Abstract
The development of hybrid inorganic–organic films with well-controlled properties is important for many applications. Molecular layer deposition (MLD) allows the deposition of these hybrid films using sequential, self-limiting reactions, similar to atomic layer deposition (ALD). In this paper, we use first principles density functional theory (DFT) to investigate the growth mechanism of titanium-containing hybrid organic–inorganic MLD films, known as “titanicones”. We investigate in detail the chemistry between the most common Ti precursors, namely titanium tetrachloride (TiCl4) and tetrakis(dimethylamido)titanium (Ti(DMA)4), and ethylene glycol (EG) and glycerol (GL) as the organic precursors. We analyse the impact of the substrate on the initial MLD reactions in titanicone film growth using three different surface models: anatase TiO2, rutile TiO2 and Al2O3. Calculated energetics show that while TiCl4 is reactive towards the anatase and rutile TiO2 surfaces, it is not reactive towards the Al2O3 surface. Ti(DMA)4 is reactive towards all surfaces. This is attributed to the stronger Ti–Cl bonds in TiCl4 compared to Ti–N bonds in Ti(DMA)4. Ti(DMA)4 also shows high reactivity to the organics compared to TiCl4. Double reactions of EG and GL with the TiCl3 species from TiCl4 and TiDMA species from Ti(DMA)4 are also explored to better understand the origin of the different thicknesses of EG–titanicone and GL–titanicone films observed in experimental work. We find that EG and GL coupled with TiCl4 can orient in a flat lying configuration on anatase while on rutile, the preferred orientation is upright. When combined with Ti(DMA)4, EG and GL prefer the flat lying configuration on all surfaces. This work shows that the choice of the surface and the metallic precursor has a major impact on the behaviour of organic species. DFT findings provide motivation to develop a low temperature rutile TiO2/titanicone film suggesting that the desired film growth could be achieved.
Introduction
Molecular layer deposition (MLD), a thin film deposition technique, has attracted significant attention in recent years as a suitable approach for the deposition of organic–inorganic hybrid films for applications in several technological application areas, including packaging/encapsulation, electronics, batteries and biomedical applications [1-4]. MLD is very similar to the widely used atomic layer deposition (ALD) technique, which involves the fabrication of inorganic films used extensively in photovoltaics, (nano)electronics, energy storage and catalysis [5-8]. Similarly to ALD, MLD is based on sequential self-limiting reactions of readily vaporized inorganic precursors but the second reactant is a highly volatile organic species. Thus, in contrast to ALD, in MLD the deposition chemistry can be extended by including organic precursors, leading to the deposition of hybrid organic–inorganic MLD films [3,9]. Two organic precursors can also be employed to deposit pure organic MLD films, such as polymers [10-12]. Similar to ALD, MLD enables the deposition of conformal and smooth films with a controlled thickness at Angstrom level [3,9].
The advantages offered by hybrid organic–inorganic MLD films are that they are ultrathin films with high flexibility, have tunable properties and excellent mechanical and electronic properties resulting from the combination of the individual properties of the organic and inorganic components that are incorporated into the film [1,2,13-15].
A special class of hybrid organic–inorganic films are so-called metalcones, which are fabricated via MLD using organometallic precursors and organic alcohols. These films are described as O–M–O–(CHx)y–O–M–O. Metalcones are known to be flexible in nature due to the flexible organic backbones present in their architectures and with excellent mechanical properties at the atomic and molecular level arising from the organometallic precursors [16,17]. The first metalcone films were Al based and were known as “alucones” [18-20]. Soon after, other metalcone groups were developed, such as the Ti, Zn, Hf, Mg and V based metalcones known as “titanicones”, “zincones” [21,22], “hafnicones” [23], “magnesicones” [24] and “vanadicones” [25], respectively. While other organic backbones, e.g., aromatic rings, have been used, the field tends to use the term “metalcone” as a general description for these hybrid materials.
One of the most extensively researched metalcones are titanicones. Titanicones are deposited by coupling a titanium inorganic precursor, such as titanium tetrachloride (TiCl4), with organic alcohols such as ethylene glycol (EG) [26-31], glycerol (GL) [29-31] or fumaric acid (FC) [32]. Tetrakis(dimethylamino)titanium (Ti(N(CH3)2)4), henceforth denoted Ti(DMA)4) was also successfully employed as Ti source and combined with EG and GL to deposit titanicone films [33].
In ref [30] titanicone films were deposited using TiCl4 as metal source and EG or GL as organic precursors. TiCl4–EG films and TiCl4–GL films were deposited on Si(100) wafers. XRR analysis found a growth per cycle (GPC) of ≈4.6 Å/cycle for TiCl4–EG films at 90 °C which decreases to 1.5 Å/cycle at 135 °C. It was assumed that this drop could be related to the desorption of unreacted TiCl4 species at 135 °C. In addition, it was proposed that the reduction in growth rate could also be caused by the desorption of Ti(–O(CH2)2O–)2 species or double reactions of the organic precursor in which the molecule lies flat with both termini binding to Ti sites.
The thickness and thickness reduction with temperature increment observed for TiCl4–EG films was similar to the thickness and thickness reduction observed for alucone films grown using trimethylaluminium (TMA) and EG (4.0 Å/cycle at 85 °C to 0.4 Å/cycle at 175 °C) [18] and zincone films grown using diethylzinc (DEZ) and EG (4.0 Å/cycle at 90 °C to 0.25 Å/cycle at 170 °C) [21].
For TiCl4–GL films the growth rate was 2.8 Å/cycle at 130 °C and it decreased to 2.1 Å/cycle at 210 °C. The thickness of TiCl4–GL films was similar to the thickness of alucone films grown using TMA and GL (2.5 Å/cycle) [34]. Similar growth rates of TiCl4–EG and TiCl4–GL films were achieved also in ref. [31] (TiCl4–EG: 4.5 Å/cycle at 115 °C; TiCl4–GL: 2.2 Å/cycle at 150 °C).
Compared to EG, GL has an extra OH group that increases the bridging between the polymer chains in the film and this also improves the stability of the film. Nanoindentation experiments [30] showed also that GL-based titanicones have much higher elastic modulus and higher hardness compared to EG-based titanicones. Annealing experiments indicated a higher thermal stability for GL-based titanicones as well [30].
Ti(DMA)4 is another Ti-based inorganic precursor that was investigated as an alternative to TiCl4 as the titanium precursor. Ti(DMA)4 was combined with EG and GL to deposit titanicone films in a temperature range from 80 °C to 160 °C on an Si substrate with 100 nm thermal oxide. In situ ellipsometry revealed for EG-based titanicones that the growth initiates but terminates after only 5 to 10 cycles, while for GL-based titanicones the growth proceeds and films have a GPC of 0.9 Å/cycle to 0.2 Å/cycle as the temperature increases. This is due to the double reaction phenomenon, which for EG removes both the reactive OH groups required to couple with the Ti precursor, while even with a double reaction, GL has a third OH group available to couple to the Ti(DMA)4 precursor in the next cycle [33]. Ti(DMA)4 inorganic precursor is well known and widely used in ALD for the deposition of TiO2 and TiN films by thermal and plasma-enhanced processes [35,36].
As described above, TiCl4 and Ti(DMA)4 were successfully employed in MLD of titanicone films [30,33]. While TiCl4 is a small halide molecule that has Ti–Cl bonds, Ti(DMA)4 is a bulkier molecule with Ti–N bonds. Bond dissociation energy values for the breakage of the Ti–Cl and Ti–N bonds serve as a measure of the stability of these precursors and these are 494 kJ/mol and 464 kJ/mol for Ti–Cl and Ti–N, respectively. Bond dissociation energies indicate that a lower energy is needed to break the Ti–N bond compared to the Ti–Cl bond and hence a higher reactivity for the Ti(DMA)4 precursor towards the surface and towards the co-reactant is expected.
In addition, due to the bulky DMA ligands of Ti(DMA)4, the “reservior” effect, which is very common for small molecules such as TiCl4 [30], DEZ [21] and TMA [37] and which leads to uncontrolled growth is avoided. Moreover, the ligand elimination reactions of the Ti(DMA)4 process yield H–N(CH3)2 by-products. In contrast this is non-corrosive compared to HCl by-products released during the TiCl4 process. For TiCl4 this is considered a significant drawback [38].
Fumaric acid (FC) is another alcohol organic precursor that was used to deposit titanicone films using TiCl4 on an Si substrate in a temperature range of 180 °C to 350 °C. A temperature-dependent growth characteristic was observed with the growth rate decreasing from 1.10 Å/cycle at 180 °C to 0.49 Å/cycle at 300 °C. This reduction was attributed to the increased thermal motion and desorption of molecules on the growth surface. FTIR spectra indicated that the hybrid film shows a stable bridging bonding mode between Ti and the acid group at temperatures under 200 °C and a high bridging/bidentate mixed bonding mode at temperatures over 250 °C and 300 °C [32].
Many studies show that the desired properties and the target thickness of a metalcone MLD film are not actually achieved. To help understand this, first principles density functional theory (DFT) calculations have been employed to explore the reaction mechanisms, energetics and the role of the organic precursors on the growth of hybrid films [24,39-41]. DFT studies show that the aliphatic diol precursors, namely EG and GL, when combined with TMA, undergo double reactions, binding with the surface fragments through the two terminal OH groups, and this phenomenon reduces the number of active sites and terminates the surface growth [39].
However, DFT results also show that this scheme depends on the metal precursor and the surface as the same aliphatic precursors behave differently when combined with Mg(Cp)2; EG still prefers to orient in a flat configuration and react twice with the surface, while GL prefers to lie in an upright configuration. This yields thicker GL-based magnesicone films, consistent with the experimental observations [24].
DFT studies and experimental work show that aromatic organics can be a better option for the growth of thicker, more flexible and more stable hybrid films. This because aromatic molecules, due to their stiff backbone prefer to orient in a vertical configuration and avoid the double reactions [40,42]. Such hybrid films are known as “metal–organic” films. These also offer the advantage of allowing the facile tuning of the properties of the organic component through ring functionalisation, without impacting on the stability of the resulting films.
Many Ti–organic MLD processes have been developed by using different aromatic molecules as organic precursors. In ref [43], TiCl4 was coupled with 4-aminophenol (AP) to deposit Ti–(O–C6H4–N=) thin films with an essentially ideal growth rate of 10–11 Å/cycle at a temperature range of 120–160 °C. The deposited films were of high quality and stable in air. The high growth rates are attributed to the rigid structure of AP and the fact that Cl ligands of TiCl4 are small and do not cause steric hindrance.
