Abstract
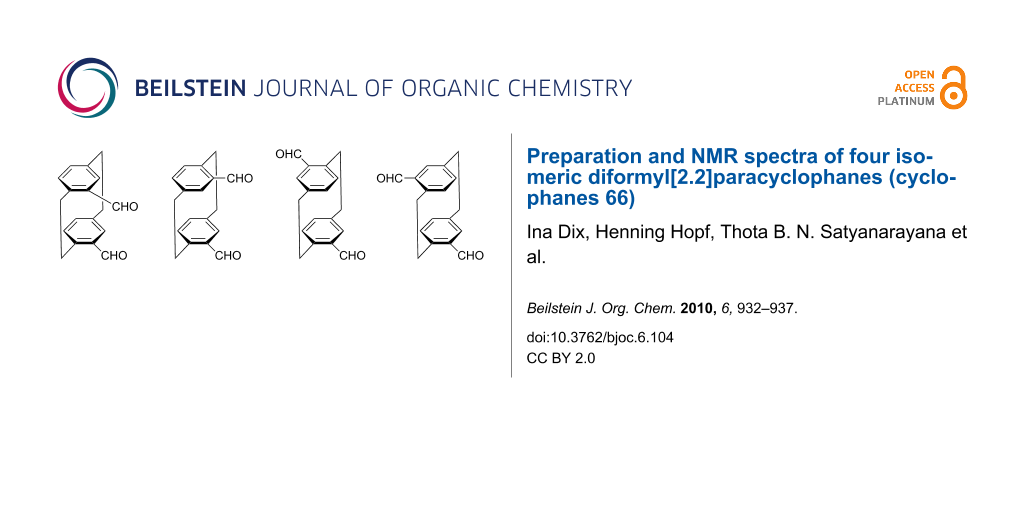
Four isomeric dialdehydes 4, readily available from cycloaddition of propiolic aldehyde (2) to 1,2,4,5-hexatetraene (1), were separated by chromatography and recrystallization, and were characterized by their spectroscopic data. The individual isomers can now be easily identified from their 1H NMR spectra even if only one of them is present.

Graphical Abstract
Introduction
Previously, we reported that the [2 + 4] cycloaddition of 1,2,4,5-hexatetraene (1) to propiolic aldehyde (2) produced a mixture of four [2.2]paracyclophane dialdehydes 4. This result is in agreement with the generation of the p-xylylene intermediate 3 in the first step of the reaction, which can dimerize by four different modes (Scheme 1) [2]. Depending on the amounts and the quality of the solutions containing the tetraene 1 and the dienophile 2, more than 60 g of the dialdehyde mixture 4 can be obtained in a single run, corresponding to maximum yields of 46%. Both starting compounds are very reactive and can decompose, even under refrigeration.
![[1860-5397-6-104-i1]](/bjoc/content/inline/1860-5397-6-104-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the four [2.2]paracyclophane dialdehydes 4 by cycloaddition (ps - pseudo).
Scheme 1: Preparation of the four [2.2]paracyclophane dialdehydes 4 by cycloaddition (ps - pseudo).
Although we have separated the four isomers and used them many times for the preparation of numerous [2.2]paracyclophane derivatives (inter alia annelated derivatives [2], metal complexes [3-5], diethynyl derivatives [6,7] and preparation of ligands for chiral reagents [8,9]), we have neither described the separation of these versatile starting materials nor explained their spectroscopic and analytical data in full. With this short communication, we would finally like to remedy this omission, in particular since these compounds are also beginning to attract the attention of other research groups [6,7,10-13].
Results and Discussion
Separation of the diformyl[2.2]paracyclophanes 4
We have separated the isomer mixture 4 by different methods. The easiest way is by middle pressure liquid chromatography (MPLC), which readily affords all isomers in gram amounts. To obtain larger amounts of specific isomers, e.g. the pseudo-para compound, it might be advisable to use the combination of column chromatography and MPLC or recrystallization described in the experimental section. In alcoholic solvents, pseudo-para-4 is the least soluble of all isomers and may hence be easily separated.
