. V.; Balenkova, E. S.; Nenajdenko, V. G. J. Org. Chem. 2007, 72, 7878–7885. doi:10.1021/jo071030o
Return to citation in text: [1] -
Nenajdenko, V. G.; Gulevich, A. V.; Chernichenko, K. Y.; Sokolova, N. V.; Balenkova, E. S. Mendeleev Commun. 2011, 21, 245–246. doi:10.1016/j.mencom.2011.09.004
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Cerulli, V.; Rocca, V.; Riva, R. Beilstein J. Org. Chem. 2011, 7, 976–979. doi:10.3762/bjoc.7.109
Return to citation in text: [1] -
Banfi, L.; Basso, A.; Chiappe, C.; De Moliner, F.; Riva, R.; Sonaglia, L. Org. Biomol. Chem. 2012, 10, 3819–3829. doi:10.1039/c2ob25060h
Return to citation in text: [1] -
Zhu, D.; Xia, L.; Pan, L.; Li, S.; Chen, R.; Mou, Y.; Chen, X. J. Org. Chem. 2012, 77, 1386–1395. doi:10.1021/jo2021967
Return to citation in text: [1] -
Caputo, S.; Basso, A.; Moni, L.; Riva, R.; Rocca, V.; Banfi, L. Beilstein J. Org. Chem. 2016, 12, 139–143. doi:10.3762/bjoc.12.15
Return to citation in text: [1] -
Lambruschini, C.; Moni, L.; Banfi, L. Eur. J. Org. Chem. 2020, 3766–3778. doi:10.1002/ejoc.202000016
Return to citation in text: [1] -
Hashimoto, T.; Kimura, H.; Kawamata, Y.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 7279–7281. doi:10.1002/anie.201201905
Return to citation in text: [1] -
Zhao, W.; Huang, L.; Guan, Y.; Wulff, W. D. Angew. Chem., Int. Ed. 2014, 53, 3436–3441. doi:10.1002/anie.201310491
Return to citation in text: [1] -
Zhang, J.; Yu, P.; Li, S.-Y.; Sun, H.; Xiang, S.-H.; Wang, J.; Houk, K. N.; Tan, B. Science 2018, 361, eaas8707. doi:10.1126/science.aas8707
Return to citation in text: [1] -
Zhang, J.; Wang, Y.-Y.; Sun, H.; Li, S.-Y.; Xiang, S.-H.; Tan, B. Sci. China: Chem. 2020, 63, 47–54. doi:10.1007/s11426-019-9606-2
Return to citation in text: [1] -
Nechaev, A. A.; Peshkov, A. A.; Peshkov, V. A.; Van der Eycken, E. V. Synthesis 2016, 48, 2280–2286. doi:10.1055/s-0035-1561422
Return to citation in text: [1] -
Peshkov, A. A.; Peshkov, V. A.; Li, Z.; Pereshivko, O. P.; Van der Eycken, E. V. Eur. J. Org. Chem. 2014, 6390–6393. doi:10.1002/ejoc.201402723
Return to citation in text: [1] -
Wei, H.; Wang, G.; Wang, Y.; Li, B.; Huang, J.; Kashtanov, S.; Van Hecke, K.; Pereshivko, O. P.; Peshkov, V. A. Chem. – Asian J. 2017, 12, 825–829. doi:10.1002/asia.201700051
Return to citation in text: [1] -
Hasan, M.; Zaman, M.; Peshkov, A. A.; Amire, N.; Les, A.; Nechaev, A. A.; Wang, Y.; Kashtanov, S.; Van der Eycken, E. V.; Pereshivko, O. P.; Peshkov, V. A. Eur. J. Org. Chem. 2020, 3378–3389. doi:10.1002/ejoc.202000015
Return to citation in text: [1] -
Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014
Return to citation in text: [1] -
Lectard, S.; Hamashima, Y.; Sodeoka, M. Adv. Synth. Catal. 2010, 352, 2708–2732. doi:10.1002/adsc.201000624
Return to citation in text: [1] -
Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944–2947. doi:10.1021/ol201007e
Return to citation in text: [1] [2] -
Shibata, N.; Suzuki, E.; Takeuchi, Y. J. Am. Chem. Soc. 2000, 122, 10728–10729. doi:10.1021/ja002732x
Return to citation in text: [1] -
Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t
Return to citation in text: [1] -
Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Roques, N. Org. Lett. 2000, 2, 3699–3701. doi:10.1021/ol006610l
Return to citation in text: [1] -
Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Toupet, L.; Roques, N. Tetrahedron Lett. 2001, 42, 1867–1869. doi:10.1016/s0040-4039(01)00017-x
Return to citation in text: [1] -
Mohar, B.; Baudoux, J.; Plaquevent, J.-C.; Cahard, D. Angew. Chem., Int. Ed. 2001, 40, 4214–4216. doi:10.1002/1521-3773(20011119)40:22<4214::aid-anie4214>3.0.co;2-b
Return to citation in text: [1] -
Baudequin, C.; Plaquevent, J.-C.; Audouard, C.; Cahard, D. Green Chem. 2002, 4, 584–586. doi:10.1039/b208817g
Return to citation in text: [1] -
Baudequin, C.; Loubassou, J.-F.; Plaquevent, J.-C.; Cahard, D. J. Fluorine Chem. 2003, 122, 189–193. doi:10.1016/s0022-1139(03)00085-x
Return to citation in text: [1] -
Thierry, B.; Audouard, C.; Plaquevent, J.-C.; Cahard, D. Synlett 2004, 856–860. doi:10.1055/s-2004-817781
Return to citation in text: [1] -
Mohar, B.; Sterk, D.; Ferron, L.; Cahard, D. Tetrahedron Lett. 2005, 46, 5029–5031. doi:10.1016/j.tetlet.2005.05.074
Return to citation in text: [1] -
Fukuzumi, T.; Shibata, N.; Sugiura, M.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2006, 127, 548–551. doi:10.1016/j.jfluchem.2006.01.004
Return to citation in text: [1] -
Ramírez, J.; Huber, D. P.; Togni, A. Synlett 2007, 1143–1147. doi:10.1055/s-2007-973897
Return to citation in text: [1] -
Yi, W.-B.; Huang, X.; Zhang, Z.; Zhu, D.-R.; Cai, C.; Zhang, W. Beilstein J. Org. Chem. 2012, 8, 1233–1240. doi:10.3762/bjoc.8.138
Return to citation in text: [1] -
Wang, M.; Wang, B. M.; Shi, L.; Tu, Y. Q.; Fan, C.-A.; Wang, S. H.; Hu, X. D.; Zhang, S. Y. Chem. Commun. 2005, 5580–5582. doi:10.1039/b510004f
Return to citation in text: [1] -
Shibata, N.; Ishimaru, T.; Suzuki, E.; Kirk, K. L. J. Org. Chem. 2003, 68, 2494–2497. doi:10.1021/jo026792s
Return to citation in text: [1] -
Zoute, L.; Audouard, C.; Plaquevent, J.-C.; Cahard, D. Org. Biomol. Chem. 2003, 1, 1833–1834. doi:10.1039/b303113f
Return to citation in text: [1] -
Shibata, N.; Ishimaru, T.; Nakamura, M.; Toru, T. Synlett 2004, 2509–2512. doi:10.1055/s-2004-834810
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] -
Huber, D. P.; Stanek, K.; Togni, A. Tetrahedron: Asymmetry 2006, 17, 658–664. doi:10.1016/j.tetasy.2006.01.035
Return to citation in text: [1] -
Mang, J.-Y.; Kim, D.-Y. Bull. Korean Chem. Soc. 2008, 29, 2091–2092. doi:10.5012/bkcs.2008.29.11.2091
Return to citation in text: [1] -
Mang, J. Y.; Kwon, D. G.; Kim, D. Y. J. Fluorine Chem. 2009, 130, 259–262. doi:10.1016/j.jfluchem.2008.11.001
Return to citation in text: [1] -
Han, X.; Zhong, F.; Lu, Y. Adv. Synth. Catal. 2010, 352, 2778–2782. doi:10.1002/adsc.201000562
Return to citation in text: [1] -
Cui, H.-F.; Li, P.; Wang, X.-W.; Chai, Z.; Yang, Y.-Q.; Cai, Y.-P.; Zhu, S.-Z.; Zhao, G. Tetrahedron 2011, 67, 312–317. doi:10.1016/j.tet.2010.11.041
Return to citation in text: [1] -
Kwiatkowski, J.; Lu, Y. Org. Biomol. Chem. 2015, 13, 2350–2359. doi:10.1039/c4ob02486a
Return to citation in text: [1] -
Crystallographic data for rac-12e (CCDC 2015756) and (S)-12e (CCDC 2015755) have been deposited with the Cambridge Crystallographic Data Centre.