In another study, Ti–organic films were deposited using TiCl4 and the homo/heterobifunctional aromatic molecules hydroquinone (HQ), 4-aminophenol (AP), p-phenylenediamine (PDA) and 4,4′-oxydianiline (ODA). All films were deposited on a Si surface. All four processes yielded amorphous Ti–organic films. A growth rate of 10–11 Å/cycle was achieved for the TiCl4–AP process, which is higher when compared to the growth rates 4.3 Å/cycle, 1.2 Å/cycle and 1.4 Å/cycle for TiCl4–HQ, TiCl4–PDA and TiCl4–ODA processes, respectively. The results are attributed the higher reactivity of the OH group with TiCl4 in comparison to the NH2 group and the higher tendency of the heterobifunctional organic precursor to orientate in an upright configuration and avoid unwanted double reactions on the surface [42].
TiCl4 was also coupled with 4,4′-oxydianiline (ODA) to deposit the (–Ti–N–C6H4–O–C6H4–N–)n thin films. Films were deposited on an Si surface in two temperature ranges, 160–230 °C and 250–490 °C. The growth rate increases with increasing temperature, from 0.3 Å/cycle at 160 °C to 1.1 Å/cycle at 490 °C. Films deposited at a higher temperature were also more stable in atmosphere compared to films deposited at low temperatures [44].
Because of the similarity with TiO2 ALD films, titanicone films are promising for electronics and solar applications [45] and may be used for biological implants as well. As TiO2 films have catalytic and photocatalytic properties [46] porous TiO2 frameworks formed by the annealing of titanicone films may serve as catalytic supports [47]. Titanicone films can also be pyrolyzed under Ar to yield conducting TiO2/carbon composite films with important electrochemical applications as electrodes for Li ion batteries or pseudocapacitance supercapacitors [31]. These films were also employed as coatings of nano Si electrodes and successfully improved their performance [48].
As described above, different titanicone and Ti–organic MLD processes have been developed and although first principles density functional theory (DFT) simulations have recently been used to explore the reaction mechanism in Ti–organic MLD film growth [42], such a study that explores the surface reactions and the precursor chemistry in titanicone MLD film growth and the role of the precursor chemistries is still lacking.
In this study we investigate the molecular mechanism of formation of titanicone films on anatase TiO2, rutile TiO2 and Al2O3 surfaces using TiCl4 or Ti(DMA)4 as Ti source and EG or GL as organic components. Calculated energetics suggest a higher reactivity of Ti(DMA)4 towards the selected surfaces and the organic molecules compared to TiCl4. This is due to the stronger Ti–Cl bonds in TiCl4 compared to Ti–N bonds in Ti(DMA)4. We also found that the choice of surface and inorganic precursors can influence the behavior of organic molecules by allowing or preventing undesirable double reactions. Based on the calculated energetics we propose that a low temperature rutile TiO2/TiCl4–EG process and rutile TiO2/TiCl4–GL process can lead to thicker hybrid films.
Computational Methods
All DFT calculations in this work were performed using the Vienna Ab initio Simulation Package (VASP) version 5.4 [49]. Titanicone films were modelled on an anatase TiO2 (101) surface with a coverage of 1 ML OH, rutile TiO2 (110) surface with a coverage of 0.75 ML OH, and an Al2O3 (0001) surface at a coverage 0.50 ML OH [50,51]. These surface models interact with titanium tetrachloride (TiCl4) and tetrakis(dimethylamido)titanium (Ti(DMA)4) inorganic precursors and then ethylene glycol (EG) and glycerol (GL) organic co-reactants.
The valence electron–core electron interactions are described by projector augmented wave potentials (PAW) [52] and the valance electron configurations are: Ti: 3d34s1, Al: 3s23p1, O: 2s22p4, Cl: 3s²3p⁵, C: 2s22p2 and H: 1s1. Calculations were performed using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional [53]. The employed convergence criterion for the energy was 1 × 10−4 eV while that for the forces was −2 × 10−2 eV/Å. The geometry was optimized by relaxing the ionic positions using an energy cut-off of 400 eV as well as a Monkhorst−Pack k-point sampling grid of (3 × 3 × 1). The lattice parameters are a = 20.612, b = 15.164, c = 24.399 and α = β = γ = 90° for the anatase TiO2 surface model, a = 11.846, b = 13.051, c = 33.870 and α = β = γ = 90° for the rutile TiO2 surface model and a = b = 19.228, c = 40.627 and α = β = 90°, γ = 120° for the Al2O3 surface model. The surfaces are 2, 4 and 5 layers thick for anatase (101), rutile (110) and Al2O3 (0001).
Reaction energetics were calculated using:
![[Graphic 1]](/bjnano/content/inline/2190-4286-13-103-i4.svg?max-width=637&scale=1.18182)
Here Ep is the energy of products and Er is the energy of reactants.
For the example of TiCl4 adsorbing on the hydroxylated surfaces the energy was calculated as follows:
![[2190-4286-13-103-i1]](/bjnano/content/inline/2190-4286-13-103-i1.svg?max-width=590&scale=1.18182)
where ![[Graphic 2]](/bjnano/content/inline/2190-4286-13-103-i5.svg?max-width=637&scale=1.18182) is the total energy of TiCl4 adsorbed on hydroxylated surface,
is the total energy of TiCl4 adsorbed on hydroxylated surface, ![[Graphic 3]](/bjnano/content/inline/2190-4286-13-103-i6.svg?max-width=637&scale=1.18182) is the total energy of free TiCl4 molecule and EHO–surface is the total energy of the hydroxylated surface. Energetics of ligand elimination can be calculated from:
is the total energy of free TiCl4 molecule and EHO–surface is the total energy of the hydroxylated surface. Energetics of ligand elimination can be calculated from:
![[2190-4286-13-103-i2]](/bjnano/content/inline/2190-4286-13-103-i2.svg?max-width=590&scale=1.18182)
where a HCl molecule is eliminated. A similar equation is used for elimination of another HCl to give TiCl2 on the TiO2 surface.
For the example of EG interacting on a TiCl3-terminated surface the energy was calculated as follows:
![[2190-4286-13-103-i3]](/bjnano/content/inline/2190-4286-13-103-i3.svg?max-width=590&scale=1.18182)
where ![[Graphic 4]](/bjnano/content/inline/2190-4286-13-103-i7.svg?max-width=637&scale=1.18182) is the total energy of EG bound to TiCl4 on the hydroxylated surface, EHCl is the total energy of a free HCl molecule released as by-product, EEG is the total energy of the EG molecule and
is the total energy of EG bound to TiCl4 on the hydroxylated surface, EHCl is the total energy of a free HCl molecule released as by-product, EEG is the total energy of the EG molecule and ![[Graphic 5]](/bjnano/content/inline/2190-4286-13-103-i8.svg?max-width=637&scale=1.18182) is the total energy of TiCl3-terminated hydroxylated surface. In computing the energies in Equation 1 and Equation 2, we also employ van der Waals interactions using the DFT-D3 parameterisation [54]. Similar energy expressions are used for the Ti(DMA)4 precursor.
is the total energy of TiCl3-terminated hydroxylated surface. In computing the energies in Equation 1 and Equation 2, we also employ van der Waals interactions using the DFT-D3 parameterisation [54]. Similar energy expressions are used for the Ti(DMA)4 precursor.
The underlying data files are available at https://github.com/ArbreshaMuriqi/Titanicone.
Results and Discussion
Surface models of TiCl3/TiCl2-terminated anatase/rutile TiO2 and Al2O3 after the TiCl4 pulse
As oxide/metalcone films are of high interest, we explored the feasibility of anatase TiO2/titanicone, rutile TiO2/titanicone and Al2O3/titanicone film formation. This is a common approach in modelling MLD chemistry [24,39-41]. We performed fundamental investigation on the interactions between the hydroxylated anatase TiO2, hydroxylated rutile TiO2 and hydroxylated Al2O3 surfaces with TiCl4 and EG or GL precursors. The hydroxylated surfaces that results from the interactions with water and before the introduction of TiCl4 are taken from previous studies [50,51].
In the first calculations we have calculated the interaction energies of the TiCl4 molecule on the selected surfaces. These energies are −0.29 eV for the anatase TiO2, −0.22 eV for the rutile TiO2 and 0.87 eV for the Al2O3. These energies indicate that TiCl4 will adsorb favourably, although with a small energy gain, on the anatase TiO2 surface and rutile TiO2 surface and will not adsorb on the Al2O3 surface. A previous study reports that TiO2 films grow well using TiCl4 and H2O on amorphous Al2O3 [55]. However, in our case, the Al2O3 surface model we use is crystalline. It is well known that in an amorphous surface the molecular mobility is significantly higher than in any corresponding crystalline form and there is a lower coordination number for atoms in the surface which gives rise to enhanced chemical reactivity of the amorphous surface. These results show that the choice of the surface can have a major impact on the initial deposition steps.
As we found that the adsorption of TiCl4 was not favourable on the Al2O3 surface, we continued with only the anatase TiO2 surface models and the rutile TiO2 surface models.
Next, we investigated the first and second ligand loss reactions of TiCl4, which involve the proton transfer from the surface OH groups to the Cl ligands of TiCl4 to form HCl as by-product, and the formation of new Ti–O bonds between the TiCl4 molecule and surface oxygen. During the first ligand loss reaction, TiCl4 forms one new Ti–O bond with the surface and one HCl molecule is released. This reaction leaves the surface covered with three Cl ligands, TiO2–TiCl3. During the second ligand loss reaction TiCl4 forms a second new Ti–O bond with the surface and a second HCl molecule is released. After this reaction the surface is left covered with two Cl ligands, TiO2–TiCl2. The coordination number of Ti in the TiCl4 molecule is four and remains unchanged during the first and second ligand loss reactions. Atomic structures of the anatase TiO2 surface and rutile TiO2 surface after adsorption of one TiCl4 molecule and the elimination of the first and second ligand of TiCl4 are presented in Figure 1. Calculated interaction and ligand loss energies of TiCl4 on the anatase TiO2 surface and rutile TiO2 surface are presented in Table 1. Energetics for the ligand loss reactions are calculated relative to the first model of TiCl4 interacting with the surface, and present an overall reaction energy. The overall energy for TiCl4 to lose Cl ligands and bind on either TiO2 surface is negative, although there is a notable energy cost for losing the second Cl ligand from surface bound TiCl3. We do not explore further loss of HCl to give adsorbed TiCl since XPS analysis from previous experimental work showed that chlorine impurities are still present in the film [33], indicating that some Ti–Cl bonds have reminded unreacted.
