Abstract
The previously reported concept of using mixtures of monodentate ligands in a combinatorial manner in order to influence enantio- or regioselectivity of transition metal catalyzed processes has been extended to include diastereoselectivity. Accordingly, 1,2- and 1,3-asymmetric induction in the Rh-catalyzed hydrogenation of a chiral allylic alcohol and a chiral homo-allylic alcohol has been studied by using mixtures of monodentate P-ligands. It was found that appropriate 1:1 mixtures of two different P-ligands enhance the degree of diastereoselectivity relative to the use of the respective pure ligands themselves. Here, as in the previous cases regarding enantio- or regioselectivity, this type of combinatorial catalysis leads to improved catalytic profiles without the need to prepare new ligands.

Graphical Abstract
The application of combinatorial chemical methods in asymmetric catalysis has emerged as a promising area of research, and indeed several reviews covering the subject have appeared.[1-8] It is based on the preparation of libraries of chiral metal complexes or metal-free catalysts[9] followed by medium- or high-throughput screening.[5] Catalyst diversity is achieved by the design and synthesis of modular ligands comprised of several building blocks which can be varied at will and easily assembled covalently. Another strategy involves the use of additives, catalyst poisons or catalyst activators.[10,11]
Based on the discovery that monodentate BINOL-derived phosphites,[12,13] phosphonites[14,15] and phosphoramidites [16-18] are excellent ligands in enantioselective Rh-catalyzed olefin-hydrogenation, we proposed in 2003 a new approach in the area of combinatorial asymmetric transition metal catalysis:[19,20] The use of mixtures of two chiral monodentate ligands La and Lb in metal catalysts of the type ML2, leading to two so-called homo-combinations and one hetero-combination which are in equilibrium with one another (Scheme 1). If the hetero-combination dominates due to enhanced activity and enantioselectivity, a superior catalytic profile can be expected. A mechanistic study of Rh-catalyzed olefin-hydrogenation using BINOL-derived monodentate phosphites (homo-combination) has shown that two such ligands are bound to the metal in the transition state of the reaction,[13] and it is certain that in the case of mixtures analogous species are involved.[19,20]
![[1860-5397-1-3-i1]](/bjoc/content/inline/1860-5397-1-3-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
This new strategy was first employed in Rh-catalyzed olefin-hydrogenation using mixtures of BINOL-derived phosphites and phosphonites[19,20] and was then generalized.[21-23] Following our initial discovery,[19] Feringa and deVries reported related results using BINOL-derived phosphoramidites.[24] In all of these studies it was found that the mixture, and consequently the hetero-combination MLaLb, is more active and more enantioselective than either of the homo-combinations MLaLa or MLbLb which are also present in the reaction vessel.
In further research we discovered that it is also possible to employ a mixture composed of a chiral and an achiral monodentate P-ligand in asymmetric Rh-catalyzed olefin-hydrogenation, enhanced activity and reversal of enantioselectivity being observed in some cases.[25] Moreover, appropriate pairs of chiral and achiral monodentate P-ligands in other systems result in enhanced activity and nearly complete enantioselectivity.[26] Since it is currently not straightforward to predict which particular pair of chiral/chiral or chiral/achiral ligands is optimal for a given asymmetric transformation, a trial and error strategy is necessary.[27] Although this may not appear intellectually appealing, such an empirical process has an important advantage: Once a library of monodenate ligands has put on the shelf by synthesis or commercial acquisition, simply mixing them pairwise in all possible permutational modes generates high catalyst diversity without the need to synthesize new ligands. Of course, once a hit has been identified in the form of MLaLb, structural and mechanistic considerations may help in designing the optimal bidentate ligand composed of building blocks derived from the particular ligands La and Lb.
