Abstract
A facile, solvothermal synthesis of mesoporous cerium oxide nanospheres is reported for the purpose of the photocatalytic degradation of organic dyes and future applications in sustainable energy research. The earth-abundant, relatively affordable, mixed valence cerium oxide sample, which consists of predominantly Ce7O12, has been characterized by powder X-ray diffraction, X-ray photoelectron and UV–vis spectroscopy, and transmission electron microscopy. Together with N2 sorption experiments, the data confirms that the new cerium oxide material is mesoporous and absorbs visible light. The photocatalytic degradation of rhodamin B is investigated with a series of radical scavengers, suggesting that the mechanism of photocatalytic activity under visible-light irradiation involves predominantly hydroxyl radicals as the active species.
Introduction
The degradation of organic pollutants by affordable and effective chemical methods is an acute problem that has been tackled by advanced oxidation processes [1]. The photocatalytic production of reactive oxygen species by using semiconductor technology has emerged as a sustainable and promising route for such advanced oxidation processes [2-6]. In these photocatalytic processes, based on TiO2 for example, radiation larger than the band gap is absorbed to promote an electron from the valence to the conduction band [2-4]. The resultant strongly oxidizing, valence band holes (h+) and reducing, conduction band electrons (e–) are short-lived under ambient conditions and react with water and air to form reactive oxygen species such as •OH, •OOH, H2O2, and O2− for example [1,2,4-6]. These reactive oxygen species can subsequently decompose organic pollutants. Recent developments in nanotechnology have enhanced the performance of photocatalytic and solar energy absorption processes by providing higher surface areas and more effective charge separation in semiconductor materials on the nanoscale. In fact, the commercially available Degussa P25 mixed-phase TiO2 is commonly employed as a benchmark in photocatalysis for applications ranging from dye-sensitized solar cells to the oxidative degradation of pollutants [7-11]. Despite being cheap, chemically robust, and generally non-toxic, TiO2 has a wide band gap of more than 3.0 eV, which means that photocatalytic processes that use TiO2 as the sensitizer can only absorb UV radiation (≈5% of the solar spectrum) [2-4,7,8]. Moreover, the valence band of TiO2 is strongly oxidizing whereas the conduction band level is only mildly reducing, which results in a low energy-conversion efficiency since most of the oxidation potential is wasted thermally. A number of other metal oxide semiconductors have been explored for the visible-light driven photocatalytic degradation of pollutants and microbes, such as bismuth oxides [5,6] and cerium oxides [12,13]. CeO2 specifically has been applied in a number of sustainable energy applications lately, including oxidative catalysis, hydrogen storage, and solar thermal dissociation of water and CO2 [14-18].
Cerium oxides with oxygen vacancies represent an underexplored area of nanotechnology with the potential to provide visible-light absorbing photocatalysts [13,19-21]. Cerium is relatively earth-abundant and the oxides, including Ce2O3 and Ce7O12, are known to have band gaps in the visible region [13,19-21]. Our team has maintained a keen interest in alternative affordable, earth-abundant, visible light absorbing metal oxides to be used in two-photon ‘Z-schemes’ for dye-sensitized photoelectrosynthesis cells (DSPECs) [22-24]. To be employed in DSPECs, high surface areas for dye adsorption and an efficient charge conduction are critical properties [22-24]. As part of the preliminary investigations into this field, we communicate herein the preparation of high surface area, mesoporous cerium oxide nanospheres, which is a mixed phase of Ce7O12 and CeO2, and can absorb visible light to photocatalytically degrade dyes such as rhodamine B (RhB). The materials characterization of the cerium oxide nanospheres and some mechanistic insights into the photocatalytic process are presented.
Findings
Polycrystalline Ce7O12 samples have been previously synthesized, but harsh conditions (up to 1030 °C) by reduction of CeO2 with CO were employed [25,26]. Instead, mild, surfactant-free solvothermal conditions were used to prepare mesoporous cerium oxide with oxygen vacancies. A solution of ceric ammonium nitrate (CAN) in ethylene glycol and isopropanol as the solvent and reductant was heated up to 130 °C to yield mesoporous cerium oxide nanospheres after work-up. The powder X-ray diffraction (XRD) pattern (Figure 1a) indicates that the as-prepared cerium oxide material can be indexed to a superposition of hexagonal Ce7O12 (JCPDS File No. 71-0567) and cubic CeO2 (JCPDS File No. 81-0792) phases [14,26,27]. The peaks cannot be attributed to Ce(OH)3 or Ce2O3 phases [13,28], and confirm that the material contains a mixed phase. The considerable broadening of the peaks suggest that the domain sizes of the nanocrystalites are small, and has been estimated to be 4.8 nm ((211) plane, 2θ = 28.3°) by the Scherrer equation [29].
![[2190-4286-5-60-1]](/bjnano/content/figures/2190-4286-5-60-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Powder XRD pattern of cerium oxide nanospheres. (b) Wide-scan XPS survey spectrum. (c) High-resolution XPS spectrum of mesoporous cerium oxide (black circles) with the overall fit (black) and the fits to Ce4+ (blue) and Ce3+ (red).