![[2190-4286-13-103-1]](/bjnano/content/figures/2190-4286-13-103-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The plotted ligand loss reactions of TiCl4 on the anatase/rutile TiO2 surfaces. Optimised atomic structure of a) TiCl4 interacting with the anatase TiO2 surface, b) TiCl3-terminated anatase TiO2 surface, c) TiCl2-terminated anatase TiO2 surface, d) TiCl4 interacting with the rutile TiO2 surface, e) TiCl3-terminated rutile TiO2 surface and f) TiCl2-terminated rutile TiO2 surface. Light grey: Ti, red: O, green: Cl, white: H. Figure coding is the same for all figures.
Figure 1: The plotted ligand loss reactions of TiCl4 on the anatase/rutile TiO2 surfaces. Optimised atomic st...
Table 1: Computed adsorption and ligand loss energies of TiCl4 on anatase/rutile TiO2 surfaces. In this and all tables in this paper A: anatase TiO2, R: rutile TiO2.
| Structure | Interaction energy (eV) | Structure | Interaction energy (eV) |
| A–TiCl4 | −0.29 | R–TiCl4 | −0.22 |
| A–TiCl3 | −0.97 | R–TiCl3 | −1.19 |
| A–TiCl2 | −0.33 | R–TiCl2 | −0.08 |
The length of the Ti–O bonds formed between TiCl4 and both TiO2 surfaces during the first and second ligand loss reactions is presented in Table S1 (Supporting Information File 1). From Table S1 we can see that Ti–O bonds formed between anatase TiO2 surface and TiCl4 are about 0.02–0.03 Å shorter when compared to the Ti–O bonds formed between rutile TiO2 surface and TiCl4. Generally, shorter Ti–O bonds tend to be stronger, and this can also influence the stability of the system.
In addition to the surface models that result from the adsorption of a single TiCl4 molecule, in order to investigate the double reaction phenomenon of EG and GL, we have also built models in which two TiCl4 molecules are adsorbed on the anatase TiO2 and rutile TiO2 surfaces. Calculated energies for the adsorption of the second TiCl4 molecule are −0.81 eV on the anatase TiO2 surface and −0.03 eV on the rutile TiO2 surface. This difference may arise from the higher stability of rutile TiO2 compared to anatase TiO2. The resulting surfaces after the adsorption of the second TiCl4 molecule are terminated with two TiCl3 species, anatase–2TiCl3 and rutile–2TiCl3, and the distance between the Ti atoms of the two TiCl3 species is 6.0 Å on the anatase TiO2 surface and 6.9 Å on rutile TiO2 surface.
Reactions between organic precursors and TiCl3/TiCl2-terminated anatase/rutile TiO2 surface
With these post-TiCl4 pulse models of anatase TiO2 and rutile TiO2 available, the interactions between the TiO2–TiCl3 and TiO2–TiCl2 species and the organic precursors are investigated by analysing the formation of MLD products with EG and GL. Reactions with EG and GL involve the transfer of one proton from a terminal OH group of the organic molecule to a Cl ligand of TiCl3/TiCl2 to release a HCl molecule as by-product and form one new Ti–O bond between the TiCl3/TiCl2 species and the organic molecules. A schematic illustration of titanicone MLD films based on the reactions between the hydroxylated surface and TiCl4 and the reactions between the TiCl3/TiCl2 surface species with EG and GL is presented in Figure 2. The resulting atomic structures of the MLD reaction products with EG and GL are shown in Figure 3.
![[2190-4286-13-103-2]](/bjnano/content/figures/2190-4286-13-103-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Schematic representation of titanicone MLD using titanium tetrachloride (TiCl4) as inorganic precursor and ethylene glycol (EG) or glycerol (GL) as organic precursors.
Figure 2: Schematic representation of titanicone MLD using titanium tetrachloride (TiCl4) as inorganic precur...
![[2190-4286-13-103-3]](/bjnano/content/figures/2190-4286-13-103-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Optimised atomic structure of a) anatase TiCl3–EG, b) anatase TiCl3–GL, c) anatase TiCl2–EG, d) anatase TiCl2–GL, e) rutile TiCl3–EG, f) rutile TiCl3–GL, g) rutile TiCl2–EG and h) rutile TiCl2–GL.
Figure 3: Optimised atomic structure of a) anatase TiCl3–EG, b) anatase TiCl3–GL, c) anatase TiCl2–EG, d) ana...
The overall energy change for the reactions between the TiCl3/TiCl2-terminated anatase TiO2 and rutile TiO2 surfaces with EG and GL is presented in Table 2 and these results show that the calculated energies for the reactions between the TiCl3-terminated surfaces with EG and GL, associated with the release of one HCl molecule, are exothermic and therefore favourable. There are also clear differences when EG and GL bind with TiCl3 and TiCl2-terminated TiO2 surfaces. The calculated energies for the reactions between the TiCl2-terminated surfaces with EG and GL, associated with the release of one HCl molecule, are endothermic, meaning that these reactions are not favourable. We found a similar difference for the reaction of TiCl3/TiCl2 with aromatic precursors [42].
Table 2: Computed interaction energies of EG and GL on the TiCl3/TiCl2-terminated anatase/rutile TiO2 surface.
| Structure | Interaction energy (eV) | Structure | Interaction energy (eV) |
| A–TiCl3–EG | −0.79 | R–TiCl3–EG | −0.96 |
| A–TiCl3–GL | −0.66 | R–TiCl3–GL | −1.77 |
| A–TiCl2–EG | 0.08 | R–TiCl2–EG | 0.49 |
| A–TiCl2–GL | 0.15 | R–TiCl2–GL | 0.81 |
We also consider the Ti–O distances formed between the EG and GL with TiCl3/TiCl2 species as well as changes in the Ti-surface distances between TiCl3 and TiCl2 and surface oxygens after the introduction of EG and GL, Table S2 (Supporting Information File 1). We notice that Ti–O bonds formed between EG and GL and the TiCl3 terminated anatase TiO2 and rutile TiO2 surfaces are 0.02 Å shorter compared to Ti–O bonds formed between EG and GL with the TiCl2 terminated anatase TiO2 and rutile TiO2 surfaces. Ti–O distances between TiCl4 and the surface oxygens also change after the introduction of EG and GL. After the introduction of EG and GL, Ti–O bonds to the surface are lengthen for 0.02 Å to 0.03 Å in the TiCl3 terminated anatase TiO2 and rutile TiO2 surfaces and 0.02 Å to 0.07 Å in the TiCl2 terminated anatase TiO2 and rutile TiO2 surfaces. As DFT results suggest that the formation of Ti–O bonds between EG and GL and the TiCl2 species on the anatase TiO2 and rutile TiO2 surfaces is energetically unfavourable, in the next calculations we exclude the TiCl2 terminated surfaces.
Comparison of upright and flat-lying reactions of ethylene glycol (EG) and glycerol (GL) on the TiCl3-terminated anatase/rutile TiO2 surface
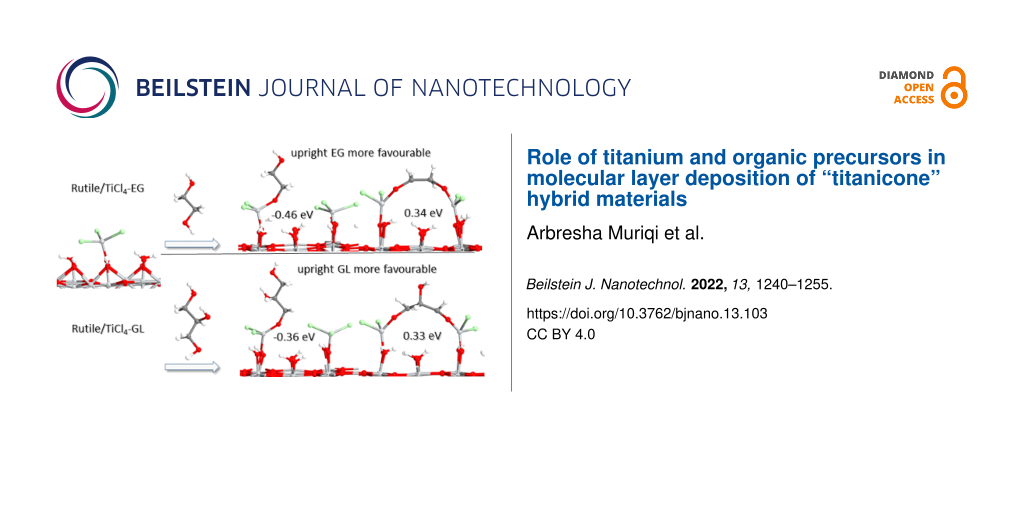
Double reactions of EG and GL are investigated by examining the interactions between the TiO2 surface terminated with 2TiCl3 and 2TiCl3 with EG and GL in the upright configuration and in the flat configuration. In the upright configuration, EG and GL bind to Ti sites through one terminal OH group and one HCl molecule is released. In the flat configuration, EG and GL bind through two terminal OH groups with two neighbouring Ti sites and two HCl molecules are released.
Optimised atomic structures of the MLD reaction products of TiCl3-terminated anatase TiO2 and rutile TiO2 surfaces with upright and flat lying EG and GL are shown in Figure 4. The computed energies when EG and GL bind with one Ti site in the upright configuration and with two Ti sites in the flat lying configuration are shown in Table 3. The calculated energy for the upright configuration of EG and GL is calculated with reference to the energy of the adsorbed Ti precursor on the relevant surface, while the calculated energy for the flat configuration of EG and GL is with reference to the energy of the corresponding upright structure. This allows us to assess if the double reactions of EG and GL are thermodynamically favourable when reacting with TiCl3 on anatase TiO2 and rutile TiO2 surfaces.
![[2190-4286-13-103-4]](/bjnano/content/figures/2190-4286-13-103-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Optimised atomic structure of a) upright and flat EG on the anatase 2TiCl3, b) upright and flat GL on the anatase 2TiCl3, c) upright and flat EG on the rutile 2TiCl3, d) upright and flat GL on the rutile 2TiCl3.
Figure 4: Optimised atomic structure of a) upright and flat EG on the anatase 2TiCl3, b) upright and flat GL ...