We have recently extended this combinatorial principle beyond enantioselectivity[19-26] to include regioselectivity,[28] specifically in the hydroformylation of methacrylic acid tert-butyl ester. By using a 1:1 mixture of triphenylphosphine and a certain phosphinine (substituted phospha-benzene), 95% regioselectivity in favor of C-C bond formation at the higher substituted C-atom was achieved, whereas the use of the pure ligands as traditional homo-combinations led to low degrees of regioselectivity, but in the opposite sense.[28]
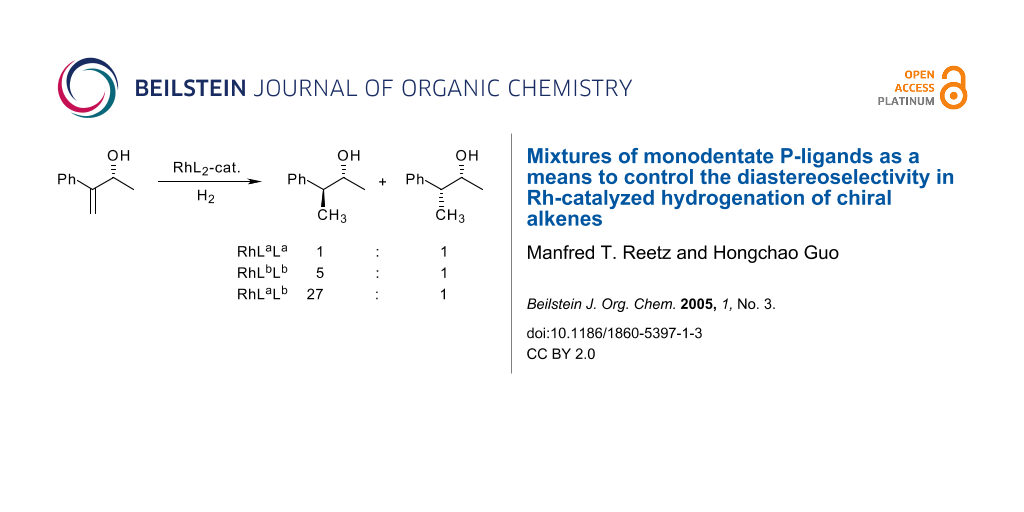
We now show for the first time that mixtures of monodentate P-ligands can also be used to influence diastereoselectivity, specifically in the Rh-catalyzed hydrogenation of chiral olefins. The literature contains several reports of 1,2- and 1,3-asymmetric induction in the hydrogenation of chiral olefins,[29,30] the underlying principle being Rh-complexation of functional groups present in the olefin (e.g., hydroxyl moieties) and/or 1,3-allylic strain[31] within the framework of substrate-directed stereoselectivity. Generally, a racemate was used to study diastereoselectivity. Catalyst ("reagent") control based on optically active ligands in the hydrogenation of chiral olefins has also been studied, in which case the substrates need to be used in enantiomerically pure form.[30] In some cases the role of the achiral ligands at the metal was studied, although not in an extensive manner. For example, Brown reported the Rh-catalyzed hydrogenation of the racemic allylic alcohol 1 with formation of diastereomers 2 and 3, ketone 4 forming as a side product due to undesired isomerization.[29] From a small collection of mono-phosphines and bidentate diphosphines, diphos-4 led to the highest degree of diastereoselectivity in favor of the anti-product 2 (32:1). In our study we also chose this transformation as the model reaction. All reactions were performed with the racemate of 1, although only one enantiomeric form is shown here for simplicity.
![[1860-5397-1-3-i2]](/bjoc/content/inline/1860-5397-1-3-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
As a second model reaction the analogous Rh-catalyzed hydrogenation of the homo-allylic alcohol 5 with formation of diastereomers 6 and 7 was considered. Here again one enantiomeric form is shown arbitrarily, although a racemate was used.
![[1860-5397-1-3-i3]](/bjoc/content/inline/1860-5397-1-3-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Monodentate P-ligands P1 – P23 were employed in the hydrogenation reactions, the Rh:ligand ratio being 1:2 in all cases. These ligands were not chosen on the basis of structural or mechanistic considerations; rather, they happened to be available in our laboratory, and indeed many of them are commercially available (P1–P6, P8, and P11–P14). Most of the ligands are achiral, but some contain one or even two stereogenic centers. Ligands P18, P20 and P21[32] were used in the (S)-form. In the case of the menthyl-derivative P15, a single stereoisomer was employed, although the configuration at phosphorus is unknown.[33] The menthol-derived ligand P23 having the (S)-configuration at phosphorus was described earlier.[34] Since the substrates 1 and 5 were used as racemates, kinetic resolution may be involved when the chiral ligands are employed, i.e., diastereoselectivity may change with conversion. This aspect was not a subject of the present study. In all hydrogenation reactions the standard precursor Rh(nbd)2BF4 (nbd = norbornadiene) was treated with the ligands to form pre-catalysts of the type RhL2(nbd)BF4 and free nbd. Dichloromethane served as the solvent.