Figure 1: (a) Powder XRD pattern of cerium oxide nanospheres. (b) Wide-scan XPS survey spectrum. (c) High-res...
In order to confirm the valence states of Ce and quantify their relative ratios in the prepared cerium oxide, X-ray photoelectron spectroscopy (XPS) experiments with monochromatic Al Kα radiation (hν = 1486.7 eV) were conducted. Unlike CeO2, in which the Ce atoms are all in the oxidation state 4+, the Ce atoms in Ce7O12 consist of both Ce3+ and Ce4+ valence states. The wide-scan survey spectrum in Figure 1b only shows Ce 3d, O 1s, and C 1s signals, and no other signals. The presence of the C 1s signal is probably from residual organic solvents or from air. This C 1s signal was used to calibrate the binding energy of the Ce 3d peaks. The high-resolution spectrum of the Ce 3d core states is illustrated in Figure 1c. Neither Ce4+ nor Ce3+ alone could give a satisfactory fitting to the spectrum in Figure 1c. Instead, the fitting of the Ce 3d spectrum required five components derived from both Ce3+ and Ce4+. There are two components (red) from Ce3+. The principal peak is at 886.4 eV and a 4f0 to 4f1v (v denotes valence hole) shake-down peak is at 879.9 eV [30]. The Ce4+ component consists of three peaks (blue). The peaks at 889.3 eV and 883.0 eV are the principal and 4f1v to 4f2v2 shake-down peaks from the 4f1v electronic configuration. The highest binding energy peak at 898.7 eV is from the 4f0 electronic configuration [30]. The binding energy of these peaks is in good agreement with those found in the literature [19,20,31]. However, by integrating the area under the fitted peaks, the concentration of Ce3+ is only 23%, which deviates from the predicted stoichiometric value (57%). This observation suggests that the Ce7O12 phase is mixed with some CeO2 phase on the surface. The nominal molecular formula of the material based on the XPS data is CeO1.89, comprising of around 54% Ce7O12 and 46% CeO2. The mixture of two crystalline forms is also observed in our XRD measurements and TEM results (vide infra).
The UV–vis diffuse reflectance spectra (subjected to a Kubelka–Munck transformation) of the cerium oxide nanospheres, CeO2 (commercially available 7 nm) nanopowder, and TiO2 (commercially available Degussa P25) nanoparticles are illustrated in Figure 2a. As expected, the cerium oxide sample displayed stronger visible light absorption than both commercially available 7 nm CeO2 and P25 TiO2 nanomaterials. The estimated band gap from the Tauc plot is approximately 2.7 eV (Figure 2b), which corresponds to an absorption edge in the blue region (460 nm). The reduced band gap compared to CeO2 can be attributed to the presence of oxygen vacancies, as previously reported [32]. The enhanced visible light absorption has been exploited for driving the photocatalytic degradation of RhB in aqueous solutions (vide infra).
![[2190-4286-5-60-2]](/bjnano/content/figures/2190-4286-5-60-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) UV–vis diffuse reflectance spectra of cerium oxide nanospheres (black), 7 nm CeO2 (red) nanopowder, and P25 TiO2 (blue). (b) Tauc plot for cerium oxide to obtain the band gap.
Figure 2: (a) UV–vis diffuse reflectance spectra of cerium oxide nanospheres (black), 7 nm CeO2 (red) nanopow...
The transmission electron microscopy (TEM) images of the cerium oxide sample supported the mesoporous nature and nanosphere morphology of the material (Figure 3a). The material has fairly monodisperse nanospheres with diameters of 50–70 nm. Each nanosphere consists of an irregular mesoporous structure that is an aggregate of small nanocrystalline domains. The high-resolution TEM images confirm that the cerium oxide consists of crystalline domains, 4–5 nm in size (red dotted ring), that can be indexed to Ce7O12 and CeO2 (Figure 3b). Nitrogen sorption experiments were conducted to ascertain the average surface area and pore size distribution of the material. The nitrogen adsorption–desorption isotherm of the cerium oxide sample (Figure 3c) shows a type-II curve and the surface area of the sample is 93 m2 g−1 as calculated by the Brunauer–Emmett–Teller (BET) method. The average pore size determined by a Barrett–Joyner–Halenda (BJH) analysis is 3 nm, confirming the mesoporous nature of the cerium oxide sample.
![[2190-4286-5-60-3]](/bjnano/content/figures/2190-4286-5-60-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) TEM and (b) HRTEM images of the mesoporous cerium oxide nanospheres. (c) Nitrogen adsorption–desorption isotherm of the mesoporous cerium oxide nanospheres.
Figure 3: (a) TEM and (b) HRTEM images of the mesoporous cerium oxide nanospheres. (c) Nitrogen adsorption–de...