Table 3: Computed interaction energy of EG and GL in the upright configuration with TiCl3-terminated anatase/rutile TiO2 surface. The energy change between the flat (double reaction) and upright configuration is also presented.
| Structure | Interaction energy (eV) | Structure | Interaction energy (eV) |
| A–2TiCl3 | −0.81 | R–2TiCl3 | −0.03 |
| A–2TiCl3–EG–up | −0.72 | R–2TiCl3–EG–up | −0.46 |
| A–2TiCl3–EG–flat | −0.55 | R–2TiCl3–EG–flat | 0.34 |
| A–2TiCl3–GL–up | −0.63 | R–2TiCl3–GL–up | −0.36 |
| A–2TiCl3–GL–flat | −0.33 | R–2TiCl3–GL–flat | 0.33 |
From Table 3 we see that the EG and GL molecules interact favourably in an upright configuration at the 2TiCl3-terminated anatase TiO2 surface. However, there is a further gain in energy of −0.55 eV for EG and −0.33 eV for GL when the molecules change their configuration from upright to flat lying, and a second HCl molecule is released. These energies show that EG and GL could also undergo double reactions on the anatase TiO2 surface, although for GL this difference in energy is smaller than for EG.
Similar to Al2O3, this phenomenon for EG will reduce the number of OH sites on the surface and this should make the growth of TiCl4–EG films less favourable. However, despite the reduction of OH groups on the surface, terminal oxygen sites of the flat EG can also serve as active sites and they can bind with other TiCl4 molecules in the following cycle, similar to TMA–EG alucones [39]. The GL molecule is a triol with three OH active groups, so even in the case of double reactions for GL the third OH group is available for further reactions.
The EG and GL molecules interact also favourably in an upright configuration with the 2TiCl3-terminated rutile TiO2 surface, while for their flat lying configuration the overall energy change is slightly endothermic. This shows that in a rutile surface EG and GL could prefer to orient in an upright configuration. For the 2TiCl3–EG model, the distance between Ti and O in the Ti–O–CH2CH2–O fragment that is formed during one MLD cycle (EG in the upright configuration) is ≈4.97 Å. This distance is similar with the achieved growth rate of ≈4.5 Å/cycle to 6 Å/cycle for TiCl4–EG films in a temperature range of 100–120 °C on a SiO2 membrane [27] and ≈4.6 Å/cycle at 90 °C in Si(100) wafers [30]. We note the relatively low temperature of the titanicone deposition in refs. [27,30] which may prevent the double reaction with EG.
For the TiCl3–GL model, the distance between Ti and O in the Ti–O–CH2CH2OHCH2–O fragment (GL in the upright configuration) is ≈6.26 Å. However, the achieved growth rate of TiCl4–GL films in experimental work is much smaller, 2.8 Å/cycle at 130 °C to 2.1 Å/cycle at 210 °C when deposited in Si wafers [30] and 2.2 Å/cycle at 150 °C deposited again in Si wafers in another study [31]. The distance between the surface and the available OH group of the flat GL is 4.2 Å and this distance is close to the measured growth per cycle of TiCl3–GL films in experimental work. Although the lower growth per cycle of TiCl3–GL films achieved in these experimental studies are most consistent with a flat lying orientation of GL when deposited on Si surfaces, DFT shows that the desired upright orientation of GL could be energetically favourable on a rutile TiO2 surface and this provides motivation to develop a lower temperature rutile TiO2/TiCl4–GL process where higher growth per cycle could be achieved. We note that the experimental data are not immediately comparable since different deposition temperatures are used, with significantly higher temperatures for the GL process compared to the EG process which may favour the flat-lying configuration.
In summary, DFT calculations show that while in a anatase TiO2 surface the organic molecules EG and GL can orient in both configurations, upright and lying flat, in the rutile TiO2 surface these molecules could prefer more the upright configuration, although the flat-lying configuration may also be stabilised. This indicates that the surface can also have an important role on the orientation of the organic species.
The Ti–O distances to the EG and GL in the upright and flat lying configurations are presented in Table S3 (Supporting Information File 1). The computed distances show that the Ti–O bond to the molecules does not change significantly when EG and GL change their configuration from upright to lying flat on the anatase TiO2 surface (0.01 Å longer). On the other hand, on rutile, the Ti–O bonds to the organic molecules in the flat lying configuration are 0.03 Å to 0.06 Å longer compared to Ti–O bonds of these molecules in the upright configuration. The increment of these bonds when EG and GL change their configuration from upright to lying flat on the rutile TiO2 surface also indicates the lower stability of these molecules in the flat configuration on the TiCl3 terminated rutile TiO2 surface.
For the upright EG and GL on the anatase TiO2 surface, the Ti–O distance between Ti of TiCl4 and the oxygen atom of the anatase TiO2 surface is 1.77 Å. This distance decreases to 1.75 Å when EG and GL change their configuration to lying flat. For EG in the upright and flat laying configuration on the rutile TiO2 surface the Ti–O distance between Ti of TiCl4 and the oxygen atom is 1.77 Å. For GL in the upright and flat lying configuration on the rutile TiO2 surface this distance is 1.76 Å.
Surface models of Ti(DMA)3, Ti(DMA)2 and TiDMA-terminated anatase/rutile TiO2 and Al2O3 after the Ti(DMA)4 pulse
Ti(DMA)4 is another Ti precursor that has been used for the deposition of TiO2 [35] and TiN [36] ALD films and titanicone MLD films [33]. Ti(DMA)4 is a metalorganic precursor containing Ti–N bonds to the organic ligands, dimethylamino (DMA, N(CH3)2), and with a much larger molecular size when compared to TiCl4. It offers some advantages as a precursor, including the noncorrosive nature of by-products of ligand elimination, higher reactivity due to the weak Me–N bonds and good thermal stability [38]. We performed DFT calculations to study the feasibility of the growth of titanicone films using Ti(DMA)4 and EG or GL as MLD precursors and to investigate the molecular mechanism behind the possible growth. The hydroxylated anatase TiO2 surface, hydroxylated rutile TiO2 surface and hydroxylated Al2O3 surface were selected as surface models.
In the first calculations, we calculate the reactivity of the Ti(DMA)4 precursor towards the selected surfaces. The calculated interaction energies on the anatase TiO2, rutile TiO2 and the Al2O3 surfaces are −0.88 eV, −0.74 eV and −1.47 eV, respectively. Thus, Ti(DMA)4 adsorbs favourably on the three oxide surfaces.
Next, the thermodynamics of the first, second and third ligand loss reactions of the Ti(DMA)4 precursor are examined. These reactions take place with a proton transfer from surface OH groups to the DMA ligands of Ti(DMA)4 to form protonated molecules, H–DMA, released as by-products and the formation of new Ti–O bonds between Ti(DMA)4 and oxygen atoms on the surface. After the third ligand loss reaction, Ti of Ti(DMA)4 is bonded to three surface oxygens and to one DMA ligand that can exchange with one organic molecule during the organic precursor pulse. The coordination number of Ti of the Ti(DMA)4 molecule is four and remains unchanged during the ligand loss reactions.
The ligand loss reaction mechanism of Ti(DMA)4 is presented in Figure 5. Optimized atomic structures of the anatase TiO2, rutile TiO2 and the Al2O3 surfaces interacting with Ti(DMA)4 and after the elimination of the first, second and third ligand of Ti(DMA)4 are presented in Figure 6. The computed interaction and ligand loss energies of Ti(DMA)4 are presented in Table 4. The energies for the ligand loss reactions are calculated relative to the first model of Ti(DMA)4 interacting with the surface, and present an overall reaction energy. Ligand loss energies show that the overall energy for Ti(DMA)4 to lose DMA ligands and bind on either TiO2 surface or Al2O3 surface is favourable.
![[2190-4286-13-103-5]](/bjnano/content/figures/2190-4286-13-103-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Schematic representation of ligand loss reactions of tetrakis(dimethylamido)titanium (Ti(DMA)4) inorganic precursor on a hydroxylated surface.
Figure 5: Schematic representation of ligand loss reactions of tetrakis(dimethylamido)titanium (Ti(DMA)4) ino...
![[2190-4286-13-103-6]](/bjnano/content/figures/2190-4286-13-103-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: The plotted ligand loss reactions of Ti(DMA)4 on the anatase/rutile TiO2 surface and Al2O3 surface. Optimised atomic structure of a1) Ti(DMA)4 interacting with the anatase TiO2 surface, a2) Ti(DMA)3-terminated anatase TiO2 surface, a3) Ti(DMA)2-terminated anatase TiO2 surface, a4) TiDMA-terminated anatase TiO2 surface, b1) Ti(DMA)4 interacting with the rutile TiO2 surface, b2) Ti(DMA)3-terminated rutile TiO2 surface, b3) Ti(DMA)2-terminated rutile TiO2 surface, b4) TiDMA-terminated rutile TiO2 surface, c1) Ti(DMA)4-interacting with the Al2O3 surface, c2) Ti(DMA)3-terminated Al2O3 surface, c3) Ti(DMA)2-terminated Al2O3 surface and c4) TiDMA-terminated Al2O3 surface. Dark grey: C, blue: N. Figure coding is the same for all figures.
Figure 6: The plotted ligand loss reactions of Ti(DMA)4 on the anatase/rutile TiO2 surface and Al2O3 surface....
Table 4: Computed adsorption and ligand loss energies of Ti(DMA)4 on the anatase/rutile TiO2 surface and Al2O3 surface.
| Structure | Interaction energy (eV) | Structure | Interaction energy (eV) | Structure | Interaction energy (eV) |
| A–Ti(DMA)4 | −0.88 | R–Ti(DMA)4 | −0.74 | Al2O3–Ti(DMA)4 | −1.47 |
| A–Ti(DMA)3 | −1.87 | R–Ti(DMA)3 | −4.02 | Al2O3–Ti(DMA)3 | −5.22 |
| A–Ti(DMA)2 | −1.57 | R–Ti(DMA)2 | −3.07 | Al2O3–Ti(DMA)2 | −6.05 |
| A–TiDMA | −1.36 | R–TiDMA | −2.53 | Al2O3–TiDMA | −2 |
When we compare these energies to those calculated for TiCl4 interacting and binding on the same selected surfaces, we see that Ti(DMA)4 interacts and binds more favourably with the surfaces as the calculated reaction energies are much larger. This is consistent with bond dissociation energy values for the breakage of the Ti–Cl and Ti–N bonds (494 kJ/mol and 464 kJ/mol, respectively) which show that the Ti–Cl bond is stronger and thus more difficult to break.
The overall reaction energy which includes the interaction of Ti(DMA)4 with the surface and the three ligand loss reactions is the largest on the rutile TiO2 surface, −2.53 eV, which is −0.53 eV larger when compared to the Al2O3 surface and −1.73 eV larger when compared to the anatase TiO2 surface.
The new Ti–O bonds of Ti(DMA)4 to the Al2O3 surface are 0.03–0.08 Å shorter, when compared to those formed between Ti(DMA)4 and the anatase TiO2 surface or rutile TiO2 surface, Table S4 (Supporting Information File 1).