![[1860-5397-1-3-i4]](/bjoc/content/inline/1860-5397-1-3-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the case of the hydrogenation of the allylic alcohol 1, 21 of the 23 ligands were employed, which means a total of 210 different hetero-combinations in addition to the 21 conventional reactions (homo-combinations). Not all of them were actually tested. Rather, the combinatorial search was terminated after approximately 150 reactions had been performed in an arbitrary order. This random walk through a two-fold field of combinations led to the identification of 15–20 hits, i.e., those in which the hetero-combination results in a higher diastereoselectivity than either of the two homo-combinations themselves alone. Table 1 summarizes the essential part of the data. The hetero-combination P11/P17 results in the highest diastereoselectivity found in the total search, namely 27:1 in favor of the anti-product 2 (entry 33). This contrasts with the results obtained using the homo-combination P11/P11 which shows totally sterorandom behavior (entry 11), and the other homo-combination comprising the phosphinine P17/P17[35] which results in a diastereoselectivity of only 5:1 (entry 17). Another noteworthy catalyst system is composed of the bulky phosphite P22. This ligand alone delivers a moderate diasteroselectivity of 4:1 (entry 20), whereas various hetero-combinations based on P22 and other P-ligands induce markedly enhanced selectivities (entries 23, 25, 26, 27, 28, 29, 30, 32, 34, 36). It is of no surprise that many hetero-combinations fail to induce significant improvements (data not shown), and that some result in diastereoselectivities which are actually lower than the respective homo-combinations. An example of the latter is hetero-combination P18/P22 leading to a 2:3 ratio of only 2:1, while P18/P18 and P22/P22 are both more selective (each 4:1; entries 18 and 20, respectively).
Table 1: Diastereoselective hydrogenation of the allylic alcohol 1.
| Entry | Ligand | Conversion (%) | Diastereoselectivity 2:3 | Side product 4 (%) |
|---|---|---|---|---|
| Homo-combinations | ||||
| 1 | P1/P1 | 100 | 10:1 | 47 |
| 2 | P2/P2 | 100 | 17:1 | 33 |
| 3 | P3/P3 | 100 | 14:1 | 51 |
| 4 | P4/P4 | 100 | 16:1 | 30 |
| 5 | P5/P5 | 100 | 10:1 | 49 |
| 6 | P6/P6 | 7 | 2:1 | 4 |
| 7 | P7/P7 | 78 | 1:1 | 8 |
| 8 | P8/P8 | 100 | 13:1 | 64 |
| 9 | P9/P9 | 100 | 16:1 | 58 |
| 10 | P10/P10 | 100 | 14:1 | 47 |
| 11 | P11/P11 | 93 | 1:1 | 7 |
| 12 | P12/P12 | 44 | 3:1 | 27 |
| 13 | P13/P13 | 100 | 17:1 | 15 |
| 14 | P14/P14 | 74 | 2:1 | 8 |
| 15 | P15/P15 | 100 | 12:1 | 1 |
| 16 | P16/P16 | 79 | 1.5:1 | 18 |
| 17 | P17/P17 | 100 | 5:1 | 98 |
| 18 | P18/P18 | 51 | 4:1 | 15 |
| 19 | P20/P20 | 64 | 4:1 | 24 |
| 20 | P22/P22 | 45 | 4:1 | 7 |
| Hetero-combinations (hits) | ||||
| 21 | P1/P12 | 100 | 19:1 | 45 |
| 22 | P1/P15 | 100 | 18:1 | 24 |
| 23 | P1/P22 | 100 | 17:1 | 45 |
| 24 | P2/P12 | 100 | 21:1 | 40 |
| 25 | P2/P22 | 100 | 19:1 | 33 |
| 26 | P4/P22 | 100 | 18:1 | 32 |
| 27 | P5/P22 | 100 | 18:1 | 47 |
| 28 | P6/P22 | 25 | 8:1 | 6 |
| 29 | P8/P22 | 100 | 20:1 | 65 |
| 30 | P9/P22 | 100 | 20:1 | 59 |
| 31 | P10/P15 | 100 | 20:1 | 30 |
| 32 | P10/P22 | 100 | 19:1 | 50 |
| 33 | P11/P17 | 56 | 27:1 | 48 |
| 34 | P12/P22 | 74 | 9:1 | 4 |
| 35 | P17/P20 | 100 | 7:1 | 70 |
| 36 | P17/P22 | 100 | 8:1 | 56 |
Diastereoselectivity in the hydrogenation of the homo-allylic alcohol 5 involves 1,3-asymmetric induction, which is usually considered to be more difficult.[30,31] In this case the combinatorial search was restricted to ligands P1-P17, P19 and P21–P23, although again not all permutations were actually tested. Table 2 reveals the diastereoselectivities resulting from the use of the homo-combinations and of those hetero-combinations which exert a substantial stereochemical influence (hits). Inspection of the best results arising from the homo-combinations shows that diastereoselectivity in favor of the anti-product 6 is moderate (e.g., entries 6, 12, 16, 19, 20). Since in the present study the racemic form of 5 was employed, the degree of "reagent control" in the case of the chiral ligands P16, P18 and P20 was not ascertained. The highest syn-selectivity favoring 7 amounts to only 1:1.8 when using ligand P4 (entry 4). From the list of hetero-combinations showing a stereochemical influence relative to the use of the respective pure ligands, three lead to substantial syn-selectivity (6:7 = 1:5), namely P4/P23, P9/P23 and P10/P23 (entries 24, 29 and 31, respectively). In all three cases the chiral phosphinite 23 is involved, which as a homo-combination actually favors the opposite diastereomer (6:7 = 3:1; entry 21). With regard to maximizing anti-selectivity, the hetero-combination P7/P21 results in a diastereoselectivity of 18:1 in favor of 6 (entry 26) which is clearly better than P21 alone or any other homo-combination listed in Table 2.