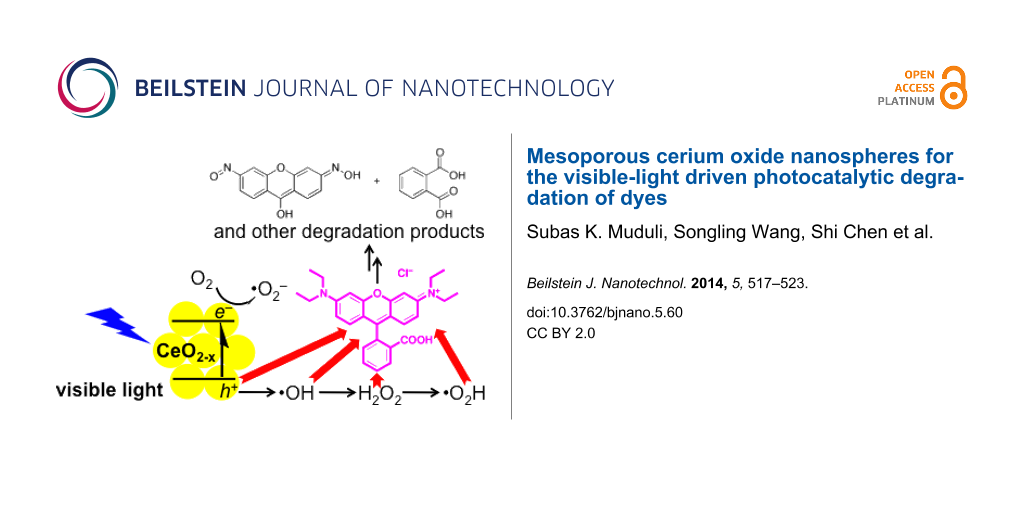
The photocatalytic behavior at visible-light irradiation of the cerium oxide sample has been probed by the photodegradation of the suspected carcinogenic dye rhodamine B (RhB). A colloidal mixture of cerium oxide and RhB has been stirred and irradiated with AM 1.5 solar intensity light after equilibration in the dark for 30 min. A standard glass filter has been applied to transmit only wavelengths larger than 420 nm, to demonstrate photocatalytic properties under ambient conditions. The UV–vis spectral changes of the colloidal mixture illustrated in Figure 4 clearly shows the degradation of RhB over time, with the dye being completely decomposed within 6 h. In comparison, RhB is only decomposed to 50% or less after irradiation under the same conditions with the commercially available P25 TiO2 and 7 nm CeO2 nanopowder. The visible-light photocatalytic degradation of organic compounds with wide band-gap materials, by a ligand-to-metal charge-transfer mechanism after adsorption [33], has been reported and may be a contributing factor for the activity of TiO2 and CeO2. In the absence of light, some of the RhB adsorbs on the cerium oxide (green). The mesoporous cerium oxide sample is patently a more effective agent for the photocatalytic degradation of RhB under visible light and ambient conditions after equilibration, and the activity cannot be accounted to the presence of CeO2 or adsorption alone. Gas chromatography mass spectrometry (GC–MS) and electrospray ionisation mass spectrometry (ESI–MS) were used to identify some of the organic products during the course of the 6 h irradiation (see Supporting Information File 1). These included N-hydroxylated desethyl rhodamine B, phthalic acid, and even ring-opened products [6]. The composition of the degradation products alludes to oxidative decomposition by reactive oxygen species, such as hydroxyl radicals.
![[2190-4286-5-60-4]](/bjnano/content/figures/2190-4286-5-60-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Comparison of RhB concentrations over time at 554 nm, after photocatalytic degradation with mesoporous cerium oxide under light (black) and in the dark with no equilibration (green), 7 nm CeO2 (red), P25 TiO2 (blue), and with no catalyst (grey).
Figure 4: Comparison of RhB concentrations over time at 554 nm, after photocatalytic degradation with mesopor...
Chemical scavengers were employed to investigate the mechanism of the photocatalytic processes and to identify the major contributors to the photocatalytic processes. The concentration of RhB, monitored at 554 nm, was used as the proxy to identify the active agent in the decomposition of RhB. Control experiments were performed in the absence of scavengers (black line, Figure 5a). The established scavengers used include sodium oxalate for h+ (red), CrO3 for e− (green), isopropanol for •OH (blue), and 1,4-benzoquinone for •OOH/•O2− (grey, Figure 5a) [5]. The inhibition of photocatalytic activity is most pronounced in the presence of the hole scavenger, with impaired activity in the presence of both •OH and •OOH/•O2− scavengers. Interestingly, the electron scavenger does not significantly affect the photodegradation experiments. The participation of •OH radicals was confirmed with the use of sodium terephthalate as a fluorescence probe [34]. Over the course of 6 h, the fluorescence intensity due to formation of 2-hydroxyterephthalate grew [35], with a blue shift possibly due to coordination to the mesoporous cerium oxide nanoparticles (Figure 5b). These results indicate that the photocatalytic mechanism can be summarized as depicted in Figure 5c, in which h+, and downstream reactive oxygen species •OH and •OOH/•O2−, are the active agents for the chemical destruction of RhB. Superoxide radicals from the reduction of O2 or direct reduction by electrons from the cerium oxide appear to play secondary roles in the photocatalytic destruction of RhB.