NMR spectra of the diformyl[2.2]paracyclophanes 4
As the 1H and 13C NMR spectra of [2.2]paracyclophane-4-carbaldehyde have previously been fully assigned and, hence, the influence of the substituent upon the 1H and 13C NMR chemical shifts of all positions of [2.2]paracyclophane are known [14], the assignment of the dicarbaldehyde isomers could, in principle, be derived from the comparison of their experimental chemical shifts with those calculated by assuming substituent chemical shift (SCS) additivity. However, there is no certainty that such an assumption is justified, as it is difficult to judge whether steric and/or electronic substituent interactions could cause significant deviations from SCS additivity. Furthermore, the SCSs exerted by the formyl group upon the nuclei of the distant ring are relatively small, |ΔδH| < 0.11 ppm, |ΔδC| < 0.88 ppm. We therefore decided to seek independent experimental proof for the four isomeric structures.
For this purpose, consideration of their symmetry is helpful. The ps-gem and the ps-meta isomer both have a symmetry element (plane and two-fold axis, respectively), which causes each CH2CH2 bridge to have only two different 1H chemical shifts. The protons of the bridge close to the formyl substituents form an AA´XX´ spin system, see Scheme 2. These protons are labelled Ha or Hs depending on whether they are anti or syn with respect to the 4-CHO group.
![[1860-5397-6-104-i2]](/bjoc/content/inline/1860-5397-6-104-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Spin systems of the CH2CH2 protons in the isomeric dialdehydes 4. Protons at C-9 and C-10 in ps-gem- and ps-meta-4 form YY´ZZ´ spin systems, which need not be considered here. Protons at C-9 and C-10 in ps-ortho- and ps-para-4 form AKRX spin systems equivalent to those of the C-1 and C-2 protons.
Scheme 2: Spin systems of the CH2CH2 protons in the isomeric dialdehydes 4. Protons at C-9 and C-10 in ps-gem...
The most characteristic and most easily recognized feature of an AA´XX´ spectrum is the distance N, defined as |J(AX)+J(AX´)|. In the ps-gem isomer, N equals |Jgem+Jtrans| with Jgem ≈ –13 Hz and Jtrans ≈ +4 Hz [14]. Hence, N is expected to be near 9 Hz. In the ps-meta isomer, N equals |Jgem+Jcis| with Jgem as above and Jcis ≈ +10 Hz, hence N ≈ 3 Hz. Spectra a and b in Figure 1 have N = 9.3 and 3.5 Hz, respectively, and therefore correspond to the ps-gem and the ps-meta isomer in this order. The ps-ortho and the ps-para isomer have a 2-fold axis and a centre of symmetry, respectively, which render the two bridges in each compound equivalent. So each of these isomers gives rise to a single AKRX spin system for their bridges, see spectra c and d in Figure 1.
![[1860-5397-6-104-1]](/bjoc/content/figures/1860-5397-6-104-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: CH2CH2 regions of the 600 MHz 1H NMR spectra of the isomeric dialdehydes 4, a: ps-gem, b: ps-meta, c: ps-ortho, d: ps-para isomer.
Figure 1: CH2CH2 regions of the 600 MHz 1H NMR spectra of the isomeric dialdehydes 4, a: ps-gem, b: ps-meta, ...
The distinction of the ps-ortho from the ps-para isomer is made possible with the help of an NOE experiment. In the isomer belonging to the spectrum shown in Figure 1c, the multiplet at δ = 3.11 ppm experiences a strong NOE when the resonance of 5-H (ortho to the 4-CHO group) is saturated, and vice versa. Its coupling constants show that the 3.11 ppm multiplet belongs to 9-Hs (cis to the deshielded proton 10-Hs, which itself is syn to the second formyl group, see Scheme 3).
![[1860-5397-6-104-i3]](/bjoc/content/inline/1860-5397-6-104-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: NOEs observed for ps-ortho-4 when the 5-H resonance is irradiated.
Scheme 3: NOEs observed for ps-ortho-4 when the 5-H resonance is irradiated.
This proves the ps-ortho arrangement of the two aldehyde groups. Then, by default, the spectrum in Figure 1d is that of ps-para-4. The 1H and 13C NMR data of the isomers are given in Table 1 and Table 2.