Table 2: Diastereoselective hydrogenation of the homo-allylic alcohol 5.
| Entry | Ligand | Conversion (%) | Diastereoselectivity 6:7 |
|---|---|---|---|
| Homo-combinations | |||
| 1 | P1 | 100 | 1:1.4 |
| 2 | P2 | 100 | 1:1.5 |
| 3 | P3 | 100 | 1:1 |
| 4 | P4 | 100 | 1:1.8 |
| 5 | P5 | 100 | 1.2:1 |
| 6 | P6 | 100 | 7:1 |
| 7 | P7 | 6 | 3:1 |
| 8 | P8 | 100 | 2:1 |
| 9 | P9 | 100 | 1:1 |
| 10 | P10 | 100 | 1:1 |
| 11 | P11 | 2 | 2:1 |
| 12 | P12 | 100 | 7:1 |
| 13 | P13 | 100 | 1.2:1 |
| 14 | P14 | 1 | 1.6:1 |
| 15 | P15 | 100 | 2.6:1 |
| 16 | P16 | 100 | 9:1 |
| 17 | P17 | 32 | 3:1 |
| 18 | P19 | 89 | 3:1 |
| 19 | P21 | 2 | 6:1 |
| 20 | P22 | 100 | 10:1 |
| 21 | P23 | 100 | 3:1 |
| Hetero-combinations (hits) | |||
| 22 | P3/P19 | 100 | 1:2 |
| 23 | P4/P19 | 100 | 1:2.6 |
| 24 | P4/P23 | 100 | 1:5 |
| 25 | P5/P19 | 100 | 1:1.8 |
| 26 | P7/P21 | 26 | 18:1 |
| 27 | P8/P19 | 100 | 1:1.8 |
| 28 | P9/P19 | 100 | 1:2 |
| 29 | P9/P23 | 100 | 1:5 |
| 30 | P10/P19 | 100 | 1:2 |
| 31 | P10/P23 | 100 | 1:5 |
| 32 | P11/P14 | 14 | 6:1 |
| 33 | P11/P15 | 100 | 7:1 |
| 34 | P14/P15 | 86 | 9:1 |
| 35 | P14/P19 | 77 | 5:1 |
| 36 | P17/P19 | 100 | 1:3 |
The purpose of the present study was to demonstrate that mixtures of two monodentate P-ligands can influence the diastereoselectivity of transition metal catalyzed reactions of chiral substrates. This was accomplished by studying the diastereoselective Rh-catalyzed hydrogenation of the chiral allylic alcohol 1 and the homo-allylic alcohol 5. Since the goal was proof-of-principle, only 23 randomly chosen monodentate P-ligands that happened to be available in our laboratory were employed, and not even all combinations were actually tested. We do not claim that the hits reported here constitute the optimal catalyst systems. If in the present reactions (or in other processes) higher diastereoselectivities are strived for, the combinatorial search needs to be systematized and the library of ligands extended. Indeed, numerous analogs of the present ligands are known, many of which are commercially available. Moreover, important classes of P-ligands not considered here can also be included in future studies of this kind.