![[2190-4286-5-60-5]](/bjnano/content/figures/2190-4286-5-60-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: (a) Photocatalytic degradation of RhB over time at 554 nm, in the absence of scavengers (black), and the presence of h+ (red), •OH (blue), e– (green), and •OOH/•O2− scavengers (grey). (b) Growth of the fluorescence intensity of 2-hydroxyterephthlate as a probe for hydroxyl radicals. (c) Proposed pathway for photocatalytic RhB degradation.
Figure 5: (a) Photocatalytic degradation of RhB over time at 554 nm, in the absence of scavengers (black), an...
Conclusion
In summary, we have presented a facile, solvothermal synthesis of new mesoporous cerium oxide nanospheres, with isopropanol and ethylene glycol as the solvents and reducing agents. No expensive surfactants and templates have been used in the preparation of the earth-abundant, relatively affordable, mixed valence cerium oxide. The cerium oxide has been characterized with a suite of structural, spectroscopic, and electron microscopy techniques, confirming the high surface area, mesoporous nature, and visible-light absorption properties of the material. The visible-light photocatalytic activity in the degradation of RhB surpasses that of the commercially available CeO2 and P25 TiO2 nanopowders. With a series of radical scavengers, the mechanism of the photocatalytic activity is proposed to involve a prominent role of •OH radicals as the active species in RhB degradation. This new material is a promising candidate as a robust, earth-abundant, visible-light absorbing metal oxide scaffold to be used in DSPECs and other sustainable energy applications.
Supporting Information
The Supporting Information provides details about the synthesis of the nanospheres as well as additional experimental data.
| Supporting Information File 1: Synthesis procedure, characterization, and dye degradation studies. | ||
| Format: PDF | Size: 999.5 KB | Download |
Acknowledgements
H. S. Soo is supported by a NTU start-up grant (M4081012) and the Nanyang Assistant Professorship (M4081154). T. C. Sum is supported by a NTU start-up grant (M4080514), and the SPMS collaborative Research Award (M4080536). H. S. Soo and T. C. Sum also acknowledge the funding support from the Singapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE) CREATE Programme. This research programme/project is funded by the National Research Foundation (NRF), Prime Minister’s Office, Singapore under its Campus for Research Excellence and Technological Enterprise (CREATE) programme. S.K. Muduli and H.S. Soo thank Nanyang Asst. Prof. Ling Xing Yi for the use of her solar simulator, and Nanyang Asst. Prof. Zhao Yanli for the use of his gas sorption analyzer. The authors also thank Dr. Wei Fengxia for her assistance with HRTEM measurements and Dr. Sarifuddin Gazi for his help with EPR experiments.
References
-
Andreozzi, R.; Caprio, V.; Insola, A.; Marotta, R. Catal. Today 1999, 53, 51–59. doi:10.1016/S0920-5861(99)00102-9
Return to citation in text: [1] [2] -
Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W. Chem. Rev. 1995, 95, 69–96. doi:10.1021/cr00033a004
Return to citation in text: [1] [2] [3] [4] -
Chalasani, R.; Vasudevan, S. ACS Nano 2013, 7, 4093–4104. doi:10.1021/nn400287k
Return to citation in text: [1] [2] [3] -
Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200
Return to citation in text: [1] [2] [3] [4] -