Table 1: 1H NMR data of the isomeric dialdehydes (CDCl3/TMS).
| ps-gem-4a,b | ps-ortho-4a,c | ps-meta-4a,d | ps-para-4a,e | |
|---|---|---|---|---|
| 5-H (d) | 6.99 | 6.87 | 7.04 | 7.05 |
| 7-H (dd) | 6.75 | 6.79 (ddd) | 6.65 | 6.64 |
| 8-H (d) | 6.66 | 6.61 | 6.49 | 6.53 |
| CHO (s) | 9.81 | 9.82 | 9.98 | 9.94 |
| 1-Ha | 3.12 (m) | 3.32 (ddd) | 3.03 (m) | 3.29 (ddd) |
| 1-Hs | 4.13 (m) | 3.11 (ddd) | 4.12 (m) | 3.16 (ddd) |
| 2-Ha | 2.93 (ddd) | 3.02 (ddd) | ||
| 2-Hs | 4.17 (ddd) | 4.14 (ddd) | ||
| 9-Ha | ≈3.19 (m) | 3.27 (m) | ||
| 9-Hs | ≈3.19 (m) | 3.10 (m) | ||
a J(5,7) = 2.0, J(7,8) = 7.8 Hz.
b N = |J(1a,1s) + J(1a,2s)| = 9.3 Hz.
c J(1a,1s) = (–)13.3 Hz, J(1a,2a) = 10.1 Hz, J(1a,2s) = 1.3 Hz, J(1s,2a) = 7.4 Hz, J(1s,2s) = 10.0 Hz, J(2a,2s) = (–)13.1 Hz, J(7,9a) = 0.8 Hz.
d N = |J(1a,1s) + J(1a,2a)| = 3.5 Hz.
e J(1a,1s) = (–)13.2 Hz, J(1a,2a) = 10.7 Hz, J(1a,2s) = 2.4 Hz, J(1s,2a) = 5.8 Hz, J(1s,2s) = 10.4 Hz, J(2a,2s) = (–)13.1 Hz.
Table 2: 13C NMR data of the isomeric dialdehydes (CDCl3, δ = 77.01 ppm).
| ps-gem-4 | ps-ortho-4 | ps-meta-4 | ps-para-4 | |
|---|---|---|---|---|
| CHO | 191.36 | 192.00 | 191.68 | 191.92 |
| C-3 (Cq) | 142.75 | 142.52 | 143.19 | 142.92 |
| C-4 (Cq) | 137.00 | 136.41 | 136.94 | 136.79 |
| C-5 (CH) | 134.63 | 136.16 | 136.12 | 136.55 |
| C-6 (Cq) | 140.42 | 140.69 | 140.57 | 140.54 |
| C-7 (CH) | 137.94 | 137.86 | 137.47 | 136.98 |
| C-8 (CH) | 136.07 | 136.21 | 134.89 | 135.24 |
| C-1 (CH2) | 31.91 | 34.19 | 33.71 | 34.36 |
| C-2 (CH2) | 33.87 | 32.81 | ||
| C-9 (CH2) | 34.72 | 34.66 | ||
The question of SCS additivity posed above can now be answered as follows. In the 1H NMR spectra, additivity is satisfied with, in general, |Σ SCSobs–Σ SCScalc| ≤ 0.06 ppm, except for 2-Ha and 2-Hs in ps-meta-4 where one finds 0.11 and 0.17 ppm, respectively. In the 13C NMR spectra, SCS additivity is less strict, namely |Σ SCSobs–Σ SCScalc| ≤ 0.7 ppm, except for C-2, C-4 and C-5 in ps-gem-4 with 1.2, 1.1 and 0.9 ppm, respectively. We also find that SCS additivity was distinctly better in the previously studied analogous bis(dimethoxycarbonyl)[2.2]paracyclophanes [15] compared to the present dialdehydes. This may be attributed to the larger magnitudes of the SCS of the aldehyde compared to those of the methyl ester functional group.