At this time we refrain from proposing any hypotheses regarding the structural and mechanistic reasons for enhanced diastereoselectivity when using certain combinations of monodentate P-ligands. The same applies to the observed differences in rate. The combinatorial search is purely empirical. However, in addition to the inherent different degrees of diastereoselectivity arising from the homo- and hetero-combinations, their relative amounts present in catalytically active form and the relative reaction rates dictate the stereochemical outcome. Since the relative amount of catalytically active forms is thermodynamically controlled, changing the La : Lb ratio may affect diastereoselectivity and can thus be used as a tool in future studies. It also remains to be seen if mixtures of chiral ligands affect diastereoselectivity in reactions of chiral substrates when they are used in enantiomerically pure form.
In conclusion, the previous concept of using mixtures of monodentate ligands in order to influence enantio-[19-26] and regioselectivity [28] of transition metal catalyzed reactions has been extended to include diastereoselectivity. The idea of using mixtures of monodentate ligands in a combinatorial manner is rapidly emerging as a powerful method to enhance activity and selectivity of transition metal-catalyzed reactions.
References
-
Gennari, C.; Piarulli, U. Chem. Rev. 2003, 103, 3071–3100.
Return to citation in text: [1] -
Dahmen, S.; Bräse, S. Synthesis 2001, 1431–1449.
Return to citation in text: [1] -
Hoveyda, A. H. Diversity-based identification of efficient homochiral organometallic catalysts for enantioselective synthesis. In Handbook of Combinatorial Chemistry; Nicolaou, K. C.; Hanko, R.; Hartwig, W., Eds.; Wiley-VCH: Weinheim, 2002; Vol. 2, pp 991–1016.
Return to citation in text: [1] -
de Vries, J. G.; de Vries, A. H. M. Eur. J. Org. Chem. 2003, 799–811.
Return to citation in text: [1] -
Reetz, M. T. Angew. Chem. 2001, 113, 292–320.
Angew. Chem., Int. Ed. 2001, 40, 284–310.
Return to citation in text: [1] [2] -
Francis, M. B.; Jacobsen, E. N. Angew. Chem. 1999, 111, 987–991.
Angew. Chem., Int. Ed. 1999, 38, 937–941.
Return to citation in text: [1] -
Berkessel, A.; Riedl, R. J. Comb. Chem. 2000, 2, 215–219.
Return to citation in text: [1] -
Ding, K.; Du, H.; Yuan, Y.; Long, J. Chem.–Eur. J. 2004, 10, 2872–2884.
Return to citation in text: [1] -
Sigman, M. S.; Jacobsen, E. N. J. Am. Chem. Soc. 1998, 120, 4901–4902.
Return to citation in text: [1] -
Mikami, K.; Yamanaka, M. Chem. Rev. 2003, 103, 3369–3400.
Return to citation in text: [1] -
Faller, J. W.; Lavoie, A. R.; Parr, J. Chem. Rev. 2003, 103, 3345–3367.
Return to citation in text: [1] -
Reetz, M. T.; Mehler, G. Angew. Chem. 2000, 112, 4047–4049.
Angew. Chem., Int. Ed. 2000, 39, 3889–3890.
Return to citation in text: [1] -
Reetz, M. T.; Meiswinkel, A.; Mehler, G.; Angermund, K.; Graf, M.; Thiel, W.; Mynott, R.; Blackmond, D. G. J. Am. Chem. Soc. 2005, 127, 10305–10313.
For a study regarding the mechanism and source of enantioselectivity in Rh-catalyzed olefin-hydrogenation using BINOL-derived monodentate phosphites.
Return to citation in text: [1] [2] -
Reetz, M. T.; Sell, T. Tetrahedron Lett. 2000, 41, 6333–6336.
Return to citation in text: [1] -
Claver, C.; Fernandez, E.; Gillon, A.; Heslop, K.; Hyett, D. J.; Martorell, A.; Orpen, A. G.; Pringle, P. G. Chem. Commun. 2000, 961–962.
Return to citation in text: [1] -
van den Berg, M.; Minnaard, A. J.; Schudde, E. P.; van Esch, J.; de Vries, A. H. M.; de Vries, J. G.; Feringa, B. L. J. Am. Chem. Soc. 2000, 122, 11539–11540.
Return to citation in text: [1] -
van den Berg, M.; Minnaard, A. J.; Haak, R. M.; Leeman, M.; Schudde, E. P.; Meetsma, A.; Feringa, B. L.; de Vries, A. H. M.; Maljaars, C. E. P.; Willans, C. E.; Hyett, D.; Boogers, J. A. F.; Henderickx, H. J. W.; de Vries, J. G. Adv. Synth. Catal. 2003, 345, 308–323.