In order to investigate double reactions of EG and GL we built also the surface models after the adsorption of two Ti(DMA)4 molecules. The reactions when a second Ti(DMA)4 molecule is adsorbed are associated with the release of three H-DMA molecules as by-products and the surfaces are left covered with two TiDMA species, anatase–2TiDMA, rutile–2TiDMA and Al2O3–2TiDMA. Calculated energetics for the adsorption of the second Ti(DMA)4 precursors are −1.05 eV for the anatase TiO2 surface, −1.29 eV for the rutile TiO2 surface, −2.06 eV for the Al2O3 surface. The distance between Ti atoms of the two TiDMA species is 7.2 Å on the anatase TiO2 surface and rutile TiO2 surface and 7.1 Å on the Al2O3 surface.
Reactions between organic precursors and TiDMA-terminated anatase/rutile TiO2 and Al2O3 surfaces
Next, we analysed MLD reactions using EG and GL as organic reactants where the organic molecules are modelled in both upright and flat lying configurations. Similar to the reaction between TiCl4 and EG or GL, the reaction between Ti(DMA)4 and EG or GL requires the transfer of one proton from the terminal OH group of the organic molecule to one DMA ligand of Ti(DMA)4 to release a H–DMA molecule as a by-product coupled with the formation of a new Ti–O bond between Ti and the organic molecule. Figure 7 shows the optimized atomic structures after the introduction of one EG and GL molecule and the loss of one H–DMA by-product.
![[2190-4286-13-103-7]](/bjnano/content/figures/2190-4286-13-103-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Optimised atomic structure of a) anatase TiDMA–EG, b) anatase TiDMA–GL, c) rutile TiDMA–EG, d) rutile TiDMA–EG, e) Al2O3 TiDMA–EG and f) Al2O3 TiDMA–GL.
Figure 7: Optimised atomic structure of a) anatase TiDMA–EG, b) anatase TiDMA–GL, c) rutile TiDMA–EG, d) ruti...
The overall energy change for the reactions between the TiDMA-terminated anatase TiO2, rutile TiO2 and Al2O3 surfaces with EG and GL is presented in Table 5. Calculated energies show that the Ti–O bond formation with EG and GL, with release of H–DMA, is favourable on all surfaces. The overall interaction energy is largest on anatase TiO2 followed by rutile TiO2.
Table 5: Computed interaction energies of EG and GL on the TiDMA-terminated anatase/rutile TiO2 and Al2O3 surfaces.
| Structure | Interaction energy (eV) | Structure | Interaction energy (eV) | Structure | Interaction energy (eV) |
| A–TiDMA–EG | −3.42 | R–TiDMA–EG | −2.98 | Al2O3–TiDMA–EG | −1.75 |
| A–TiDMA–GL | −3.31 | R–TiDMA–GL | −2.89 | Al2O3–TiDMA–GL | −1.60 |
Ti–O distances between Ti and the organic molecules are shown in Table S5 (Supporting Information File 1). Ti–O bonds formed between EG and GL with the TiDMA-terminated anatase TiO2 and rutile TiO2 surfaces are 0.06 Å to 0.08 Å shorter when compared to those formed between the EG and GL and the TiDMA-terminated Al2O3 surface, which is consistent with the more favourable interaction energies.
We also examined the change in the Ti–O distances to the surface oxygens after the introduction of the EG and GL, and these are presented in Table S6 (Supporting Information File 1). The computed Ti–O distances show that after the introduction of the EG and GL Ti–O bonds to the surface oxygens undergo negligible changes, indicating that the EG and GL will not affect the stability of the systems.
Comparison of upright and flat-lying reactions of ethylene glycol (EG) and glycerol (GL) on the 2TiDMA-terminated anatase/rutile TiO2 and Al2O3 surfaces
We investigated also the double reactions of EG and GL on the anatase-2TiDMA surface, rutile–2TiDMA surface and Al2O3–2DMA, where EG and GL are modelled in the upright and flat lying configurations. In the upright configuration organic molecules bind to Ti sites through one terminal OH group and one H–DMA molecule is released while in the flat configuration organic molecules will bind through two terminal OH groups with two neighbouring Ti sites and two H–DMA molecules are released. Figure 8 shows the optimised atomic structures of the MLD reaction products of 2TiDMA-terminated anatase TiO2, rutile TiO2 and Al2O3 surfaces with the upright and flat lying EG and GL. Calculated energetics for the reactions of EG and GL in the upright and flat laying configuration with the 2TiDMA-terminated surfaces are shown in Table 6.
![[2190-4286-13-103-8]](/bjnano/content/figures/2190-4286-13-103-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Optimised atomic structure of a) upright and flat EG on the anatase–2TiDMA, b) upright and flat GL on the anatase–2TiDMA, c) upright and flat EG on the rutile–2TiDMA, d) upright and flat GL on the rutile–2TiDMA, e) upright and flat EG on the Al2O3–2TiDMA and f) upright and flat GL on the Al2O3–2TiDMA.
Figure 8: Optimised atomic structure of a) upright and flat EG on the anatase–2TiDMA, b) upright and flat GL ...
Table 6: Computed interaction energies of EG and GL in the upright configuration with the 2TiDMA-terminated anatase/rutile TiO2 and Al2O3 surfaces. The energy change between the flat (double reaction) and upright configuration is also presented.
| Structure | Interaction energy (eV) | Structure | Interaction energy (eV) | Structure | Interaction energy (eV) |
| A– 2TiDMA | −1.05 | R–2TiDMA | −1.29 | Al2O3–2TiDMA | −2.06 |
| A–2TiDMA–EG–up | −1.44 | R–2TiDMA–EG–up | −1.65 | Al2O3–2TiDMA–EG–up | −1.73 |
| A–2TiDMA–EG–flat | −0.32 | R–2TiDMA–EG–flat | −0.20 | Al2O3–2TiDMA–EG–flat | −1.6 |
| A–2TiDMA–GL–up | −1.40 | R–2TiDMA–GL–up | −1.61 | Al2O3–2TiDMA–GL–up | −1.81 |
| A–2TiDMA–GL–flat | −0.68 | R–2TiDMA–GL–flat | −0.90 | Al2O3–2TiDMA–GL–flat | −1.3 |
Calculated energetics for the reactions between upright EG and GL with the 2TiDMA-terminated anatase TiO2, rutile TiO2 and Al2O3 surfaces are negative, meaning that the reactions are exothermic and therefore favourable. The energies for the reactions between the upright EG and GL and the 2TiDMA-terminated anatase TiO2, rutile TiO2 and Al2O3 surfaces are calculated relative to the energy for the adsorption of two Ti(DMA)4 molecules on the relevant surface. The energies for the flat configuration are calculated relative to the upright structures of EG and GL. A negative energy gain was calculated for all reactions when EG and GL change their configuration from upright to flat lying and one H–DMA molecule is released, regardless of the surface.
Calculated energetics show that while the EG and GL can bind to the Ti sites via formation of Ti–O bonds and loss of H–DMA, it is most favourable for the organic precursors to lie flat and create the double reactions through the two terminal OH groups. This phenomenon removes all active sites for EG and the growth will be less favourable while GL has an extra OH group compared to EG which reacts with Ti(DMA)4 in the next cycle and the growth proceeds.
Calculations energetics suggest that organic molecules EG and GL combined with Ti(DMA)4 in a anatase TiO2, rutile TiO2 and Al2O3 surface behave similarly as when combined with Ti(DMA)4 on a Si substrate [33]. Experimental data in ref [33] show that for Ti(DMA)4–EG films deposited on a Si substrate in a temperature range of 80–150 °C the growth starts but it stops after 5–10 cycles, and this most probably due to the favourable double reactions of EG molecules with the Ti species. For Ti(DMA)4–GL films the growth proceeds even when the molecule reacts twice with the surface due to the extra OH group which reacts with Ti(DMA)4 in the next cycle. However, although the growth of Ti(DMA)4–GL films proceeds, these double reactions lead to small GPCs, 0.9 Å/cycle to 0.2 Å/cycle when deposited in a Si substrate in a temperature range of 80–160 °C. The stability and GPC of the Ti(DMA)4–GL-based film may also depend on the strength of interaction with GL, which is less favourable than with EG.
The Ti–O distances to the EG and GL molecules in the upright and flat lying configurations are presented in Table S7 (Supporting Information File 1). The computed Ti–O bonds to the organic molecules in the anatase TiO2 and rutile TiO2 surfaces are lengthen from 0.05 Å to 0.25 Å when EG and GL change their configuration from upright to lying flat.
In summary, DFT calculations show that the chemistry of the organic molecules will depend on the inorganic precursor used and on the surface onto which the films are deposited. We see that EG and GL prefer to lie flat when combined with Ti(DMA)4 on the anatase TiO2, rutile TiO2 and Al2O3 surfaces and with TiCl4 in a anatase TiO2 surface but they do not prefer to lie flat when combined with TiCl4 on a rutile TiO2 surface. The interactions with TiCl4 precursor are also less favourable than with Ti(DMA)4, partially explained by the strong Ti–Cl bond compared to Ti–N.
Conclusion
In this study, we used first principles density functional theory (DFT) to investigate the atomistic mechanism of the growth of titanium containing hybrid films known as “titanicones” deposited by MLD. We investigated in detail the chemistry of the MLD process between the TiCl4 or Ti(DMA)4 inorganic precursors and EG or GL organic molecules. We used anatase TiO2, rutile TiO2 and Al2O3 surface models.
Through DFT we calculated the reactivity of TiCl4 or Ti(DMA)4 inorganic precursors towards the selected surfaces and towards EG and GL organic molecules and predicted the preferred orientation of these organic molecules when combined with TiCl4 and Ti(DMA)4 in a anatase TiO2, rutile TiO2 and Al2O3 surface.
Calculated energetics show that while TiCl4 interacts and binds favourably with the anatase TiO2 and rutile TiO2 surfaces via Ti–O bonds and release of HCl, the interaction with the Al2O3 surface is not favourable. Ligand loss reactions of TiCl4 on the anatase TiO2 and rutile TiO2 surface are favourable, although there is a notable energy cost for losing the second Cl ligand from surface bound TiCl3. Double reactions of EG and GL molecules with TiCl3 species are also closely explored. DFT findings show that these molecules can bind with TiCl3 species in the anatase TiO2 surface in the upright configuration with one terminal OH group and in the flat configuration with two terminal OH groups. In the rutile TiO2 surface on the other hand, the preferred orientation of EG and GL molecules is upright. Therefore, we consider the rutile TiO2 surface as a suitable surface for the growth of TiCl4–EG and TiCl4–GL titanicone films where the desired GPCs could be achieved.