Conclusion
Although the present method of preparing the dialdehydes 4 involves extensive separation work (chromatography and recrystallization), it offers certain advantages compared to alternative (more specific) routes [10-13]. It is much shorter than many of the alternatives, which involve the rather expensive starting material, [2.2]paracyclophane. This method also provides four different substitution patterns in one step, two of which are interesting chiral derivatives (pseudo-ortho- and pseudo-meta-4). This leads directly to derivatives with a preparatively very useful functional group. The four individual isomers can now be easily identified, even if only one isomer is present, because each one shows a characteristic multiplet for the proton 2-Hs at δH = 4.1–4.2 ppm: i.e., ps-gem-4 and ps-meta-4 give an AA´XX´ pattern with N = 9.3 and 3.5 Hz, respectively; to a good approximation, ps-ortho-4 has a dd pattern with J = 13 and 10 Hz; and ps-para-4 displays a ddd pattern with the two inner lines coinciding (rel. intensities 1:1:1:2:1:1:1, J = 13, 10.5 and 2.5 Hz). Another characteristic is the small chemical shift difference δ5-H−δ7-H of 0.08 ppm for ps-ortho-4, whereas this difference is 0.24–0.41 ppm for all other isomers.
Experimental
Separation of the isomers 4
After the cycloaddition has been carried out [3], the precipitate formed was removed by filtration. The mother liquor was concentrated to half its volume and the solution stored in the refrigerator overnight. The newly formed crop was filtered off and united with the first precipitate. The combined solid reaction products were treated with chloroform in a Soxhlet extractor to remove any polymeric material produced. The solvent was removed and the residual solid could be directly separated by MPLC chromatography (silica gel) with dichloromethane/ethyl acetate (98:2, v/v) to provide the analytically pure isomers 4 (product ratio: pseudo-gem- : pseudo-ortho- : pseudo-meta- : pseudo-para-4: 1.8 : 1.1 : 1.0 : 2.8); (Rf = 0.47, 0.55, 0.60, 0.63). To reduce the separation effort, the above product mixture can be enriched first by standard silica gel chromatography with dichloromethane/ethyl acetate (98:2, v/v). This procedure provided three fractions: a fast moving one consisting of pseudo-para- and pseudo-meta-4, a middle fraction containing all four isomers, and a third fraction consisting of the pseudo-ortho- and the pseudo-geminal isomers. If necessary, this process can be repeated, thus allowing separation of the four dialdehydes into two fractions each containing two isomers.
These may then be subjected to MPLC chromatography or repeated normal pressure column chromatography, allowing, for example, the separation of pure pseudo-ortho-4. Alternatively, the pseudo-meta/para-mixture could be purified by recrystallization from methanol, in which the pseudo-para isomer is only poorly soluble. On refluxing this mixture in methanol, followed by separation of the precipitate, practically pure pseudo-para compound was obtained. To remove the last traces of isomeric impurities from pseudo-para-4, it was recrystallized from n-butanol.
Spectroscopic properties
The NMR spectra were measured at 600 MHz (1H, CDCl3, TMS, δ = 0.00 ppm) and at 151 MHz (13C, CDCl3, δ = 77.01 ppm) on a Bruker Avance II 600 instrument. Assignments were carried out using H,H-COSY, H,C-HSQC, H,C-HMBC and NOEDIF techniques.
4,15-Diformyl[2.2]paracyclophane (ps-gem-4)
1H NMR: see Table 1. 13C NMR: see Table 2. IR (KBr): ![[Graphic 1]](/bjoc/content/inline/1860-5397-6-104-i4.svg?max-width=637&scale=1.18182) = 3029 (w), 2926 (w), 1676 (vs), 1588 (m), 1482 (m), 1225 (m), 1141 (m), 720 cm–1 (m). UV (acetonitrile): λmax (lg ε) = 214 (4.57), 262 (4.13), 320 nm (3.41). MS (70 eV): m/z (%) = 264 (100) [M+], 236 (45), 207 (11), 132 (87), 104 (95).
= 3029 (w), 2926 (w), 1676 (vs), 1588 (m), 1482 (m), 1225 (m), 1141 (m), 720 cm–1 (m). UV (acetonitrile): λmax (lg ε) = 214 (4.57), 262 (4.13), 320 nm (3.41). MS (70 eV): m/z (%) = 264 (100) [M+], 236 (45), 207 (11), 132 (87), 104 (95).