Return to citation in text: [1] -
Bernsmann, H.; van den Berg, M.; Hoen, R.; Minnaard, A. J.; Mehler, G.; Reetz, M. T.; de Vries, J. G.; Feringa, B. L. J. Org. Chem. 2005, 70, 943–951.
Return to citation in text: [1] -
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793.
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Reetz, M. T. Chim. Oggi 2003, 21 (10/11), 5–8.
Return to citation in text: [1] [2] [3] [4] [5] -
Reetz, M. T.; Li, X. Tetrahedron 2004, 60, 9709–9714.
Return to citation in text: [1] [2] [3] -
Reetz, M. T.; Mehler, G.; Meiswinkel, A.; Sell, T. Tetrahedron: Asymmetry 2004, 15, 2165–2167.
Return to citation in text: [1] [2] [3] -
Reetz, M. T.; Ma, J.-A.; Goddard, R. Angew. Chem. 2005, 117, 416–419.
Angew. Chem., Int. Ed. 2005, 44, 412–415.
Return to citation in text: [1] [2] [3] -
Peña, D.; Minnaard, A. J.; Boogers, J. A. F.; de Vries, A. H. M.; de Vries, J. G.; Feringa, B. L. Org. Biomol. Chem. 2003, 1, 1087–1089.
Return to citation in text: [1] [2] [3] -
Reetz, M. T.; Mehler, G. Tetrahedron Lett. 2003, 44, 4593–4596.
Return to citation in text: [1] [2] [3] -
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3019–3021.
Angew. Chem., Int. Ed. 2005, 44, 2959–2962.
Return to citation in text: [1] [2] [3] -
Sharpless, K. B. Angew. Chem. 2002, 114, 2126–2135.
Angew. Chem., Int. Ed. 2002, 41, 2024–2032. The situation is somewhat related to such systems as the Sharpless asymmetric epoxidation of allylic alcohols using Ti(OiPr)4, (CH3)3COOH and chiral ligands (tartrate being the hit). The system contains a number of Ti-species which in principle catalyze epoxidation, including achiral tert-butyl hydroperoxy-Ti-species, but it is a catalyst comprising tartrate and tert-butyl hydroperoxide which is most active and most enantioselective.
Return to citation in text: [1] -
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3022–3024.
Angew. Chem., Int. Ed. 2005, 44, 2962–2964.
Return to citation in text: [1] [2] [3] -
Brown, J. M.; Naik, R. J. Chem. Soc., Chem. Commun. 1982, 348–350.
Return to citation in text: [1] [2] -
Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307–1370.
Review of substrate-directable chemical reactions including hydrogenation of chiral olefins.
Return to citation in text: [1] [2] [3] -
Hoffmann, R. W. Chem. Rev. 1989, 89, 1841–1860.
Review of 1,3-allylic strain in diastereoselective reactions of chiral compounds.
Return to citation in text: [1] [2] -
Ligand P21 was prepared in our laboratory by J.-A. Ma by phosphorylating the (S)-form of the known naphthyl-naphthol using ClP(Ph)2.
Return to citation in text: [1] -
Ligand P15 was first prepared by G. Wilke and J. Neuffer in 1974; private communication by G. Wilke (July 19, 2005).
Return to citation in text: [1] -
Neuffer, J.; Richter, W. J. J. Organomet. Chem. 1986, 301, 289–297.
Return to citation in text: [1] -
Breit, B. Chem. Commun. 1996, 2071–2072.
Return to citation in text: [1]
| 30. |
Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307–1370.
Review of substrate-directable chemical reactions including hydrogenation of chiral olefins. |
| 31. |
Hoffmann, R. W. Chem. Rev. 1989, 89, 1841–1860.
Review of 1,3-allylic strain in diastereoselective reactions of chiral compounds. |
| 19. |
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793. |
| 20. | Reetz, M. T. Chim. Oggi 2003, 21 (10/11), 5–8. |
| 21. | Reetz, M. T.; Li, X. Tetrahedron 2004, 60, 9709–9714. |
| 22. | Reetz, M. T.; Mehler, G.; Meiswinkel, A.; Sell, T. Tetrahedron: Asymmetry 2004, 15, 2165–2167. |
| 23. |
Reetz, M. T.; Ma, J.-A.; Goddard, R. Angew. Chem. 2005, 117, 416–419.