DFT calculations show that Ti(DMA)4 also interacts and binds favourably with the anatase TiO2, rutile TiO2 and the Al2O3 surface via Ti–O bonds. The ligand loss reactions of Ti(DMA)4 are associated with the release of H–DMA (H–N(CH3)2) by-products. A higher reactivity of Ti(DMA)4 compared to TiCl4 was calculated and this is most probably due to the stronger Ti–Cl bonds present in TiCl4 compared to Ti–N bonds present in Ti(DMA)4. We show that EG and GL bind favourably with Ti species of Ti(DMA)4 via Ti–O bonds and release of H–DMA by-products. However, reaction energetics indicate that these molecules can lie flat and create the unwanted double reactions through the reaction of the two terminal OH groups with the surface fragments. For EG this phenomenon removes active OH groups from the surface and the growth will be less favourable while for GL the third OH group is available and growth proceeds. This analysis supports experimental data on Ti(DMA)4–EG and Ti(DMA)4–GL film growth and clarifies why for the Ti(DMA)4–EG process, the growth stops after 5–10 cycles while for the Ti(DMA)4–GL process, the growth proceeds [33].
Temperature is a very important factor for the successful deposition of MLD films. It significantly affects the chemistry between the MLD precursors and especially the behaviour of the organic molecules in the hybrid films. Therefore, we consider this aspect very interesting and worthy of investigating in future work.
Supporting Information
| Supporting Information File 1: Additional data on the geometry of the structures. | ||
| Format: PDF | Size: 1.2 MB | Download |
Funding
We acknowledge support from Science Foundation Ireland for computational resources at Tyndall National Institute. We acknowledge support from H2020 MSCA-ITN Network HYCOAT, Grant Number 765378 and from Science Foundation Ireland, through the SFI-NSFC Partnership Program, project NITRALD 17/NSFC/5279
References
-
Sundberg, P.; Karppinen, M. Beilstein J. Nanotechnol. 2014, 5, 1104–1136. doi:10.3762/bjnano.5.123
Return to citation in text: [1] [2] -
Meng, X. J. Mater. Chem. A 2017, 5, 18326–18378. doi:10.1039/c7ta04449f
Return to citation in text: [1] [2] -
Gregorczyk, K.; Knez, M. Prog. Mater. Sci. 2016, 75, 1–37. doi:10.1016/j.pmatsci.2015.06.004
Return to citation in text: [1] [2] [3] -
Zhao, Y.; Sun, X. ACS Energy Lett. 2018, 3, 899–914. doi:10.1021/acsenergylett.8b00145
Return to citation in text: [1] -
Leskelä, M.; Ritala, M. Thin Solid Films 2002, 409, 138–146. doi:10.1016/s0040-6090(02)00117-7
Return to citation in text: [1] -
Cremers, V.; Puurunen, R. L.; Dendooven, J. Appl. Phys. Rev. 2019, 6, 021302. doi:10.1063/1.5060967
Return to citation in text: [1] -
Johnson, R. W.; Hultqvist, A.; Bent, S. F. Mater. Today 2014, 17, 236–246. doi:10.1016/j.mattod.2014.04.026
Return to citation in text: [1] -
Lim, B. S.; Rahtu, A.; Gordon, R. G. Nat. Mater. 2003, 2, 749–754. doi:10.1038/nmat1000
Return to citation in text: [1] -
King, D. M.; Liang, X.; Weimer, A. W. Powder Technol. 2012, 221, 13–25. doi:10.1016/j.powtec.2011.12.020
Return to citation in text: [1] [2] -
Loscutoff, P. W.; Zhou, H.; Clendenning, S. B.; Bent, S. F. ACS Nano 2010, 4, 331–341. doi:10.1021/nn901013r
Return to citation in text: [1] -
Adamczyk, N. M.; Dameron, A. A.; George, S. M. Langmuir 2008, 24, 2081–2089. doi:10.1021/la7025279
Return to citation in text: [1] -
Loscutoff, P. W.; Lee, H.-B.-R.; Bent, S. F. Chem. Mater. 2010, 22, 5563–5569. doi:10.1021/cm1016239
Return to citation in text: [1] -
Al Zoubi, W.; Kamil, M. P.; Fatimah, S.; Nashrah, N.; Ko, Y. G. Prog. Mater. Sci. 2020, 112, 100663. doi:10.1016/j.pmatsci.2020.100663
Return to citation in text: [1] -
Gomez-Romero, P. Adv. Mater. (Weinheim, Ger.) 2001, 13, 163–174. doi:10.1002/1521-4095(200102)13:3<163::aid-adma163>3.0.co;2-u
Return to citation in text: [1] -
Judeinstein, P.; Sanchez, C. J. Mater. Chem. 1996, 6, 511–525. doi:10.1039/jm9960600511
Return to citation in text: [1] -
Lee, B. H.; Anderson, V. R.; George, S. M. Meet. Abstr. 2011, MA2011-02, 1846. doi:10.1149/ma2011-02/26/1846
Return to citation in text: [1] -
Lee, B. H.; Yoon, B.; Abdulagatov, A. I.; Hall, R. A.; George, S. M. Adv. Funct. Mater. 2013, 23, 532–546. doi:10.1002/adfm.201200370
Return to citation in text: [1] -
Dameron, A. A.; Seghete, D.; Burton, B. B.; Davidson, S. D.; Cavanagh, A. S.; Bertrand, J. A.; George, S. M. Chem. Mater. 2008, 20, 3315–3326. doi:10.1021/cm7032977
Return to citation in text: [1] [2] -
Lidor-Shalev, O.; Leifer, N.; Ejgenberg, M.; Aviv, H.; Perelshtein, I.; Goobes, G.; Noked, M.; Rosy. Batteries Supercaps 2021, 4, 1739–1748. doi:10.1002/batt.202100152
Return to citation in text: [1] -
Park, Y.-S.; Kim, H.; Cho, B.; Lee, C.; Choi, S.-E.; Sung, M. M.; Lee, J. S. ACS Appl. Mater. Interfaces 2016, 8, 17489–17498. doi:10.1021/acsami.6b01856
Return to citation in text: [1] -
Yoon, B.; O'Patchen, J. L.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Vap. Deposition 2009, 15, 112–121. doi:10.1002/cvde.200806756
Return to citation in text: [1] [2] [3] -
Peng, Q.; Gong, B.; VanGundy, R. M.; Parsons, G. N. Chem. Mater. 2009, 21, 820–830. doi:10.1021/cm8020403
Return to citation in text: [1] -
Lee, B. H.; Anderson, V. R.; George, S. M. ACS Appl. Mater. Interfaces 2014, 6, 16880–16887. doi:10.1021/am504341r
Return to citation in text: [1] -
Kint, J.; Mattelaer, F.; Vandenbroucke, S. S. T.; Muriqi, A.; Minjauw, M. M.; Nisula, M.; Vereecken, P. M.; Nolan, M.; Dendooven, J.; Detavernier, C. Chem. Mater. 2020, 32, 4451–4466. doi:10.1021/acs.chemmater.9b05116
Return to citation in text: [1] [2] [3] [4] -
Van de Kerckhove, K.; Mattelaer, F.; Dendooven, J.; Detavernier, C. Dalton Trans. 2017, 46, 4542–4553. doi:10.1039/c7dt00374a
Return to citation in text: [1] -
Song, Z.; Fathizadeh, M.; Huang, Y.; Chu, K. H.; Yoon, Y.; Wang, L.; Xu, W. L.; Yu, M. J. Membr. Sci. 2016, 510, 72–78. doi:10.1016/j.memsci.2016.03.011
Return to citation in text: [1] -
Ishchuk, S.; Taffa, D. H.; Hazut, O.; Kaynan, N.; Yerushalmi, R. ACS Nano 2012, 6, 7263–7269. doi:10.1021/nn302370y
Return to citation in text: [1] [2] [3] -
Sarkar, D.; Ishchuk, S.; Taffa, D. H.; Kaynan, N.; Berke, B. A.; Bendikov, T.; Yerushalmi, R. J. Phys. Chem. C 2016, 120, 3853–3862. doi:10.1021/acs.jpcc.5b11795
Return to citation in text: [1] -
Brown, J. J.; Hall, R. A.; Kladitis, P. E.; George, S. M.; Bright, V. M. ACS Nano 2013, 7, 7812–7823. doi:10.1021/nn402733g
Return to citation in text: [1] [2] -
Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Abdulagatov, A. I.; Terauds, K. E.; Travis, J. J.; Cavanagh, A. S.; Raj, R.; George, S. M. J. Phys. Chem. C 2013, 117, 17442–17450. doi:10.1021/jp4051947
Return to citation in text: [1] [2] [3] [4] [5] -
Cao, Y.-Q.; Zhu, L.; Li, X.; Cao, Z.-Y.; Wu, D.; Li, A.-D. Dalton Trans. 2015, 44, 14782–14792. doi:10.1039/c5dt00384a
Return to citation in text: [1] [2] -
Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Son, S.-B.; Wang, Y.; Xu, J.; Li, X.; Groner, M.; Stokes, A.; Yang, Y.; Cheng, Y.-T.; Ban, C. ACS Appl. Mater. Interfaces 2017, 9, 40143–40150. doi:10.1021/acsami.7b08960
Return to citation in text: [1] -
Xie, Q.; Jiang, Y.-L.; Detavernier, C.; Deduytsche, D.; Van Meirhaeghe, R. L.; Ru, G.-P.; Li, B.-Z.; Qu, X.-P. J. Appl. Phys. 2007, 102, 083521. doi:10.1063/1.2798384
Return to citation in text: [1] [2] -
Elam, J. W.; Schuisky, M.; Ferguson, J. D.; George, S. M. Thin Solid Films 2003, 436, 145–156. doi:10.1016/s0040-6090(03)00533-9
Return to citation in text: [1] [2] -
Yoon, B.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2009, 21, 5365–5374. doi:10.1021/cm9013267
