Abstract
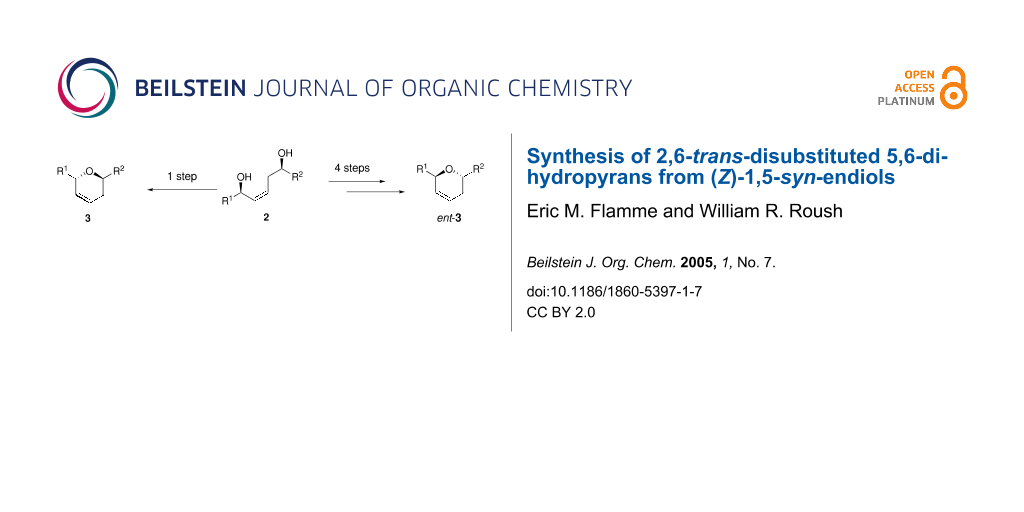
Certain (Z)-1,5-syn-diols 2 may be converted into 2,6-trans-5,6-dihydropyrans by using phosphonium salt 4 or phosphorane 5 as dehydrating agents. A more general four step procedure converts the (Z)-1,5-syn-endiols into enantiomeric dihydropyrans ent-3 via regioselective silylation of the allylic alcohol unit followed by mesylate formation and base-promoted nucleophilic displacement.

Graphical Abstract
Findings
We recently reported a one-pot double allylboration reaction sequence which provides (Z)-1,5-syn-endiols from simple aldehyde starting materials with excellent diastereo- and enantioselectivity.[1] In connection with an ongoing natural product synthesis project, we were interested in developing methods to transform diols 2 into dihydropyrans 3 or the enantiomeric dihydropyrans ent-3 through complementary, regioselective cyclodehydration processes (Scheme 1).
![[1860-5397-1-7-i1]](/bjoc/content/inline/1860-5397-1-7-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
2,6-Disubstituted dihydropyrans are common structural elements of many biologically active natural products.[2,3] A number of methods have been developed to synthesize substituted dihydropyrans including: (i) hetero-Diels-Alder cycloadditions,[4-7] (ii) electrophile-initiated alkylation of glycals,[8-11] (iii) ring closing metathesis,[12,13] (iv) vinylsilane cyclization of oxocarbenium ions,[14] and (v) intramolecular allylations.[15,16] However, we were unaware of any reports that describe the direct conversion of 1,5-diols containing an internal olefin such as 2 directly to 2,6-trans-disubstituted 5,6-dihydropyrans. Furthermore, there are only limited reports describing the stereoselective synthesis of related tetrahydropyrans through cyclodehydration of enantiopure 1,5-diols substrates.[17]
The challenge of synthesizing dihydropyrans 3 or ent-3 from 1,5-diols such as 2 lies in the differentiation of the two hydroxyl groups. Selective activation of the allylic alcohol in 2 as a leaving group followed by nucleophilic attack by the homoallylic alcohol will lead to dihydropyran 3. However, activation of the homoallylic alcohol followed by nucleophilic attack by the allylic alcohol will provide the enantiomeric dihydropyan ent-3. Cyclic ethers of various ring size have been synthesized by the cyclodehydration of diols through the use of oxyphosphonium salts and phosphorane reagents.[18-23] We reasoned that because the rate determining step of these cyclizations is believed to be the nucleophilic substitution step, selective formation of 3 should be possible owing to the superior leaving group ability of the activated allylic hydroxyl.[21]
Diol 2a (R1 = R2 = CH2CH2Ph) was used initially in the development of a suitable cyclodehydration protocol (Figure 1). Because the R1 and R2 substituents of 2a are identical, steric effects on the activation of the two hydroxyl groups are eliminated. Therefore, the enantioselectivity of the ring closing step will depend only on the relative rates of the competing cyclization processes leading to 3a and ent-3a. Initial attempts at cyclization of 2a using Ph3P and diethyl azodicarboxylate or Ph3P-CCl4 were low yielding (entries 1, 2).[22] Interestingly, small amounts of the 2,6-cis-disubstituted dihydropyran 6a were detected under these conditions, suggesting the intervention of a competitive double inversion process or a carbocation-mediated cyclization process. In order to avoid the presence of nucleophilic counter ions, we turned to the use of phosphonium salt 4 and phosphorane 5 as the cyclodehydration reagents (Scheme 2).[24,25]