Return to citation in text: [1]
| 79. | Shibata, N.; Ishimaru, T.; Suzuki, E.; Kirk, K. L. J. Org. Chem. 2003, 68, 2494–2497. doi:10.1021/jo026792s |
| 80. | Zoute, L.; Audouard, C.; Plaquevent, J.-C.; Cahard, D. Org. Biomol. Chem. 2003, 1, 1833–1834. doi:10.1039/b303113f |
| 81. | Shibata, N.; Ishimaru, T.; Nakamura, M.; Toru, T. Synlett 2004, 2509–2512. doi:10.1055/s-2004-834810 |
| 82. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 83. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 84. | Huber, D. P.; Stanek, K.; Togni, A. Tetrahedron: Asymmetry 2006, 17, 658–664. doi:10.1016/j.tetasy.2006.01.035 |
| 85. | Mang, J.-Y.; Kim, D.-Y. Bull. Korean Chem. Soc. 2008, 29, 2091–2092. doi:10.5012/bkcs.2008.29.11.2091 |
| 86. | Mang, J. Y.; Kwon, D. G.; Kim, D. Y. J. Fluorine Chem. 2009, 130, 259–262. doi:10.1016/j.jfluchem.2008.11.001 |
| 87. | Han, X.; Zhong, F.; Lu, Y. Adv. Synth. Catal. 2010, 352, 2778–2782. doi:10.1002/adsc.201000562 |
| 88. | Cui, H.-F.; Li, P.; Wang, X.-W.; Chai, Z.; Yang, Y.-Q.; Cai, Y.-P.; Zhu, S.-Z.; Zhao, G. Tetrahedron 2011, 67, 312–317. doi:10.1016/j.tet.2010.11.041 |
| 89. | Kwiatkowski, J.; Lu, Y. Org. Biomol. Chem. 2015, 13, 2350–2359. doi:10.1039/c4ob02486a |
| 1. | Bienaymé, H.; Hulme, C.; Oddon, G.; Schmitt, P. Chem. – Eur. J. 2000, 6, 3321–3329. doi:10.1002/1521-3765(20000915)6:18<3321::aid-chem3321>3.0.co;2-a |
| 2. | Zhu, J.; Bienaymé, H., Eds. Multicomponent Reactions, 1st ed.; Wiley-VCH: Weinheim, Germany, 2005. doi:10.1002/3527605118 |
| 3. | Sunderhaus, J. D.; Martin, S. F. Chem. – Eur. J. 2009, 15, 1300–1308. doi:10.1002/chem.200802140 |
| 4. | Ganem, B. Acc. Chem. Res. 2009, 42, 463–472. doi:10.1021/ar800214s |
| 5. | Orru, R. V. A.; Ruijter, E., Eds. Synthesis of Heterocycles via Multicomponent Reactions I; Topics in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2010. doi:10.1007/978-3-642-12675-8 |
| 6. | Orru, R. V. A.; Ruijter, E., Eds. Synthesis of Heterocycles via Multicomponent Reactions II; Topics in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2010. doi:10.1007/978-3-642-15455-3 |
| 7. | Jiang, B.; Rajale, T.; Wever, W.; Tu, S.-J.; Li, G. Chem. – Asian J. 2010, 5, 2318–2335. doi:10.1002/asia.201000310 |
| 8. | Dömling, A.; Wang, W.; Wang, K. Chem. Rev. 2012, 112, 3083–3135. doi:10.1021/cr100233r |
| 9. | Zhu, J.; Wang, Q.; Wang, M.-X., Eds. Multicomponent Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. doi:10.1002/9783527678174 |
| 10. | Banfi, L.; Basso, A.; Moni, L.; Riva, R. Eur. J. Org. Chem. 2014, 2005–2015. doi:10.1002/ejoc.201301715 |
| 11. | Cioc, R. C.; Ruijter, E.; Orru, R. V. A. Green Chem. 2014, 16, 2958–2975. doi:10.1039/c4gc00013g |