Wang, W.; Yu, Y.; An, T.; Li, G.; Yip, H. Y.; Yu, J. C.; Wong, P. K. Environ. Sci. Technol. 2012, 46, 4599–4606. doi:10.1021/es2042977
Return to citation in text: [1] [2] [3] [4] -
Yu, K.; Yang, S.; He, H.; Sun, C.; Gu, C.; Ju, Y. J. Phys. Chem. A 2009, 113, 10024–10032. doi:10.1021/jp905173e
Return to citation in text: [1] [2] [3] [4] -
Chen, H.; Nanayakkara, C. E.; Grassian, V. H. Chem. Rev. 2012, 112, 5919–5948. doi:10.1021/cr3002092
Return to citation in text: [1] [2] -
Chen, X.; Mao, S. S. Chem. Rev. 2007, 107, 2891–2959. doi:10.1021/cr0500535
Return to citation in text: [1] [2] -
Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Chem. Rev. 2010, 110, 6595–6663. doi:10.1021/cr900356p
Return to citation in text: [1] -
Hodes, G.; Cahen, D. Acc. Chem. Res. 2012, 45, 705–713. doi:10.1021/ar200219h
Return to citation in text: [1] -
Kubacka, A.; Fernández-García, M.; Colón, G. Chem. Rev. 2012, 112, 1555–1614. doi:10.1021/cr100454n
Return to citation in text: [1] -
Pouretedal, H. R.; Kadkhodaie, A. Chin. J. Catal. 2010, 31, 1328–1334. doi:10.1016/S1872-2067(10)60121-0
Return to citation in text: [1] -
Vuppala, V.; Motappa, M. G.; Venkata, S. S.; Sadashivaiah, P. H. Eur. J. Chem. 2012, 3, 191–195. doi:10.5155/eurjchem.3.2.191-195.564
Return to citation in text: [1] [2] [3] [4] -
Chueh, W. C.; Falter, C.; Abbott, M.; Scipio, D.; Furler, P.; Haile, S. M.; Steinfeld, A. Science 2010, 330, 1797–1801. doi:10.1126/science.1197834
Return to citation in text: [1] [2] -
Chueh, W. C.; Haile, S. M. ChemSusChem 2009, 2, 735–739. doi:10.1002/cssc.200900138
Return to citation in text: [1] -
Scheffe, J. R.; Steinfeld, A. Energy Fuels 2012, 26, 1928–1936. doi:10.1021/ef201875v
Return to citation in text: [1] -
Zhang, D.; Du, X.; Shi, L.; Gao, R. Dalton Trans. 2012, 41, 14455–14475. doi:10.1039/c2dt31759a
Return to citation in text: [1] -
Muhich, C. L.; Evanko, B. W.; Weston, K. C.; Lichty, P.; Liang, X.; Martinek, J.; Musgrave, C. B.; Weimer, A. W. Science 2013, 341, 540–542. doi:10.1126/science.1239454
Return to citation in text: [1] -
Ghoshal, T.; Fleming, P. G.; Holmes, J. D.; Morris, M. A. J. Mater. Chem. 2012, 22, 22949–22957. doi:10.1039/c2jm35073d
Return to citation in text: [1] [2] [3] -
Stetsovych, V.; Pagliuca, F.; Dvořák, F.; Duchoň, T.; Vorokhta, M.; Aulická, M.; Lachnitt, J.; Schernich, S.; Matolinová, I.; Veltruská, K.; Skála, T.; Mazur, D.; Mysliveček, J.; Libuda, J.; Matolín, V. J. Phys. Chem. Lett. 2013, 4, 866–871. doi:10.1021/jz400187j
Return to citation in text: [1] [2] [3] -
Wilkens, H.; Schuckmann, O.; Oelke, R.; Gevers, S.; Schaefer, A.; Bäumer, M.; Zoellner, M. H.; Schroeder, T.; Wollschläger, J. Appl. Phys. Lett. 2013, 102, 111602. doi:10.1063/1.4795867
Return to citation in text: [1] [2] -
Luo, H.; Song, W.; Hoertz, P. G.; Hanson, K.; Ghosh, R.; Rangan, S.; Brennaman, M. K.; Concepcion, J. J.; Binstead, R. A.; Bartynski, R. A.; Lopez, R.; Meyer, T. J. Chem. Mater. 2013, 25, 122–131. doi:10.1021/cm3027972
Return to citation in text: [1] [2] -
Song, W.; Chen, Z.; Glasson, C. R. K.; Hanson, K.; Luo, H.; Norris, M. R.; Ashford, D. L.; Concepcion, J. J.; Brennaman, M. K.; Meyer, T. J. ChemPhysChem 2012, 13, 2882–2890. doi:10.1002/cphc.201200100
Return to citation in text: [1] [2] -
Alibabaei, L.; Luo, H.; House, R. L.; Hoertz, P. G.; Lopez, R.; Meyer, T. J. J. Mater. Chem. A 2013, 1, 4133–4145. doi:10.1039/c2ta00935h
Return to citation in text: [1] [2] -
Ray, S. P.; Cox, D. E. J. Solid State Chem. 1975, 15, 333–343. doi:10.1016/0022-4596(75)90289-3
Return to citation in text: [1] -