![[1860-5397-1-7-1]](/bjoc/content/figures/1860-5397-1-7-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-1-7-i2]](/bjoc/content/inline/1860-5397-1-7-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Treatment of diol 2a with 4 in acetonitrile (0.05 M) at 0°C and warming slowly to 23°C provided a 9 : 1 mixture of trans and cis dihydropyrans 3a and 6a in 65% combined yield (entry 3). We speculated that the cis isomer might arise via formation of an allylic cation (perhaps facilitated by acid formed by competitive elimination of the oxyphosphonium salt during the reaction). Accordingly, the trans/cis ratio was improved to 13 : 1 by addition of 2,6-tert-butyl-4-methyl-pyridine as an acid scavenger (entry 4). Ultimately, we found that use of CH2Cl2 as the solvent and Et3N as base provided 3a in 78% yield with 20:1 trans/cis selectivity (entry 5). However, chiral HPLC analysis indicated that 3a from entry 5 had an enantiomeric excess of only 25% e.e. while the enantiomeric purity of the starting diol 2a was 91% e.e.
This result suggests that the selectivity for displacement of the two hydroxyl groups of 2a is only ca. 2 : 1 under these conditions. Examination of molecular models indicates that in order for the activated allylic alcohol to be displaced in this intramolecular substitution process, the allylic C-O bond substantially deviates from coplanarity with the adjacent π-system. Therefore, the difference in relative rates of displacement of the two activated hydroxyl groups is much less than originally anticipated. The enantioselectivity could be increased to 90% by lowering the reaction temperature to -78°C, but the yield of dihydropyran 3a was reduced to less than 50% (entry 7).
Dehydration of 2a with phosphorane 5 in toluene at 80°C in the presence of triethylamine provided exclusively trans dihydropyran 3a in a yield of 71% and 50% e.e. (Figure 1, entry 9). Attempts to increase the enantioselectivity of the reaction by lowering the reaction temperature were thwarted by the poor solubility profile of 5. Changing the solvent from toluene to hexanes, THF, or NMP also did not significantly impact the %e.e.
Attempts to extend these results to other systems met with limited success (Figure 2). We anticipated that the regioselectivity of dehydration of a substrate like 2b in which the homoallylic hydroxyl is more sterically hindered than the allylic one would be improved relative to 2a. Indeed, the cyclodehydration of 2b with phosphorane 5 in toluene at 80°C proceeded with ca. 9 : 1 regioselectivity (entry 4), and use of the phosphonium salt 4 at -35°C also gave reasonably good results (84% e.e.). However, all attempts to dehdyrate the α,α'-oxygenated diol 7 with either 4 or 5 were unsuccessful.
![[1860-5397-1-7-2]](/bjoc/content/figures/1860-5397-1-7-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyclodehydration of other diol substrates
Figure 2: Cyclodehydration of other diol substrates
Given that the cyclodehydration reactions of diol substrates were complicated by selectivity and reactivity issues (Figure 1 and Figure 2), we turned to an alternative strategy which would not rely on chemoselectivity in the cyclodehydration step. To this end, mesylates 11 were synthesized (Scheme 3).
![[1860-5397-1-7-i3]](/bjoc/content/inline/1860-5397-1-7-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Treatment of 1,5-diols 2 with TBS-Cl and imidazole effects selective protection of the allylic alcohol (Scheme 4). The mono-TBS protected 9 is the major product in all cases except when 2f was used as the substrate; in this case, the allylic hydroxyl is more hindered than the homoallylic hydroxyl, which is silylated preferentially. The homoallylic TBS ethers 12 and the bis-TBS ethers 13 can be conveniently recycled. The origin of the regioselectivity of the selective silylation of 1,5-diols 2 is unknown at present.