Return to citation in text: [1] -
Kim, B.; Lee, N.; Park, S.; Park, T.; Song, J.; Han, S.; Park, H.; Lee, D.; Kim, H.; Jeon, H. J. Alloys Compd. 2021, 857, 157931. doi:10.1016/j.jallcom.2020.157931
Return to citation in text: [1] [2] -
Muriqi, A.; Nolan, M. Dalton Trans. 2020, 49, 8710–8721. doi:10.1039/d0dt01376e
Return to citation in text: [1] [2] [3] [4] -
Muriqi, A.; Karppinen, M.; Nolan, M. Dalton Trans. 2021, 50, 17583–17593. doi:10.1039/d1dt03195c
Return to citation in text: [1] [2] [3] -
Kaur, P.; Muriqi, A.; Wree, J.-L.; Ghiyasi, R.; Safdar, M.; Nolan, M.; Karppinen, M.; Devi, A. Dalton Trans. 2022, 51, 5603–5611. doi:10.1039/d2dt00353h
Return to citation in text: [1] [2] -
Tanskanen, A.; Sundberg, P.; Nolan, M.; Karppinen, M. Thin Solid Films 2021, 736, 138896. doi:10.1016/j.tsf.2021.138896
Return to citation in text: [1] [2] [3] [4] -
Sundberg, P.; Karppinen, M. Eur. J. Inorg. Chem. 2014, 968–974. doi:10.1002/ejic.201301560
Return to citation in text: [1] -
Sood, A.; Sundberg, P.; Malm, J.; Karppinen, M. Appl. Surf. Sci. 2011, 257, 6435–6439. doi:10.1016/j.apsusc.2011.02.022
Return to citation in text: [1] -
Bach, U.; Lupo, D.; Comte, P.; Moser, J. E.; Weissörtel, F.; Salbeck, J.; Spreitzer, H.; Grätzel, M. Nature 1998, 395, 583–585. doi:10.1038/26936
Return to citation in text: [1] -
Linsebigler, A. L.; Lu, G.; Yates, J. T., Jr. Chem. Rev. 1995, 95, 735–758. doi:10.1021/cr00035a013
Return to citation in text: [1] -
Grätzel, M. Nature 2001, 414, 338–344. doi:10.1038/35104607
Return to citation in text: [1] -
Huertas, Z. C.; Settipani, D.; Flox, C.; Morante, J. R.; Kallio, T.; Biendicho, J. J. Sci. Rep. 2022, 12, 137. doi:10.1038/s41598-021-04105-x
Return to citation in text: [1] -
Kresse, G.; Furthmüller, J. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. doi:10.1103/physrevb.54.11169
Return to citation in text: [1] -
Liu, J.; Saedy, S.; Verma, R.; van Ommen, J. R.; Nolan, M. ChemRxiv 2020, 1–44. doi:10.26434/chemrxiv.12320855
Return to citation in text: [1] [2] -
Jenness, G. R.; Seiter, J.; Shukla, M. K. Phys. Chem. Chem. Phys. 2018, 20, 18850–18861. doi:10.1039/c8cp02590h
Return to citation in text: [1] [2] -
Blöchl, P. E. Phys. Rev. B 1994, 50, 17953–17979. doi:10.1103/physrevb.50.17953
Return to citation in text: [1] -
Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Return to citation in text: [1] -
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344
Return to citation in text: [1] -
Ali, S.; Juntunen, T.; Sintonen, S.; Ylivaara, O. M. E.; Puurunen, R. L.; Lipsanen, H.; Tittonen, I.; Hannula, S.-P. Nanotechnology 2016, 27, 445704. doi:10.1088/0957-4484/27/44/445704
Return to citation in text: [1]
| 21. | Yoon, B.; O'Patchen, J. L.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Vap. Deposition 2009, 15, 112–121. doi:10.1002/cvde.200806756 |
| 37. | Yoon, B.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2009, 21, 5365–5374. doi:10.1021/cm9013267 |
| 38. | Kim, B.; Lee, N.; Park, S.; Park, T.; Song, J.; Han, S.; Park, H.; Lee, D.; Kim, H.; Jeon, H. J. Alloys Compd. 2021, 857, 157931. doi:10.1016/j.jallcom.2020.157931 |
| 42. | Tanskanen, A.; Sundberg, P.; Nolan, M.; Karppinen, M. Thin Solid Films 2021, 736, 138896. doi:10.1016/j.tsf.2021.138896 |
| 44. | Sood, A.; Sundberg, P.; Malm, J.; Karppinen, M. Appl. Surf. Sci. 2011, 257, 6435–6439. doi:10.1016/j.apsusc.2011.02.022 |
| 40. | Muriqi, A.; Karppinen, M.; Nolan, M. Dalton Trans. 2021, 50, 17583–17593. doi:10.1039/d1dt03195c |
| 42. | Tanskanen, A.; Sundberg, P.; Nolan, M.; Karppinen, M. Thin Solid Films 2021, 736, 138896. doi:10.1016/j.tsf.2021.138896 |
| 43. | Sundberg, P.; Karppinen, M. Eur. J. Inorg. Chem. 2014, 968–974. doi:10.1002/ejic.201301560 |
| 39. | Muriqi, A.; Nolan, M. Dalton Trans. 2020, 49, 8710–8721. doi:10.1039/d0dt01376e |
| 24. | Kint, J.; Mattelaer, F.; Vandenbroucke, S. S. T.; Muriqi, A.; Minjauw, M. M.; Nisula, M.; Vereecken, P. M.; Nolan, M.; Dendooven, J.; Detavernier, C. Chem. Mater. 2020, 32, 4451–4466. doi:10.1021/acs.chemmater.9b05116 |
| 32. | Cao, Y.-Q.; Zhu, L.; Li, X.; Cao, Z.-Y.; Wu, D.; Li, A.-D. Dalton Trans. 2015, 44, 14782–14792. doi:10.1039/c5dt00384a |
| 24. | Kint, J.; Mattelaer, F.; Vandenbroucke, S. S. T.; Muriqi, A.; Minjauw, M. M.; Nisula, M.; Vereecken, P. M.; Nolan, M.; Dendooven, J.; Detavernier, C. Chem. Mater. 2020, 32, 4451–4466. doi:10.1021/acs.chemmater.9b05116 |
| 39. | Muriqi, A.; Nolan, M. Dalton Trans. 2020, 49, 8710–8721. doi:10.1039/d0dt01376e |
| 40. | Muriqi, A.; Karppinen, M.; Nolan, M. Dalton Trans. 2021, 50, 17583–17593. doi:10.1039/d1dt03195c |
| 41. | Kaur, P.; Muriqi, A.; Wree, J.-L.; Ghiyasi, R.; Safdar, M.; Nolan, M.; Karppinen, M.; Devi, A. Dalton Trans. 2022, 51, 5603–5611. doi:10.1039/d2dt00353h |
| 45. | Bach, U.; Lupo, D.; Comte, P.; Moser, J. E.; Weissörtel, F.; Salbeck, J.; Spreitzer, H.; Grätzel, M. Nature 1998, 395, 583–585. doi:10.1038/26936 |
| 46. | Linsebigler, A. L.; Lu, G.; Yates, J. T., Jr. Chem. Rev. 1995, 95, 735–758. doi:10.1021/cr00035a013 |
| 53. | Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865–3868. doi:10.1103/physrevlett.77.3865 |
| 54. | Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344 |
| 50. | Liu, J.; Saedy, S.; Verma, R.; van Ommen, J. R.; Nolan, M. ChemRxiv 2020, 1–44. doi:10.26434/chemrxiv.12320855 |
| 51. | Jenness, G. R.; Seiter, J.; Shukla, M. K. Phys. Chem. Chem. Phys. 2018, 20, 18850–18861. doi:10.1039/c8cp02590h |
| 52. | Blöchl, P. E. Phys. Rev. B 1994, 50, 17953–17979. doi:10.1103/physrevb.50.17953 |
| 42. | Tanskanen, A.; Sundberg, P.; Nolan, M.; Karppinen, M. Thin Solid Films 2021, 736, 138896. doi:10.1016/j.tsf.2021.138896 |
| 49. | Kresse, G.; Furthmüller, J. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. doi:10.1103/physrevb.54.11169 |
| 31. | Abdulagatov, A. I.; Terauds, K. E.; Travis, J. J.; Cavanagh, A. S.; Raj, R.; George, S. M. J. Phys. Chem. C 2013, 117, 17442–17450. doi:10.1021/jp4051947 |
| 48. | Huertas, Z. C.; Settipani, D.; Flox, C.; Morante, J. R.; Kallio, T.; Biendicho, J. J. Sci. Rep. 2022, 12, 137. doi:10.1038/s41598-021-04105-x |
| 50. | Liu, J.; Saedy, S.; Verma, R.; van Ommen, J. R.; Nolan, M. ChemRxiv 2020, 1–44. doi:10.26434/chemrxiv.12320855 |
| 51. | Jenness, G. R.; Seiter, J.; Shukla, M. K. Phys. Chem. Chem. Phys. 2018, 20, 18850–18861. doi:10.1039/c8cp02590h |
| 55. | Ali, S.; Juntunen, T.; Sintonen, S.; Ylivaara, O. M. E.; Puurunen, R. L.; Lipsanen, H.; Tittonen, I.; Hannula, S.-P. Nanotechnology 2016, 27, 445704. doi:10.1088/0957-4484/27/44/445704 |
| 24. | Kint, J.; Mattelaer, F.; Vandenbroucke, S. S. T.; Muriqi, A.; Minjauw, M. M.; Nisula, M.; Vereecken, P. M.; Nolan, M.; Dendooven, J.; Detavernier, C. Chem. Mater. 2020, 32, 4451–4466. doi:10.1021/acs.chemmater.9b05116 |
| 39. | Muriqi, A.; Nolan, M. Dalton Trans. 2020, 49, 8710–8721. doi:10.1039/d0dt01376e |
| 40. | Muriqi, A.; Karppinen, M.; Nolan, M. Dalton Trans. 2021, 50, 17583–17593. doi:10.1039/d1dt03195c |
| 41. | Kaur, P.; Muriqi, A.; Wree, J.-L.; Ghiyasi, R.; Safdar, M.; Nolan, M.; Karppinen, M.; Devi, A. Dalton Trans. 2022, 51, 5603–5611. doi:10.1039/d2dt00353h |
| 1. | Sundberg, P.; Karppinen, M. Beilstein J. Nanotechnol. 2014, 5, 1104–1136. doi:10.3762/bjnano.5.123 |
| 2. | Meng, X. J. Mater. Chem. A 2017, 5, 18326–18378. doi:10.1039/c7ta04449f |
| 3. | Gregorczyk, K.; Knez, M. Prog. Mater. Sci. 2016, 75, 1–37. doi:10.1016/j.pmatsci.2015.06.004 |
| 4. | Zhao, Y.; Sun, X. ACS Energy Lett. 2018, 3, 899–914. doi:10.1021/acsenergylett.8b00145 |
| 3. | Gregorczyk, K.; Knez, M. Prog. Mater. Sci. 2016, 75, 1–37. doi:10.1016/j.pmatsci.2015.06.004 |