Angew. Chem., Int. Ed. 2005, 44, 412–415. |
| 24. | Peña, D.; Minnaard, A. J.; Boogers, J. A. F.; de Vries, A. H. M.; de Vries, J. G.; Feringa, B. L. Org. Biomol. Chem. 2003, 1, 1087–1089. |
| 25. | Reetz, M. T.; Mehler, G. Tetrahedron Lett. 2003, 44, 4593–4596. |
| 26. |
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3019–3021.
Angew. Chem., Int. Ed. 2005, 44, 2959–2962. |
| 1. | Gennari, C.; Piarulli, U. Chem. Rev. 2003, 103, 3071–3100. |
| 2. | Dahmen, S.; Bräse, S. Synthesis 2001, 1431–1449. |
| 3. | Hoveyda, A. H. Diversity-based identification of efficient homochiral organometallic catalysts for enantioselective synthesis. In Handbook of Combinatorial Chemistry; Nicolaou, K. C.; Hanko, R.; Hartwig, W., Eds.; Wiley-VCH: Weinheim, 2002; Vol. 2, pp 991–1016. |
| 4. | de Vries, J. G.; de Vries, A. H. M. Eur. J. Org. Chem. 2003, 799–811. |
| 5. |
Reetz, M. T. Angew. Chem. 2001, 113, 292–320.
Angew. Chem., Int. Ed. 2001, 40, 284–310. |
| 6. |
Francis, M. B.; Jacobsen, E. N. Angew. Chem. 1999, 111, 987–991.
Angew. Chem., Int. Ed. 1999, 38, 937–941. |
| 7. | Berkessel, A.; Riedl, R. J. Comb. Chem. 2000, 2, 215–219. |
| 8. | Ding, K.; Du, H.; Yuan, Y.; Long, J. Chem.–Eur. J. 2004, 10, 2872–2884. |
| 12. |
Reetz, M. T.; Mehler, G. Angew. Chem. 2000, 112, 4047–4049.
Angew. Chem., Int. Ed. 2000, 39, 3889–3890. |
| 13. |
Reetz, M. T.; Meiswinkel, A.; Mehler, G.; Angermund, K.; Graf, M.; Thiel, W.; Mynott, R.; Blackmond, D. G. J. Am. Chem. Soc. 2005, 127, 10305–10313.
For a study regarding the mechanism and source of enantioselectivity in Rh-catalyzed olefin-hydrogenation using BINOL-derived monodentate phosphites. |
| 10. | Mikami, K.; Yamanaka, M. Chem. Rev. 2003, 103, 3369–3400. |
| 11. | Faller, J. W.; Lavoie, A. R.; Parr, J. Chem. Rev. 2003, 103, 3345–3367. |
| 26. |
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3019–3021.
Angew. Chem., Int. Ed. 2005, 44, 2959–2962. |
| 5. |
Reetz, M. T. Angew. Chem. 2001, 113, 292–320.
Angew. Chem., Int. Ed. 2001, 40, 284–310. |
| 19. |
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793. |
| 24. | Peña, D.; Minnaard, A. J.; Boogers, J. A. F.; de Vries, A. H. M.; de Vries, J. G.; Feringa, B. L. Org. Biomol. Chem. 2003, 1, 1087–1089. |
| 13. |
Reetz, M. T.; Meiswinkel, A.; Mehler, G.; Angermund, K.; Graf, M.; Thiel, W.; Mynott, R.; Blackmond, D. G. J. Am. Chem. Soc. 2005, 127, 10305–10313.
For a study regarding the mechanism and source of enantioselectivity in Rh-catalyzed olefin-hydrogenation using BINOL-derived monodentate phosphites. |
| 19. |
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793. |
| 20. | Reetz, M. T. Chim. Oggi 2003, 21 (10/11), 5–8. |
| 19. |
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793. |
| 20. | Reetz, M. T. Chim. Oggi 2003, 21 (10/11), 5–8. |
| 21. | Reetz, M. T.; Li, X. Tetrahedron 2004, 60, 9709–9714. |
| 22. | Reetz, M. T.; Mehler, G.; Meiswinkel, A.; Sell, T. Tetrahedron: Asymmetry 2004, 15, 2165–2167. |
| 23. |
Reetz, M. T.; Ma, J.-A.; Goddard, R. Angew. Chem. 2005, 117, 416–419.