![[1860-5397-1-7-i4]](/bjoc/content/inline/1860-5397-1-7-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Subsequent treatment of mono-TBS ethers 9 with methanesulfonyl chloride (MsCl) followed by deprotection of the TBS ether by using 3HFEt3N afforded mesylates 11 (80 – ≥ 95% yield). Treatment of mesylate 11a with 1 equivalent of potassium tert-butoxide in tert-BuOH (0.01 M) between 40–50 °C provided ent-3a in 75% yield and 10:1 selectivity for the dihydropyran product and a diene resulting from an undesired E2 elimination (Figure 3, entry 1).[26,27] This result was particularly gratifying since Thomas has reported that attempts to cyclized suitably activated 1,5-diol substrates under basic conditions did not afford dihydropyran products.[28] It was necessary to carry out the reactions under dilute conditions in order to minimize elimination to diene 14. However, increased steric demands about the mesylate (11b) or the allylic alcohol (11c) led to increased amounts of elimination, although the isolated yields of the dihydropyrans 3b and 3c were acceptable (Figure 3). Treatment of mesylate 11e containing cyclohexyl groups flanking both the allylic alcohol and the mesylate gave a 1:1 mixture of ent-3e and 14e. α-Oxygenated substrates 11d and 11f cyclized with 3.5:1 and 3:1 selectivity under these conditions.
![[1860-5397-1-7-3]](/bjoc/content/figures/1860-5397-1-7-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Cyclization of Hydroxymesylates 11
Figure 3: Cyclization of Hydroxymesylates 11
It is known that tributylstannyl ethers are decent nucleophiles and are considerably less basic than oxanions.[29,30] Indeed, we found that elimination products 14 could be suppressed by treatment of alcohols 11 with (Bu3Sn)2O in benzene followed by heating the resulting tributylstannyl ethers in DMF at 80°C (Figure 3). In this way, selectivity for the formation of dihydropyran ent-3 versus elimination could be increased up to 99:1 for substrates 11a, 11d, and 11f. All other substrates examined also showed improved selectivity for pyran formation, and in all cases the dihydropyran ent-3 was obtained in at least 72% yield.
In summary, the scope of cyclodehydration reactions of (Z)-1,5-syn-diols 2 to give the the targeted dihydropyrans 3 using 4 and 5 as dehydrating reagents in a one-pot procedure is limited to substrates that lack oxygen substituents at positions adjacent to the leaving group. Acceptable enantioselectivity can be achieved by performing these cyclodehydration reactions at low temperatures. However, a much more general procedure for synthesis of the enantiomeric dihydropyrans ent-3 involves the stannyl ether mediated cyclization of hydroxy mesylates 11. This method relies on a selective silylation of the homoallylic alcohol, and represents a new route to access 2,6-trans-disubstituted 5,6-dihydropyrans. Application of this methodology in natural products synthesis will be reported in due course.
References
-
Flamme, E. M.; Roush, W. R. J. Am. Chem. Soc. 2002, 124, 13644–13645. doi:10.1021/ja028055j
Return to citation in text: [1] -
Faulkner, D. J. Nat. Prod. Rep. 2000, 17, 7–55. doi:10.1039/a809395d
Return to citation in text: [1] -
Norcross, R. D.; Paterson, I. Chem. Rev. 1995, 95, 2041–2114. doi:10.1021/cr00038a012
Return to citation in text: [1] -
Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2398–2400. doi:10.1002/(SICI)1521-3773(19990816)38:16<2398::AID-ANIE2398>3.0.CO;2-E
Return to citation in text: [1] -
Lubineau, A.; Auge, J.; Lubin, N. Tetrahedron 1993, 49, 4639–4650. doi:10.1016/S0040-4020(01)81292-8
Return to citation in text: [1] -
Danishefsky, S. J.; Selnick, H. G.; Zelle, R. E.; DeNinno, M. P. J. Am. Chem. Soc. 1988, 110, 4368–4378. doi:10.1021/ja00221a043
Return to citation in text: [1] -
Danishefsky, S. J.; DeNinno, M. P. Angew. Chem., Int. Ed. Engl. 1987, 26, 15–23. doi:10.1002/anie.198700151
Return to citation in text: [1] -
Steinhuebel, D. P.; Fleming, J. J.; Du Bois, J. Org. Lett. 2002, 4, 293–295. doi:10.1021/ol010273e
Return to citation in text: [1] -
Ferrier, R. J. Top. Curr. Chem. 2001, 125, 153–175.
Return to citation in text: [1] -
Levy, D. E.; Tang, C. The Chemistry of C-Glycosides; Pergamon Press: Tarrytown, 1995; Vol. 13.