| 12. | Levi, L.; Müller, T. J. J. Chem. Soc. Rev. 2016, 45, 2825–2846. doi:10.1039/c5cs00805k |
| 13. | Murlykina, M. V.; Morozova, A. D.; Zviagin, I. M.; Sakhno, Y. I.; Desenko, S. M.; Chebanov, V. A. Front. Chem. (Lausanne, Switz.) 2018, 6, 527. doi:10.3389/fchem.2018.00527 |
| 25. | Banfi, L.; Basso, A.; Riva, R. Synthesis of Heterocycles Through Classical Ugi and Passerini Reactions Followed by Secondary Transformations Involving One or Two Additional Functional Groups. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R. V. A.; Ruijter, E., Eds.; Topics in Heterocyclic Chemistry, Vol. 23; Springer: Berlin, Germany, 2010; pp 1–39. doi:10.1007/7081_2009_23 |
| 26. | Sharma, U. K.; Sharma, N.; Vachhani, D. D.; Van der Eycken, E. V. Chem. Soc. Rev. 2015, 44, 1836–1860. doi:10.1039/c4cs00253a |
| 27. | Zhang, Z.; Zheng, X.; Guo, C. Chin. J. Org. Chem. 2016, 36, 1241–1265. doi:10.6023/cjoc201511004 |
| 28. | Li, X.; Jia, X.; Yin, L. Chin. J. Org. Chem. 2017, 37, 2237–2249. doi:10.6023/cjoc201704026 |
| 29. | Banfi, L.; Basso, A.; Lambruschini, C.; Moni, L.; Riva, R. Chem. Heterocycl. Compd. 2017, 53, 382–408. doi:10.1007/s10593-017-2065-1 |
| 30. | Heravi, M. M.; Mohammadkhani, L. Synthesis of various N-heterocycles using the four-component Ugi reaction. Advances in Heterocyclic Chemistry; Elsevier: Amsterdam, Netherlands, 2020; Vol. 131, pp 351–403. doi:10.1016/bs.aihch.2019.04.001 |
| 31. | Bariwal, J.; Kaur, R.; Voskressensky, L. G.; Van der Eycken, E. V. Front. Chem. (Lausanne, Switz.) 2018, 6, 557. doi:10.3389/fchem.2018.00557 |
| 32. | Mohammadi-Khanaposhtani, M.; Jalalimanesh, N.; Saeedi, M.; Larijani, B.; Mahdavi, M. Mol. Diversity 2020, 24, 855–887. doi:10.1007/s11030-019-09975-y |
| 40. | Zhang, J.; Lin, S.-X.; Cheng, D.-J.; Liu, X.-Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 14039–14042. doi:10.1021/jacs.5b09117 |
| 23. | Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C. Angew. Chem. 1959, 71, 386. doi:10.1002/ange.19590711110 |
| 24. | Ugi, I.; Steinbrückner, C. Angew. Chem. 1960, 72, 267–268. doi:10.1002/ange.19600720709 |
| 48. | Gulevich, A. V.; Balenkova, E. S.; Nenajdenko, V. G. J. Org. Chem. 2007, 72, 7878–7885. doi:10.1021/jo071030o |
| 49. | Nenajdenko, V. G.; Gulevich, A. V.; Chernichenko, K. Y.; Sokolova, N. V.; Balenkova, E. S. Mendeleev Commun. 2011, 21, 245–246. doi:10.1016/j.mencom.2011.09.004 |
| 50. | Banfi, L.; Basso, A.; Cerulli, V.; Rocca, V.; Riva, R. Beilstein J. Org. Chem. 2011, 7, 976–979. doi:10.3762/bjoc.7.109 |
| 51. | Banfi, L.; Basso, A.; Chiappe, C.; De Moliner, F.; Riva, R.; Sonaglia, L. Org. Biomol. Chem. 2012, 10, 3819–3829. doi:10.1039/c2ob25060h |