| 9. | King, D. M.; Liang, X.; Weimer, A. W. Powder Technol. 2012, 221, 13–25. doi:10.1016/j.powtec.2011.12.020 |
| 32. | Cao, Y.-Q.; Zhu, L.; Li, X.; Cao, Z.-Y.; Wu, D.; Li, A.-D. Dalton Trans. 2015, 44, 14782–14792. doi:10.1039/c5dt00384a |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 10. | Loscutoff, P. W.; Zhou, H.; Clendenning, S. B.; Bent, S. F. ACS Nano 2010, 4, 331–341. doi:10.1021/nn901013r |
| 11. | Adamczyk, N. M.; Dameron, A. A.; George, S. M. Langmuir 2008, 24, 2081–2089. doi:10.1021/la7025279 |
| 12. | Loscutoff, P. W.; Lee, H.-B.-R.; Bent, S. F. Chem. Mater. 2010, 22, 5563–5569. doi:10.1021/cm1016239 |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 3. | Gregorczyk, K.; Knez, M. Prog. Mater. Sci. 2016, 75, 1–37. doi:10.1016/j.pmatsci.2015.06.004 |
| 9. | King, D. M.; Liang, X.; Weimer, A. W. Powder Technol. 2012, 221, 13–25. doi:10.1016/j.powtec.2011.12.020 |
| 26. | Song, Z.; Fathizadeh, M.; Huang, Y.; Chu, K. H.; Yoon, Y.; Wang, L.; Xu, W. L.; Yu, M. J. Membr. Sci. 2016, 510, 72–78. doi:10.1016/j.memsci.2016.03.011 |
| 27. | Ishchuk, S.; Taffa, D. H.; Hazut, O.; Kaynan, N.; Yerushalmi, R. ACS Nano 2012, 6, 7263–7269. doi:10.1021/nn302370y |
| 28. | Sarkar, D.; Ishchuk, S.; Taffa, D. H.; Kaynan, N.; Berke, B. A.; Bendikov, T.; Yerushalmi, R. J. Phys. Chem. C 2016, 120, 3853–3862. doi:10.1021/acs.jpcc.5b11795 |
| 29. | Brown, J. J.; Hall, R. A.; Kladitis, P. E.; George, S. M.; Bright, V. M. ACS Nano 2013, 7, 7812–7823. doi:10.1021/nn402733g |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 31. | Abdulagatov, A. I.; Terauds, K. E.; Travis, J. J.; Cavanagh, A. S.; Raj, R.; George, S. M. J. Phys. Chem. C 2013, 117, 17442–17450. doi:10.1021/jp4051947 |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 5. | Leskelä, M.; Ritala, M. Thin Solid Films 2002, 409, 138–146. doi:10.1016/s0040-6090(02)00117-7 |
| 6. | Cremers, V.; Puurunen, R. L.; Dendooven, J. Appl. Phys. Rev. 2019, 6, 021302. doi:10.1063/1.5060967 |
| 7. | Johnson, R. W.; Hultqvist, A.; Bent, S. F. Mater. Today 2014, 17, 236–246. doi:10.1016/j.mattod.2014.04.026 |
| 8. | Lim, B. S.; Rahtu, A.; Gordon, R. G. Nat. Mater. 2003, 2, 749–754. doi:10.1038/nmat1000 |
| 29. | Brown, J. J.; Hall, R. A.; Kladitis, P. E.; George, S. M.; Bright, V. M. ACS Nano 2013, 7, 7812–7823. doi:10.1021/nn402733g |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 31. | Abdulagatov, A. I.; Terauds, K. E.; Travis, J. J.; Cavanagh, A. S.; Raj, R.; George, S. M. J. Phys. Chem. C 2013, 117, 17442–17450. doi:10.1021/jp4051947 |
| 27. | Ishchuk, S.; Taffa, D. H.; Hazut, O.; Kaynan, N.; Yerushalmi, R. ACS Nano 2012, 6, 7263–7269. doi:10.1021/nn302370y |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 21. | Yoon, B.; O'Patchen, J. L.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Vap. Deposition 2009, 15, 112–121. doi:10.1002/cvde.200806756 |
| 22. | Peng, Q.; Gong, B.; VanGundy, R. M.; Parsons, G. N. Chem. Mater. 2009, 21, 820–830. doi:10.1021/cm8020403 |
| 24. | Kint, J.; Mattelaer, F.; Vandenbroucke, S. S. T.; Muriqi, A.; Minjauw, M. M.; Nisula, M.; Vereecken, P. M.; Nolan, M.; Dendooven, J.; Detavernier, C. Chem. Mater. 2020, 32, 4451–4466. doi:10.1021/acs.chemmater.9b05116 |
| 39. | Muriqi, A.; Nolan, M. Dalton Trans. 2020, 49, 8710–8721. doi:10.1039/d0dt01376e |
| 18. | Dameron, A. A.; Seghete, D.; Burton, B. B.; Davidson, S. D.; Cavanagh, A. S.; Bertrand, J. A.; George, S. M. Chem. Mater. 2008, 20, 3315–3326. doi:10.1021/cm7032977 |
| 19. | Lidor-Shalev, O.; Leifer, N.; Ejgenberg, M.; Aviv, H.; Perelshtein, I.; Goobes, G.; Noked, M.; Rosy. Batteries Supercaps 2021, 4, 1739–1748. doi:10.1002/batt.202100152 |
| 20. | Park, Y.-S.; Kim, H.; Cho, B.; Lee, C.; Choi, S.-E.; Sung, M. M.; Lee, J. S. ACS Appl. Mater. Interfaces 2016, 8, 17489–17498. doi:10.1021/acsami.6b01856 |
| 25. | Van de Kerckhove, K.; Mattelaer, F.; Dendooven, J.; Detavernier, C. Dalton Trans. 2017, 46, 4542–4553. doi:10.1039/c7dt00374a |
| 27. | Ishchuk, S.; Taffa, D. H.; Hazut, O.; Kaynan, N.; Yerushalmi, R. ACS Nano 2012, 6, 7263–7269. doi:10.1021/nn302370y |
| 16. | Lee, B. H.; Anderson, V. R.; George, S. M. Meet. Abstr. 2011, MA2011-02, 1846. doi:10.1149/ma2011-02/26/1846 |
| 17. | Lee, B. H.; Yoon, B.; Abdulagatov, A. I.; Hall, R. A.; George, S. M. Adv. Funct. Mater. 2013, 23, 532–546. doi:10.1002/adfm.201200370 |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 1. | Sundberg, P.; Karppinen, M. Beilstein J. Nanotechnol. 2014, 5, 1104–1136. doi:10.3762/bjnano.5.123 |
| 2. | Meng, X. J. Mater. Chem. A 2017, 5, 18326–18378. doi:10.1039/c7ta04449f |
| 13. | Al Zoubi, W.; Kamil, M. P.; Fatimah, S.; Nashrah, N.; Ko, Y. G. Prog. Mater. Sci. 2020, 112, 100663. doi:10.1016/j.pmatsci.2020.100663 |
| 14. | Gomez-Romero, P. Adv. Mater. (Weinheim, Ger.) 2001, 13, 163–174. doi:10.1002/1521-4095(200102)13:3<163::aid-adma163>3.0.co;2-u |
| 15. | Judeinstein, P.; Sanchez, C. J. Mater. Chem. 1996, 6, 511–525. doi:10.1039/jm9960600511 |
| 23. | Lee, B. H.; Anderson, V. R.; George, S. M. ACS Appl. Mater. Interfaces 2014, 6, 16880–16887. doi:10.1021/am504341r |
| 42. | Tanskanen, A.; Sundberg, P.; Nolan, M.; Karppinen, M. Thin Solid Films 2021, 736, 138896. doi:10.1016/j.tsf.2021.138896 |
| 21. | Yoon, B.; O'Patchen, J. L.; Seghete, D.; Cavanagh, A. S.; George, S. M. Chem. Vap. Deposition 2009, 15, 112–121. doi:10.1002/cvde.200806756 |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 18. | Dameron, A. A.; Seghete, D.; Burton, B. B.; Davidson, S. D.; Cavanagh, A. S.; Bertrand, J. A.; George, S. M. Chem. Mater. 2008, 20, 3315–3326. doi:10.1021/cm7032977 |
| 36. | Elam, J. W.; Schuisky, M.; Ferguson, J. D.; George, S. M. Thin Solid Films 2003, 436, 145–156. doi:10.1016/s0040-6090(03)00533-9 |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 31. | Abdulagatov, A. I.; Terauds, K. E.; Travis, J. J.; Cavanagh, A. S.; Raj, R.; George, S. M. J. Phys. Chem. C 2013, 117, 17442–17450. doi:10.1021/jp4051947 |
| 35. | Xie, Q.; Jiang, Y.-L.; Detavernier, C.; Deduytsche, D.; Van Meirhaeghe, R. L.; Ru, G.-P.; Li, B.-Z.; Qu, X.-P. J. Appl. Phys. 2007, 102, 083521. doi:10.1063/1.2798384 |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 35. | Xie, Q.; Jiang, Y.-L.; Detavernier, C.; Deduytsche, D.; Van Meirhaeghe, R. L.; Ru, G.-P.; Li, B.-Z.; Qu, X.-P. J. Appl. Phys. 2007, 102, 083521. doi:10.1063/1.2798384 |
| 36. | Elam, J. W.; Schuisky, M.; Ferguson, J. D.; George, S. M. Thin Solid Films 2003, 436, 145–156. doi:10.1016/s0040-6090(03)00533-9 |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 30. | Abdulagatov, A. I.; Hall, R. A.; Sutherland, J. L.; Lee, B. H.; Cavanagh, A. S.; George, S. M. Chem. Mater. 2012, 24, 2854–2863. doi:10.1021/cm300162v |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
| 34. | Son, S.-B.; Wang, Y.; Xu, J.; Li, X.; Groner, M.; Stokes, A.; Yang, Y.; Cheng, Y.-T.; Ban, C. ACS Appl. Mater. Interfaces 2017, 9, 40143–40150. doi:10.1021/acsami.7b08960 |
| 38. | Kim, B.; Lee, N.; Park, S.; Park, T.; Song, J.; Han, S.; Park, H.; Lee, D.; Kim, H.; Jeon, H. J. Alloys Compd. 2021, 857, 157931. doi:10.1016/j.jallcom.2020.157931 |
| 31. | Abdulagatov, A. I.; Terauds, K. E.; Travis, J. J.; Cavanagh, A. S.; Raj, R.; George, S. M. J. Phys. Chem. C 2013, 117, 17442–17450. doi:10.1021/jp4051947 |
| 33. | Van de Kerckhove, K.; Mattelaer, F.; Deduytsche, D.; Vereecken, P. M.; Dendooven, J.; Detavernier, C. Dalton Trans. 2016, 45, 1176–1184. doi:10.1039/c5dt03840e |
© 2022 Muriqi and Nolan; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjnano/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.