4,16-Diformyl[2.2]paracyclophane (ps-ortho-4)
1H NMR: see Table 1. 13C NMR: see Table 2. IR (KBr): = 3028 (vw), 2927 (w), 1680 (vs), 1648 (m), 1552 (m), 1232 cm–1 (m). UV (acetonitrile): λmax (lg ε) = 216 (4.53), 296 nm (3.95). – MS (70 eV): m/z (%) = 264 (100) [M+], 132 (98), 104 (92).
4,12-Diformyl[2.2]paracyclophane (ps-para-4)
1H NMR: see Table 1. 13C NMR: see Table 2. IR (KBr): = 3013 (w), 2933 (w), 1684 and 1673 (vs), 1586 (m), 1483 (m), 720 cm–1 (m). UV (acetonitrile): λmax (lg ε) = 214 (4.55), 264 (4.16), 320 nm (3.42). MS (70 eV): m/z (%) = 264 (100) [M+], 132 (94), 104 (100).
4,13-Diformyl[2.2]paracyclophane (ps-meta-4)
1H NMR: see Table 1. 13C NMR: see Table 2. IR (KBr): = 2936 (w), 1678 (vs), 1588 (m), 1487 (w), 1229 (m), 1141 (w), 900 (w), 778 (m), 719 cm–1 (m). UV (acetonitrile): λmax (lg ε) = 214 (4.54), 296 nm (3.95). MS (70 eV): m/z (%) = 264 (82) [M+], 236 (17), 207 (9), 132 (66), 104 (100).
References
-
Wullbrandt, D.; Hopf, H.; Jones, P. G. Eur. J. Org. Chem. 2010, 4046–4048. doi:10.1002/ejoc.201000114
-
Aly, A. A.; Hopf, H.; Ernst, L. Eur. J. Org. Chem. 2000, 3021–3029. doi:10.1002/1099-0690(200009)2000:17<3021::AID-EJOC3021>3.0.CO;2-9
Return to citation in text: [1] [2] -
Hopf, H.; Raulfs, F.-W.; Schomburg, D. Tetrahedron 1986, 42, 1655–1663. doi:10.1016/S0040-4020(01)87582-7
Return to citation in text: [1] [2] -
Hopf, H.; Raulfs, F.-W. Isr. J. Chem. 1985, 25, 210–216.
Return to citation in text: [1] -
El-Tamany, S.; Raulfs, F.-W.; Hopf, H. Angew. Chem., Int. Ed. Engl. 1983, 22, 633–634. doi:10.1002/anie.198306331
Angew. Chem. 1983, 95, 631. doi:10.1002/ange.19830950808
Return to citation in text: [1] -
Hopf, H.; Hinrichs, H.; Dix, I.; Bondarenko, L. Synthesis 2004, 2751–2759. doi:10.1055/s-2004-834872
Return to citation in text: [1] [2] -
Hopf, H.; Dix, I. Synlett 2006, 1416–1418.
Return to citation in text: [1] [2] -
Hillmer, J. Ph.D. Thesis, Technische Universität Braunschweig, Germany, 1991.
Return to citation in text: [1] -
Laue, T. Ph.D. Thesis, Technische Universität Braunschweig, Germany, 1991.
Return to citation in text: [1] -
Clément, S.; Guyard, L.; Knorr, M.; Däschlein, C.; Strohmann, C. Acta Crystallogr., Sect. E: Struct. Rep. Online 2009, E65, o528.