| 52. | Zhu, D.; Xia, L.; Pan, L.; Li, S.; Chen, R.; Mou, Y.; Chen, X. J. Org. Chem. 2012, 77, 1386–1395. doi:10.1021/jo2021967 |
| 53. | Caputo, S.; Basso, A.; Moni, L.; Riva, R.; Rocca, V.; Banfi, L. Beilstein J. Org. Chem. 2016, 12, 139–143. doi:10.3762/bjoc.12.15 |
| 54. | Lambruschini, C.; Moni, L.; Banfi, L. Eur. J. Org. Chem. 2020, 3766–3778. doi:10.1002/ejoc.202000016 |
| 19. | Passerini, M. Gazz. Chim. Ital. 1921, 51, 126–129. |
| 20. | Banfi, L.; Riva, R. Org. React. 2005, 65, 1–140. doi:10.1002/0471264180.or065.01 |
| 21. | Kazemizadeh, A. R.; Ramazani, A. Curr. Org. Chem. 2012, 16, 418–450. doi:10.2174/138527212799499868 |
| 22. | Chandgude, A. Multicomponent Reactions: Development, Scope, and Applications. Ph.D. Thesis, University of Groningen, Groningen, Netherlands, 2017. |
| 38. | Andreana, P. R.; Liu, C. C.; Schreiber, S. L. Org. Lett. 2004, 6, 4231–4233. doi:10.1021/ol0482893 |
| 14. | Dömling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168–3210. doi:10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u |
| 15. | Akritopoulou-Zanze, I. Curr. Opin. Chem. Biol. 2008, 12, 324–331. doi:10.1016/j.cbpa.2008.02.004 |
| 16. | Koopmanschap, G.; Ruijter, E.; Orru, R. V. A. Beilstein J. Org. Chem. 2014, 10, 544–598. doi:10.3762/bjoc.10.50 |
| 17. | Váradi, A.; Palmer, T. C.; Notis Dardashti, R.; Majumdar, S. Molecules 2016, 21, 19. doi:10.3390/molecules21010019 |
| 18. | Bode, M. L.; Gravestock, D.; Rousseau, A. L. Org. Prep. Proced. Int. 2016, 48, 89–221. doi:10.1080/00304948.2016.1138072 |
| 39. | Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Angew. Chem., Int. Ed. 2008, 47, 388–391. doi:10.1002/anie.200704315 |
| 41. | Denmark, S. E.; Fan, Y. J. Org. Chem. 2005, 70, 9667–9676. doi:10.1021/jo050549m |
| 42. | Wang, S.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Eur. J. Org. Chem. 2007, 4076–4080. doi:10.1002/ejoc.200700340 |
| 43. | Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Org. Lett. 2007, 9, 3615–3618. doi:10.1021/ol7014658 |
| 44. | Yue, T.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Angew. Chem., Int. Ed. 2008, 47, 9454–9457. doi:10.1002/anie.200804213 |
| 45. | Yue, T.; Wang, M.-X.; Wang, D.-X.; Masson, G.; Zhu, J. J. Org. Chem. 2009, 74, 8396–8399. doi:10.1021/jo9017765 |
| 46. | Mihara, H.; Xu, Y.; Shepherd, N. E.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2009, 131, 8384–8385. doi:10.1021/ja903158x |
| 47. | Zeng, X.; Ye, K.; Lu, M.; Chua, P. J.; Tan, B.; Zhong, G. Org. Lett. 2010, 12, 2414–2417. doi:10.1021/ol1007789 |
| 40. | Zhang, J.; Lin, S.-X.; Cheng, D.-J.; Liu, X.-Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 14039–14042. doi:10.1021/jacs.5b09117 |