Kümmerle, E. A.; Heger, G. J. Solid State Chem. 1999, 147, 485–500. doi:10.1006/jssc.1999.8403
Return to citation in text: [1] [2] -
Ray, S. P.; Nowick, A. S.; Cox, D. E. J. Solid State Chem. 1975, 15, 344–351. doi:10.1016/0022-4596(75)90290-X
Return to citation in text: [1] -
Suresh, R.; Ponnuswamy, V.; Mariappan, R. Appl. Surf. Sci. 2013, 273, 457–464. doi:10.1016/j.apsusc.2013.02.062
Return to citation in text: [1] -
Patterson, A. L. Phys. Rev. 1939, 56, 978–982. doi:10.1103/PhysRev.56.978
Return to citation in text: [1] -
Kotani, A.; Ogasawara, H. J. Electron Spectrosc. Relat. Phenom. 1992, 60, 257–299. doi:10.1016/0368-2048(92)80024-3
Return to citation in text: [1] [2] -
Trudeau, M. L.; Tschöpe, A.; Ying, J. Y. Surf. Interface Anal. 1995, 23, 219–226. doi:10.1002/sia.740230405
Return to citation in text: [1] -
Corma, A.; Atienzar, P.; Garcia, H.; Chane-Ching, J.-Y. Nat. Mater. 2004, 3, 394–397. doi:10.1038/nmat1129
Return to citation in text: [1] -
Liang, S.; Wen, L.; Lin, S.; Bi, J.; Feng, P.; Fu, X.; Wu, L. Angew. Chem., Int. Ed. 2014, 53, 2951–2955. doi:10.1002/anie.201311280
Return to citation in text: [1] -
Gomes, A.; Fernandes, E.; Lima, J. L. F. C. J. Biochem. Biophys. Methods 2005, 65, 45–80. doi:10.1016/j.jbbm.2005.10.003
Return to citation in text: [1] -
Ishibashi, K.; Fujishima, A.; Watanabe, T.; Hashimoto, K. J. Photochem. Photobiol., A: Chem. 2000, 134, 139–142. doi:10.1016/S1010-6030(00)00264-1
Return to citation in text: [1]
| 1. | Andreozzi, R.; Caprio, V.; Insola, A.; Marotta, R. Catal. Today 1999, 53, 51–59. doi:10.1016/S0920-5861(99)00102-9 |
| 7. | Chen, H.; Nanayakkara, C. E.; Grassian, V. H. Chem. Rev. 2012, 112, 5919–5948. doi:10.1021/cr3002092 |
| 8. | Chen, X.; Mao, S. S. Chem. Rev. 2007, 107, 2891–2959. doi:10.1021/cr0500535 |
| 9. | Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Chem. Rev. 2010, 110, 6595–6663. doi:10.1021/cr900356p |
| 10. | Hodes, G.; Cahen, D. Acc. Chem. Res. 2012, 45, 705–713. doi:10.1021/ar200219h |
| 11. | Kubacka, A.; Fernández-García, M.; Colón, G. Chem. Rev. 2012, 112, 1555–1614. doi:10.1021/cr100454n |
| 14. | Chueh, W. C.; Falter, C.; Abbott, M.; Scipio, D.; Furler, P.; Haile, S. M.; Steinfeld, A. Science 2010, 330, 1797–1801. doi:10.1126/science.1197834 |
| 26. | Kümmerle, E. A.; Heger, G. J. Solid State Chem. 1999, 147, 485–500. doi:10.1006/jssc.1999.8403 |
| 27. | Ray, S. P.; Nowick, A. S.; Cox, D. E. J. Solid State Chem. 1975, 15, 344–351. doi:10.1016/0022-4596(75)90290-X |
| 1. | Andreozzi, R.; Caprio, V.; Insola, A.; Marotta, R. Catal. Today 1999, 53, 51–59. doi:10.1016/S0920-5861(99)00102-9 |
| 2. | Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W. Chem. Rev. 1995, 95, 69–96. doi:10.1021/cr00033a004 |
| 4. | Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200 |
| 5. | Wang, W.; Yu, Y.; An, T.; Li, G.; Yip, H. Y.; Yu, J. C.; Wong, P. K. Environ. Sci. Technol. 2012, 46, 4599–4606. doi:10.1021/es2042977 |
| 6. | Yu, K.; Yang, S.; He, H.; Sun, C.; Gu, C.; Ju, Y. J. Phys. Chem. A 2009, 113, 10024–10032. doi:10.1021/jp905173e |
| 13. | Vuppala, V.; Motappa, M. G.; Venkata, S. S.; Sadashivaiah, P. H. Eur. J. Chem. 2012, 3, 191–195. doi:10.5155/eurjchem.3.2.191-195.564 |
| 28. | Suresh, R.; Ponnuswamy, V.; Mariappan, R. Appl. Surf. Sci. 2013, 273, 457–464. doi:10.1016/j.apsusc.2013.02.062 |
| 2. | Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W. Chem. Rev. 1995, 95, 69–96. doi:10.1021/cr00033a004 |