Return to citation in text: [1] -
Postema, M. H. D. C-Glycoside Synthesis; CRC Press: Boca Raton, 1995.
Return to citation in text: [1] -
Mulzer, J.; Hanbauer, M. Tetrahedron Lett. 2000, 41, 33–36. doi:10.1016/S0040-4039(99)02021-3
Return to citation in text: [1] -
Burke, S. D.; Ng, R. A.; Morrison, J. A. J. Org. Chem. 1998, 63, 3160–3161. doi:10.1021/jo980163u
Return to citation in text: [1] -
Semeyn, C.; Blaauw, R. H.; Hiemstra, H.; Speckamp, W. N. J. Org. Chem. 1997, 62, 3426–3427. doi:10.1021/jo970369f
Return to citation in text: [1] -
Huang, H.; Panek, J. S. J. Am. Chem. Soc. 2000, 122, 9836–9837. doi:10.1021/ja002087u
Return to citation in text: [1] -
Roush, W. R.; Dilley, G. J. Synlett 2001, 955–959. doi:10.1055/s-2001-14632
Return to citation in text: [1] -
Chikashita, H.; Hirao, K.; Itoh, K. Bull. Chem. Soc. Jpn. 1993, 66, 1738–1742.
A search of the literature found only one report.
Return to citation in text: [1] -
Hughes, D. L. Org. Prep. Proced. Int. 1996, 28, 127–164.
Return to citation in text: [1] -
Hendrickson, J. B.; Hussoin, M. S. Synlett 1990, 423–424. doi:10.1055/s-1990-21115
Return to citation in text: [1] -
Denney, D. B.; Denney, D. Z.; Gigantino, J. J. J. Org. Chem. 1984, 49, 2831–2832. doi:10.1021/jo00189a044
Return to citation in text: [1] -
Robinson, P. L.; Barry, C. N.; Bass, S. W.; Jarvis, S. E.; Evans, S. A., Jr. J. Org. Chem. 1983, 48, 5396–5398. doi:10.1021/jo00174a059
Return to citation in text: [1] [2] -
Carlock, J. T.; Mack, M. P. Tetrahedron Lett. 1978, 19, 5153–5156. doi:10.1016/S0040-4039(01)85836-6
Return to citation in text: [1] [2] -
Castro, B. R. Org. React. 1983, 29, 1–162.
Return to citation in text: [1] -
Phillips, D. I.; Szele, I.; Westheimer, F. H. J. Am. Chem. Soc. 1976, 98, 184–189. doi:10.1021/ja00417a029
Return to citation in text: [1] -
Kelly, J. W.; Robinson, P. L.; Evans, S. A., Jr. J. Org. Chem. 1986, 51, 4473–4475. doi:10.1021/jo00373a026
Return to citation in text: [1] -
Cink, R. D.; Forsyth, C. J. J. Org. Chem. 1997, 62, 5672–5673. doi:10.1021/jo9708755
Return to citation in text: [1] -
Boivin, T. L. B. Tetrahedron 1987, 43, 3309–3362. doi:10.1016/S0040-4020(01)81626-4
Return to citation in text: [1] -
Maguire, R. J.; Thomas, E. J. J. Chem. Soc., Perkin Trans. 1 1995, 2477–2485. doi:10.1039/p19950002477
Return to citation in text: [1] -
Davies, A. G. Organotin Chemistry; Wiley-VCH: Weinheim, 1997.