| 37. | Kusebauch, U.; Beck, B.; Messer, K.; Herdtweck, E.; Dömling, A. Org. Lett. 2003, 5, 4021–4024. doi:10.1021/ol035010u |
| 38. | Andreana, P. R.; Liu, C. C.; Schreiber, S. L. Org. Lett. 2004, 6, 4231–4233. doi:10.1021/ol0482893 |
| 39. | Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Angew. Chem., Int. Ed. 2008, 47, 388–391. doi:10.1002/anie.200704315 |
| 40. | Zhang, J.; Lin, S.-X.; Cheng, D.-J.; Liu, X.-Y.; Tan, B. J. Am. Chem. Soc. 2015, 137, 14039–14042. doi:10.1021/jacs.5b09117 |
| 37. | Kusebauch, U.; Beck, B.; Messer, K.; Herdtweck, E.; Dömling, A. Org. Lett. 2003, 5, 4021–4024. doi:10.1021/ol035010u |
| 36. | Huang, Y.; Wolf, S.; Koes, D.; Popowicz, G. M.; Camacho, C. J.; Holak, T. A.; Dömling, A. ChemMedChem 2012, 7, 49–52. doi:10.1002/cmdc.201100428 |
| 90. | Crystallographic data for rac-12e (CCDC 2015756) and (S)-12e (CCDC 2015755) have been deposited with the Cambridge Crystallographic Data Centre. |
| 33. | Wang, Q.; Wang, D.-X.; Wang, M.-X.; Zhu, J. Acc. Chem. Res. 2018, 51, 1290–1300. doi:10.1021/acs.accounts.8b00105 |
| 34. | Shaabani, S.; Dömling, A. Angew. Chem., Int. Ed. 2018, 57, 16266–16268. doi:10.1002/anie.201811129 |
| 35. | Riva, R. Science 2018, 361, 1072–1073. doi:10.1126/science.aau7497 |
| 37. | Kusebauch, U.; Beck, B.; Messer, K.; Herdtweck, E.; Dömling, A. Org. Lett. 2003, 5, 4021–4024. doi:10.1021/ol035010u |
| 38. | Andreana, P. R.; Liu, C. C.; Schreiber, S. L. Org. Lett. 2004, 6, 4231–4233. doi:10.1021/ol0482893 |
| 39. | Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Angew. Chem., Int. Ed. 2008, 47, 388–391. doi:10.1002/anie.200704315 |
| 57. | Zhang, J.; Yu, P.; Li, S.-Y.; Sun, H.; Xiang, S.-H.; Wang, J.; Houk, K. N.; Tan, B. Science 2018, 361, eaas8707. doi:10.1126/science.aas8707 |
| 33. | Wang, Q.; Wang, D.-X.; Wang, M.-X.; Zhu, J. Acc. Chem. Res. 2018, 51, 1290–1300. doi:10.1021/acs.accounts.8b00105 |
| 34. | Shaabani, S.; Dömling, A. Angew. Chem., Int. Ed. 2018, 57, 16266–16268. doi:10.1002/anie.201811129 |
| 35. | Riva, R. Science 2018, 361, 1072–1073. doi:10.1126/science.aau7497 |
| 55. | Hashimoto, T.; Kimura, H.; Kawamata, Y.; Maruoka, K. Angew. Chem., Int. Ed. 2012, 51, 7279–7281. doi:10.1002/anie.201201905 |
| 56. | Zhao, W.; Huang, L.; Guan, Y.; Wulff, W. D. Angew. Chem., Int. Ed. 2014, 53, 3436–3441. doi:10.1002/anie.201310491 |
| 67. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 68. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Roques, N. Org. Lett. 2000, 2, 3699–3701. doi:10.1021/ol006610l |
| 69. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Toupet, L.; Roques, N. Tetrahedron Lett. 2001, 42, 1867–1869. doi:10.1016/s0040-4039(01)00017-x |