| 3. | Chalasani, R.; Vasudevan, S. ACS Nano 2013, 7, 4093–4104. doi:10.1021/nn400287k |
| 4. | Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200 |
| 22. | Luo, H.; Song, W.; Hoertz, P. G.; Hanson, K.; Ghosh, R.; Rangan, S.; Brennaman, M. K.; Concepcion, J. J.; Binstead, R. A.; Bartynski, R. A.; Lopez, R.; Meyer, T. J. Chem. Mater. 2013, 25, 122–131. doi:10.1021/cm3027972 |
| 23. | Song, W.; Chen, Z.; Glasson, C. R. K.; Hanson, K.; Luo, H.; Norris, M. R.; Ashford, D. L.; Concepcion, J. J.; Brennaman, M. K.; Meyer, T. J. ChemPhysChem 2012, 13, 2882–2890. doi:10.1002/cphc.201200100 |
| 24. | Alibabaei, L.; Luo, H.; House, R. L.; Hoertz, P. G.; Lopez, R.; Meyer, T. J. J. Mater. Chem. A 2013, 1, 4133–4145. doi:10.1039/c2ta00935h |
| 2. | Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W. Chem. Rev. 1995, 95, 69–96. doi:10.1021/cr00033a004 |
| 3. | Chalasani, R.; Vasudevan, S. ACS Nano 2013, 7, 4093–4104. doi:10.1021/nn400287k |
| 4. | Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200 |
| 5. | Wang, W.; Yu, Y.; An, T.; Li, G.; Yip, H. Y.; Yu, J. C.; Wong, P. K. Environ. Sci. Technol. 2012, 46, 4599–4606. doi:10.1021/es2042977 |
| 6. | Yu, K.; Yang, S.; He, H.; Sun, C.; Gu, C.; Ju, Y. J. Phys. Chem. A 2009, 113, 10024–10032. doi:10.1021/jp905173e |
| 25. | Ray, S. P.; Cox, D. E. J. Solid State Chem. 1975, 15, 333–343. doi:10.1016/0022-4596(75)90289-3 |
| 26. | Kümmerle, E. A.; Heger, G. J. Solid State Chem. 1999, 147, 485–500. doi:10.1006/jssc.1999.8403 |
| 14. | Chueh, W. C.; Falter, C.; Abbott, M.; Scipio, D.; Furler, P.; Haile, S. M.; Steinfeld, A. Science 2010, 330, 1797–1801. doi:10.1126/science.1197834 |
| 15. | Chueh, W. C.; Haile, S. M. ChemSusChem 2009, 2, 735–739. doi:10.1002/cssc.200900138 |
| 16. | Scheffe, J. R.; Steinfeld, A. Energy Fuels 2012, 26, 1928–1936. doi:10.1021/ef201875v |
| 17. | Zhang, D.; Du, X.; Shi, L.; Gao, R. Dalton Trans. 2012, 41, 14455–14475. doi:10.1039/c2dt31759a |
| 18. | Muhich, C. L.; Evanko, B. W.; Weston, K. C.; Lichty, P.; Liang, X.; Martinek, J.; Musgrave, C. B.; Weimer, A. W. Science 2013, 341, 540–542. doi:10.1126/science.1239454 |
| 13. | Vuppala, V.; Motappa, M. G.; Venkata, S. S.; Sadashivaiah, P. H. Eur. J. Chem. 2012, 3, 191–195. doi:10.5155/eurjchem.3.2.191-195.564 |
| 19. | Ghoshal, T.; Fleming, P. G.; Holmes, J. D.; Morris, M. A. J. Mater. Chem. 2012, 22, 22949–22957. doi:10.1039/c2jm35073d |
| 20. | Stetsovych, V.; Pagliuca, F.; Dvořák, F.; Duchoň, T.; Vorokhta, M.; Aulická, M.; Lachnitt, J.; Schernich, S.; Matolinová, I.; Veltruská, K.; Skála, T.; Mazur, D.; Mysliveček, J.; Libuda, J.; Matolín, V. J. Phys. Chem. Lett. 2013, 4, 866–871. doi:10.1021/jz400187j |
| 21. | Wilkens, H.; Schuckmann, O.; Oelke, R.; Gevers, S.; Schaefer, A.; Bäumer, M.; Zoellner, M. H.; Schroeder, T.; Wollschläger, J. Appl. Phys. Lett. 2013, 102, 111602. doi:10.1063/1.4795867 |
| 12. | Pouretedal, H. R.; Kadkhodaie, A. Chin. J. Catal. 2010, 31, 1328–1334. doi:10.1016/S1872-2067(10)60121-0 |
| 13. | Vuppala, V.; Motappa, M. G.; Venkata, S. S.; Sadashivaiah, P. H. Eur. J. Chem. 2012, 3, 191–195. doi:10.5155/eurjchem.3.2.191-195.564 |
| 22. | Luo, H.; Song, W.; Hoertz, P. G.; Hanson, K.; Ghosh, R.; Rangan, S.; Brennaman, M. K.; Concepcion, J. J.; Binstead, R. A.; Bartynski, R. A.; Lopez, R.; Meyer, T. J. Chem. Mater. 2013, 25, 122–131. doi:10.1021/cm3027972 |
| 23. | Song, W.; Chen, Z.; Glasson, C. R. K.; Hanson, K.; Luo, H.; Norris, M. R.; Ashford, D. L.; Concepcion, J. J.; Brennaman, M. K.; Meyer, T. J. ChemPhysChem 2012, 13, 2882–2890. doi:10.1002/cphc.201200100 |