Return to citation in text: [1] -
David, S.; Hanessian, S. Tetrahedron 1985, 41, 643–663. doi:10.1016/S0040-4020(01)96443-9
Return to citation in text: [1]
| 1. | Flamme, E. M.; Roush, W. R. J. Am. Chem. Soc. 2002, 124, 13644–13645. doi:10.1021/ja028055j |
| 12. | Mulzer, J.; Hanbauer, M. Tetrahedron Lett. 2000, 41, 33–36. doi:10.1016/S0040-4039(99)02021-3 |
| 13. | Burke, S. D.; Ng, R. A.; Morrison, J. A. J. Org. Chem. 1998, 63, 3160–3161. doi:10.1021/jo980163u |
| 29. | Davies, A. G. Organotin Chemistry; Wiley-VCH: Weinheim, 1997. |
| 30. | David, S.; Hanessian, S. Tetrahedron 1985, 41, 643–663. doi:10.1016/S0040-4020(01)96443-9 |
| 8. | Steinhuebel, D. P.; Fleming, J. J.; Du Bois, J. Org. Lett. 2002, 4, 293–295. doi:10.1021/ol010273e |
| 9. | Ferrier, R. J. Top. Curr. Chem. 2001, 125, 153–175. |
| 10. | Levy, D. E.; Tang, C. The Chemistry of C-Glycosides; Pergamon Press: Tarrytown, 1995; Vol. 13. |
| 11. | Postema, M. H. D. C-Glycoside Synthesis; CRC Press: Boca Raton, 1995. |
| 4. | Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. Angew. Chem., Int. Ed. 1999, 38, 2398–2400. doi:10.1002/(SICI)1521-3773(19990816)38:16<2398::AID-ANIE2398>3.0.CO;2-E |
| 5. | Lubineau, A.; Auge, J.; Lubin, N. Tetrahedron 1993, 49, 4639–4650. doi:10.1016/S0040-4020(01)81292-8 |
| 6. | Danishefsky, S. J.; Selnick, H. G.; Zelle, R. E.; DeNinno, M. P. J. Am. Chem. Soc. 1988, 110, 4368–4378. doi:10.1021/ja00221a043 |
| 7. | Danishefsky, S. J.; DeNinno, M. P. Angew. Chem., Int. Ed. Engl. 1987, 26, 15–23. doi:10.1002/anie.198700151 |
| 26. | Cink, R. D.; Forsyth, C. J. J. Org. Chem. 1997, 62, 5672–5673. doi:10.1021/jo9708755 |
| 27. | Boivin, T. L. B. Tetrahedron 1987, 43, 3309–3362. doi:10.1016/S0040-4020(01)81626-4 |
| 2. | Faulkner, D. J. Nat. Prod. Rep. 2000, 17, 7–55. doi:10.1039/a809395d |
| 3. | Norcross, R. D.; Paterson, I. Chem. Rev. 1995, 95, 2041–2114. doi:10.1021/cr00038a012 |
| 28. | Maguire, R. J.; Thomas, E. J. J. Chem. Soc., Perkin Trans. 1 1995, 2477–2485. doi:10.1039/p19950002477 |
| 18. | Hughes, D. L. Org. Prep. Proced. Int. 1996, 28, 127–164. |
| 19. | Hendrickson, J. B.; Hussoin, M. S. Synlett 1990, 423–424. doi:10.1055/s-1990-21115 |
| 20. | Denney, D. B.; Denney, D. Z.; Gigantino, J. J. J. Org. Chem. 1984, 49, 2831–2832. doi:10.1021/jo00189a044 |
| 21. | Robinson, P. L.; Barry, C. N.; Bass, S. W.; Jarvis, S. E.; Evans, S. A., Jr. J. Org. Chem. 1983, 48, 5396–5398. doi:10.1021/jo00174a059 |
| 22. | Carlock, J. T.; Mack, M. P. Tetrahedron Lett. 1978, 19, 5153–5156. doi:10.1016/S0040-4039(01)85836-6 |
| 23. | Castro, B. R. Org. React. 1983, 29, 1–162. |
| 22. | Carlock, J. T.; Mack, M. P. Tetrahedron Lett. 1978, 19, 5153–5156. doi:10.1016/S0040-4039(01)85836-6 |
| 17. |
Chikashita, H.; Hirao, K.; Itoh, K. Bull. Chem. Soc. Jpn. 1993, 66, 1738–1742.
A search of the literature found only one report. |
| 24. | Phillips, D. I.; Szele, I.; Westheimer, F. H. J. Am. Chem. Soc. 1976, 98, 184–189. doi:10.1021/ja00417a029 |
| 25. | Kelly, J. W.; Robinson, P. L.; Evans, S. A., Jr. J. Org. Chem. 1986, 51, 4473–4475. doi:10.1021/jo00373a026 |
| 15. | Huang, H.; Panek, J. S. J. Am. Chem. Soc. 2000, 122, 9836–9837. doi:10.1021/ja002087u |
| 16. | Roush, W. R.; Dilley, G. J. Synlett 2001, 955–959. doi:10.1055/s-2001-14632 |
| 14. | Semeyn, C.; Blaauw, R. H.; Hiemstra, H.; Speckamp, W. N. J. Org. Chem. 1997, 62, 3426–3427. doi:10.1021/jo970369f |
| 21. | Robinson, P. L.; Barry, C. N.; Bass, S. W.; Jarvis, S. E.; Evans, S. A., Jr. J. Org. Chem. 1983, 48, 5396–5398. doi:10.1021/jo00174a059 |
© 2005 Flamme and Roush; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)