Return to citation in text: [1] [2] -
Morisaki, Y.; Lin, L.; Chujo, Y. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5979–5988. doi:10.1002/pola.23641
Return to citation in text: [1] [2] -
Nandivada, H.; Chen, H.-Y.; Bondarenko, L.; Lahann, J. Angew. Chem., Int. Ed. 2006, 45, 3360–3363. doi:10.1002/anie.200600357
Angew. Chem. 2006, 118, 3438–3441. doi:10.1002/ange.200600357
Return to citation in text: [1] [2] -
Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969
Angew. Chem. 2008, 120, 9954–9958. doi:10.1002/ange.200800969
Return to citation in text: [1] [2] -
Ernst, L. Liebigs Ann. 1995, 13–17. doi:10.1002/jlac.199519950105
Return to citation in text: [1] [2] -
Ernst, L. Fresenius' J. Anal. Chem. 1997, 357, 494–497. doi:10.1007/s002160050199
Return to citation in text: [1]
| 2. | Aly, A. A.; Hopf, H.; Ernst, L. Eur. J. Org. Chem. 2000, 3021–3029. doi:10.1002/1099-0690(200009)2000:17<3021::AID-EJOC3021>3.0.CO;2-9 |
| 8. | Hillmer, J. Ph.D. Thesis, Technische Universität Braunschweig, Germany, 1991. |
| 9. | Laue, T. Ph.D. Thesis, Technische Universität Braunschweig, Germany, 1991. |
| 6. | Hopf, H.; Hinrichs, H.; Dix, I.; Bondarenko, L. Synthesis 2004, 2751–2759. doi:10.1055/s-2004-834872 |
| 7. | Hopf, H.; Dix, I. Synlett 2006, 1416–1418. |
| 3. | Hopf, H.; Raulfs, F.-W.; Schomburg, D. Tetrahedron 1986, 42, 1655–1663. doi:10.1016/S0040-4020(01)87582-7 |
| 4. | Hopf, H.; Raulfs, F.-W. Isr. J. Chem. 1985, 25, 210–216. |
| 5. |
El-Tamany, S.; Raulfs, F.-W.; Hopf, H. Angew. Chem., Int. Ed. Engl. 1983, 22, 633–634. doi:10.1002/anie.198306331
Angew. Chem. 1983, 95, 631. doi:10.1002/ange.19830950808 |
| 2. | Aly, A. A.; Hopf, H.; Ernst, L. Eur. J. Org. Chem. 2000, 3021–3029. doi:10.1002/1099-0690(200009)2000:17<3021::AID-EJOC3021>3.0.CO;2-9 |
| 15. | Ernst, L. Fresenius' J. Anal. Chem. 1997, 357, 494–497. doi:10.1007/s002160050199 |
| 3. | Hopf, H.; Raulfs, F.-W.; Schomburg, D. Tetrahedron 1986, 42, 1655–1663. doi:10.1016/S0040-4020(01)87582-7 |
| 6. | Hopf, H.; Hinrichs, H.; Dix, I.; Bondarenko, L. Synthesis 2004, 2751–2759. doi:10.1055/s-2004-834872 |
| 7. | Hopf, H.; Dix, I. Synlett 2006, 1416–1418. |
| 10. | Clément, S.; Guyard, L.; Knorr, M.; Däschlein, C.; Strohmann, C. Acta Crystallogr., Sect. E: Struct. Rep. Online 2009, E65, o528. |
| 11. | Morisaki, Y.; Lin, L.; Chujo, Y. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5979–5988. doi:10.1002/pola.23641 |
| 12. |
Nandivada, H.; Chen, H.-Y.; Bondarenko, L.; Lahann, J. Angew. Chem., Int. Ed. 2006, 45, 3360–3363. doi:10.1002/anie.200600357
Angew. Chem. 2006, 118, 3438–3441. doi:10.1002/ange.200600357 |
| 13. |
Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969
Angew. Chem. 2008, 120, 9954–9958. doi:10.1002/ange.200800969 |
| 10. | Clément, S.; Guyard, L.; Knorr, M.; Däschlein, C.; Strohmann, C. Acta Crystallogr., Sect. E: Struct. Rep. Online 2009, E65, o528. |
| 11. | Morisaki, Y.; Lin, L.; Chujo, Y. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5979–5988. doi:10.1002/pola.23641 |
| 12. |
Nandivada, H.; Chen, H.-Y.; Bondarenko, L.; Lahann, J. Angew. Chem., Int. Ed. 2006, 45, 3360–3363. doi:10.1002/anie.200600357
Angew. Chem. 2006, 118, 3438–3441. doi:10.1002/ange.200600357 |
| 13. |
Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969
Angew. Chem. 2008, 120, 9954–9958. doi:10.1002/ange.200800969 |
© 2010 Dix et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)