| 24. | Alibabaei, L.; Luo, H.; House, R. L.; Hoertz, P. G.; Lopez, R.; Meyer, T. J. J. Mater. Chem. A 2013, 1, 4133–4145. doi:10.1039/c2ta00935h |
| 5. | Wang, W.; Yu, Y.; An, T.; Li, G.; Yip, H. Y.; Yu, J. C.; Wong, P. K. Environ. Sci. Technol. 2012, 46, 4599–4606. doi:10.1021/es2042977 |
| 6. | Yu, K.; Yang, S.; He, H.; Sun, C.; Gu, C.; Ju, Y. J. Phys. Chem. A 2009, 113, 10024–10032. doi:10.1021/jp905173e |
| 2. | Hoffmann, M. R.; Martin, S. T.; Choi, W.; Bahnemann, D. W. Chem. Rev. 1995, 95, 69–96. doi:10.1021/cr00033a004 |
| 3. | Chalasani, R.; Vasudevan, S. ACS Nano 2013, 7, 4093–4104. doi:10.1021/nn400287k |
| 4. | Kisch, H. Angew. Chem., Int. Ed. 2013, 52, 812–847. doi:10.1002/anie.201201200 |
| 7. | Chen, H.; Nanayakkara, C. E.; Grassian, V. H. Chem. Rev. 2012, 112, 5919–5948. doi:10.1021/cr3002092 |
| 8. | Chen, X.; Mao, S. S. Chem. Rev. 2007, 107, 2891–2959. doi:10.1021/cr0500535 |
| 13. | Vuppala, V.; Motappa, M. G.; Venkata, S. S.; Sadashivaiah, P. H. Eur. J. Chem. 2012, 3, 191–195. doi:10.5155/eurjchem.3.2.191-195.564 |
| 19. | Ghoshal, T.; Fleming, P. G.; Holmes, J. D.; Morris, M. A. J. Mater. Chem. 2012, 22, 22949–22957. doi:10.1039/c2jm35073d |
| 20. | Stetsovych, V.; Pagliuca, F.; Dvořák, F.; Duchoň, T.; Vorokhta, M.; Aulická, M.; Lachnitt, J.; Schernich, S.; Matolinová, I.; Veltruská, K.; Skála, T.; Mazur, D.; Mysliveček, J.; Libuda, J.; Matolín, V. J. Phys. Chem. Lett. 2013, 4, 866–871. doi:10.1021/jz400187j |
| 21. | Wilkens, H.; Schuckmann, O.; Oelke, R.; Gevers, S.; Schaefer, A.; Bäumer, M.; Zoellner, M. H.; Schroeder, T.; Wollschläger, J. Appl. Phys. Lett. 2013, 102, 111602. doi:10.1063/1.4795867 |
| 30. | Kotani, A.; Ogasawara, H. J. Electron Spectrosc. Relat. Phenom. 1992, 60, 257–299. doi:10.1016/0368-2048(92)80024-3 |
| 30. | Kotani, A.; Ogasawara, H. J. Electron Spectrosc. Relat. Phenom. 1992, 60, 257–299. doi:10.1016/0368-2048(92)80024-3 |
| 35. | Ishibashi, K.; Fujishima, A.; Watanabe, T.; Hashimoto, K. J. Photochem. Photobiol., A: Chem. 2000, 134, 139–142. doi:10.1016/S1010-6030(00)00264-1 |
| 5. | Wang, W.; Yu, Y.; An, T.; Li, G.; Yip, H. Y.; Yu, J. C.; Wong, P. K. Environ. Sci. Technol. 2012, 46, 4599–4606. doi:10.1021/es2042977 |
| 34. | Gomes, A.; Fernandes, E.; Lima, J. L. F. C. J. Biochem. Biophys. Methods 2005, 65, 45–80. doi:10.1016/j.jbbm.2005.10.003 |
| 33. | Liang, S.; Wen, L.; Lin, S.; Bi, J.; Feng, P.; Fu, X.; Wu, L. Angew. Chem., Int. Ed. 2014, 53, 2951–2955. doi:10.1002/anie.201311280 |
| 6. | Yu, K.; Yang, S.; He, H.; Sun, C.; Gu, C.; Ju, Y. J. Phys. Chem. A 2009, 113, 10024–10032. doi:10.1021/jp905173e |
| 19. | Ghoshal, T.; Fleming, P. G.; Holmes, J. D.; Morris, M. A. J. Mater. Chem. 2012, 22, 22949–22957. doi:10.1039/c2jm35073d |
| 20. | Stetsovych, V.; Pagliuca, F.; Dvořák, F.; Duchoň, T.; Vorokhta, M.; Aulická, M.; Lachnitt, J.; Schernich, S.; Matolinová, I.; Veltruská, K.; Skála, T.; Mazur, D.; Mysliveček, J.; Libuda, J.; Matolín, V. J. Phys. Chem. Lett. 2013, 4, 866–871. doi:10.1021/jz400187j |
| 31. | Trudeau, M. L.; Tschöpe, A.; Ying, J. Y. Surf. Interface Anal. 1995, 23, 219–226. doi:10.1002/sia.740230405 |
| 32. | Corma, A.; Atienzar, P.; Garcia, H.; Chane-Ching, J.-Y. Nat. Mater. 2004, 3, 394–397. doi:10.1038/nmat1129 |
© 2014 Muduli et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Nanotechnology terms and conditions: (http://www.beilstein-journals.org/bjnano)