Angew. Chem., Int. Ed. 2005, 44, 412–415. |
| 16. | van den Berg, M.; Minnaard, A. J.; Schudde, E. P.; van Esch, J.; de Vries, A. H. M.; de Vries, J. G.; Feringa, B. L. J. Am. Chem. Soc. 2000, 122, 11539–11540. |
| 17. | van den Berg, M.; Minnaard, A. J.; Haak, R. M.; Leeman, M.; Schudde, E. P.; Meetsma, A.; Feringa, B. L.; de Vries, A. H. M.; Maljaars, C. E. P.; Willans, C. E.; Hyett, D.; Boogers, J. A. F.; Henderickx, H. J. W.; de Vries, J. G. Adv. Synth. Catal. 2003, 345, 308–323. |
| 18. | Bernsmann, H.; van den Berg, M.; Hoen, R.; Minnaard, A. J.; Mehler, G.; Reetz, M. T.; de Vries, J. G.; Feringa, B. L. J. Org. Chem. 2005, 70, 943–951. |
| 28. |
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3022–3024.
Angew. Chem., Int. Ed. 2005, 44, 2962–2964. |
| 14. | Reetz, M. T.; Sell, T. Tetrahedron Lett. 2000, 41, 6333–6336. |
| 15. | Claver, C.; Fernandez, E.; Gillon, A.; Heslop, K.; Hyett, D. J.; Martorell, A.; Orpen, A. G.; Pringle, P. G. Chem. Commun. 2000, 961–962. |
| 19. |
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793. |
| 20. | Reetz, M. T. Chim. Oggi 2003, 21 (10/11), 5–8. |
| 28. |
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3022–3024.
Angew. Chem., Int. Ed. 2005, 44, 2962–2964. |
| 27. |
Sharpless, K. B. Angew. Chem. 2002, 114, 2126–2135.
Angew. Chem., Int. Ed. 2002, 41, 2024–2032. The situation is somewhat related to such systems as the Sharpless asymmetric epoxidation of allylic alcohols using Ti(OiPr)4, (CH3)3COOH and chiral ligands (tartrate being the hit). The system contains a number of Ti-species which in principle catalyze epoxidation, including achiral tert-butyl hydroperoxy-Ti-species, but it is a catalyst comprising tartrate and tert-butyl hydroperoxide which is most active and most enantioselective. |
| 19. |
Reetz, M. T.; Sell, T.; Meiswinkel, A.; Mehler, G. Angew. Chem. 2003, 115, 814–817.
Angew. Chem., Int. Ed. 2003, 42, 790–793. |
| 20. | Reetz, M. T. Chim. Oggi 2003, 21 (10/11), 5–8. |
| 21. | Reetz, M. T.; Li, X. Tetrahedron 2004, 60, 9709–9714. |
| 22. | Reetz, M. T.; Mehler, G.; Meiswinkel, A.; Sell, T. Tetrahedron: Asymmetry 2004, 15, 2165–2167. |
| 23. |
Reetz, M. T.; Ma, J.-A.; Goddard, R. Angew. Chem. 2005, 117, 416–419.
Angew. Chem., Int. Ed. 2005, 44, 412–415. |
| 24. | Peña, D.; Minnaard, A. J.; Boogers, J. A. F.; de Vries, A. H. M.; de Vries, J. G.; Feringa, B. L. Org. Biomol. Chem. 2003, 1, 1087–1089. |
| 25. | Reetz, M. T.; Mehler, G. Tetrahedron Lett. 2003, 44, 4593–4596. |
| 26. |
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3019–3021.
Angew. Chem., Int. Ed. 2005, 44, 2959–2962. |
| 33. | Ligand P15 was first prepared by G. Wilke and J. Neuffer in 1974; private communication by G. Wilke (July 19, 2005). |
| 32. | Ligand P21 was prepared in our laboratory by J.-A. Ma by phosphorylating the (S)-form of the known naphthyl-naphthol using ClP(Ph)2. |
| 31. |
Hoffmann, R. W. Chem. Rev. 1989, 89, 1841–1860.
Review of 1,3-allylic strain in diastereoselective reactions of chiral compounds. |
| 30. |
Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307–1370.
Review of substrate-directable chemical reactions including hydrogenation of chiral olefins. |
| 28. |
Reetz, M. T.; Li, X. Angew. Chem. 2005, 117, 3022–3024.
Angew. Chem., Int. Ed. 2005, 44, 2962–2964. |
| 29. | Brown, J. M.; Naik, R. J. Chem. Soc., Chem. Commun. 1982, 348–350. |
| 30. |
Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307–1370.
Review of substrate-directable chemical reactions including hydrogenation of chiral olefins. |
© 2005 Reetz and Guo; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)