| 70. | Mohar, B.; Baudoux, J.; Plaquevent, J.-C.; Cahard, D. Angew. Chem., Int. Ed. 2001, 40, 4214–4216. doi:10.1002/1521-3773(20011119)40:22<4214::aid-anie4214>3.0.co;2-b |
| 71. | Baudequin, C.; Plaquevent, J.-C.; Audouard, C.; Cahard, D. Green Chem. 2002, 4, 584–586. doi:10.1039/b208817g |
| 72. | Baudequin, C.; Loubassou, J.-F.; Plaquevent, J.-C.; Cahard, D. J. Fluorine Chem. 2003, 122, 189–193. doi:10.1016/s0022-1139(03)00085-x |
| 73. | Thierry, B.; Audouard, C.; Plaquevent, J.-C.; Cahard, D. Synlett 2004, 856–860. doi:10.1055/s-2004-817781 |
| 74. | Mohar, B.; Sterk, D.; Ferron, L.; Cahard, D. Tetrahedron Lett. 2005, 46, 5029–5031. doi:10.1016/j.tetlet.2005.05.074 |
| 75. | Fukuzumi, T.; Shibata, N.; Sugiura, M.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2006, 127, 548–551. doi:10.1016/j.jfluchem.2006.01.004 |
| 76. | Ramírez, J.; Huber, D. P.; Togni, A. Synlett 2007, 1143–1147. doi:10.1055/s-2007-973897 |
| 77. | Yi, W.-B.; Huang, X.; Zhang, Z.; Zhu, D.-R.; Cai, C.; Zhang, W. Beilstein J. Org. Chem. 2012, 8, 1233–1240. doi:10.3762/bjoc.8.138 |
| 78. | Wang, M.; Wang, B. M.; Shi, L.; Tu, Y. Q.; Fan, C.-A.; Wang, S. H.; Hu, X. D.; Zhang, S. Y. Chem. Commun. 2005, 5580–5582. doi:10.1039/b510004f |
| 63. | Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. doi:10.1016/j.jfluchem.2006.12.014 |
| 64. | Lectard, S.; Hamashima, Y.; Sodeoka, M. Adv. Synth. Catal. 2010, 352, 2708–2732. doi:10.1002/adsc.201000624 |
| 65. | Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944–2947. doi:10.1021/ol201007e |
| 65. | Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944–2947. doi:10.1021/ol201007e |
| 66. | Shibata, N.; Suzuki, E.; Takeuchi, Y. J. Am. Chem. Soc. 2000, 122, 10728–10729. doi:10.1021/ja002732x |
| 61. | Wei, H.; Wang, G.; Wang, Y.; Li, B.; Huang, J.; Kashtanov, S.; Van Hecke, K.; Pereshivko, O. P.; Peshkov, V. A. Chem. – Asian J. 2017, 12, 825–829. doi:10.1002/asia.201700051 |
| 62. | Hasan, M.; Zaman, M.; Peshkov, A. A.; Amire, N.; Les, A.; Nechaev, A. A.; Wang, Y.; Kashtanov, S.; Van der Eycken, E. V.; Pereshivko, O. P.; Peshkov, V. A. Eur. J. Org. Chem. 2020, 3378–3389. doi:10.1002/ejoc.202000015 |
| 58. | Zhang, J.; Wang, Y.-Y.; Sun, H.; Li, S.-Y.; Xiang, S.-H.; Tan, B. Sci. China: Chem. 2020, 63, 47–54. doi:10.1007/s11426-019-9606-2 |
| 59. | Nechaev, A. A.; Peshkov, A. A.; Peshkov, V. A.; Van der Eycken, E. V. Synthesis 2016, 48, 2280–2286. doi:10.1055/s-0035-1561422 |
| 60. | Peshkov, A. A.; Peshkov, V. A.; Li, Z.; Pereshivko, O. P.; Van der Eycken, E. V. Eur. J. Org. Chem. 2014, 6390–6393. doi:10.1002/ejoc.201402723 |
© 2020 Wang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)