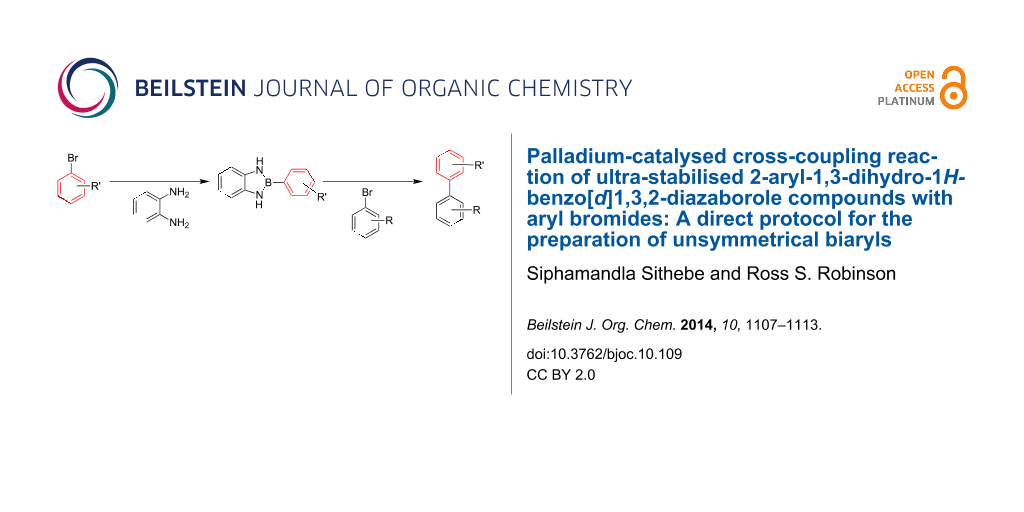
Abstract
There has been a significant interest in organoboron compounds such as arylboronic acids, arylboronate esters and potassium aryltrifluoroborate salts because they are versatile coupling partners in metal-catalysed cross-coupling reactions. On the other hand, their nitrogen analogues, namely, 1,3,2-benzodiazaborole-type compounds have been studied extensively for their intriguing absorption and fluorescence characteristics. Here we describe the first palladium-catalysed Suzuki–Miyaura cross-coupling reaction of easily accessible and ultra-stabilised 2-aryl-1,3-dihydro-1H-benzo[d]1,3,2-diazaborole derivatives with various aryl bromides. Aryl bromides bearing electron-withdrawing, electron-neutral and electron-donating substituents are reacted under the catalytic system furnishing unsymmetrical biaryl products in isolated yields of up to 96% in only 10 minutes.

Graphical Abstract
Introduction
Arylboronic acids 1, arylboronate esters 2 and potassium aryltrifluoroborate salts 3 (Figure 1) have received considerable attention and have found a special place as mild and versatile nucleophilic coupling partners for carbon–carbon bond-forming cross-coupling reactions [1-5]. Amongst them, the Suzuki–Miyaura cross-coupling reaction of aryl halides/triflates and organoboron compounds is one of the most documented and versatile cross-coupling reaction in the literature [6]. The use of organoboron compounds 1, 2 and 3 (Figure 1) as nucleophilic coupling partners in the Suzuki–Miyaura cross-coupling reaction is particularly attractive due to the non-toxicity of the byproducts, the ease with which they are transmetalated and their high stability towards air and moisture, which are the key features for coupling reactions [7].
![[1860-5397-10-109-1]](/bjoc/content/figures/1860-5397-10-109-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of organoboron compounds 1–3.
Figure 1: Structures of organoboron compounds 1–3.
On one hand, structural diverse π-conjugated organic molecules containing a three-coordinate boron moiety such as trimesitylborane (4), arylalkynyldimesitylborane 5 and 2-aryl-1,3-diethyl-1H-benzo[d]1,3,2-diazaborole 6 (Figure 2) are well known and have received considerable attention due to their interesting luminescence characteristics, fluoride ion sensing abilities, emissive as well as electron-transporting properties [8-11]. Three-coordinate boron compounds are electron-poor and strong π-electron acceptors owing to the empty boron pz-orbital, which is capable of significant delocalisation when attached to an organic π-system [11]. These compounds exhibit an unusual stability because of the bulky aryl groups, such as mesityl (2,4,6-trimethylphenyl) groups, which provide steric conjunction around the empty boron pz-orbital thereby blocking the incoming nucleophile (Figure 2, compounds 4 and 5) [12]. Alternatively, three-coordinate boron compounds functionalized with 1,3,2-benzodiazaborole units are greatly stabilised by electron back-donation from the two nitrogen atoms to the empty boron pz-orbital (Figure 2, compound 6) [13,14].
![[1860-5397-10-109-2]](/bjoc/content/figures/1860-5397-10-109-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of π-conjugated three-coordinate organoboron compounds 4 and 5.
Figure 2: Structure of π-conjugated three-coordinate organoboron compounds 4 and 5.
Despite their popularity in the organic community, their profound stability towards air and moisture, their ease with which they are accessible and their non-toxicity, these compounds have, to the best of our knowledge, never been used in any transition metal-catalysed C–C bond formation reaction as coupling partners except for their 2-alkyl/alkenyl-substituted analogues [14]. We are aware of reports describing the Suzuki–Miyaura cross-coupling reaction aryl diaminoborole containing compounds {ArB(dan)} which are structurally similar to our compounds, however, these compounds {ArB(dan)} are used as protecting groups not as coupling partners [15,16]. During the course of our studies on the syntheses, crystal structures, fluorescence and theoretical characteristics of 1,3,2-diazaborolane functionalised organic molecules, which is reported in details elsewhere [17], we were encouraged by the high yields of 2-aryl-1,3-dihydro-1H-benzo[d]1,3,2-diazaborole compounds (Scheme 1), their solubility in various organic solvents and their high stability towards air and moisture to investigate their reactivity in transition metal-catalysed cross-coupling reaction. These compounds could be left on a bench top in a basic media for weeks without any noticeable degradation [17]. Herein, we report the first palladium-catalyzed cross-coupling reaction of 2-aryl-1,3-dihydro-1H-benzo[d]1,3,2-diazaborole compounds with aryl bromides in only 10 minutes.
Results and Discussion
As shown in Scheme 1, 2-aryl-1,3-dihydro-1H-benzo[d]1,3,2-diazaborole compounds were easily prepared from aryl halides 7 via the reaction of an organomagnesium intermediate with trialkylborate solution followed by complexation with o-phenylenediamine (8) in a single pot (Scheme 1). Following this procedure, the desired products were obtained in excellent yields (81–93%) (Scheme 1).
![[1860-5397-10-109-i1]](/bjoc/content/inline/1860-5397-10-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2-aryl-1,3-dihydro-1H-benzo[d]-1,3,2-diazaborole compounds 9–12.
Scheme 1: Synthesis of 2-aryl-1,3-dihydro-1H-benzo[d]-1,3,2-diazaborole compounds 9–12.
Suzuki–Miyaura cross-coupling reaction
To find optimal reaction conditions, we initially studied the reaction of bromobenzene (13a) with compound 9 in a toluene/water mixture under different conditions as a model reaction (Table 1). Attempted cross-coupling reaction of compound 9 with bromobenzene (13a), in the absence of both the ligand and a base, gave, as expected, zero conversion of the starting materials (Table 1, entry 1).
Table 1: Initial optimisation of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-109-i2.svg?max-width=637&scale=1.0)
|
||||
| Entry | Pd cat. | Ligand | Base | Yields (%)b 13 |
|---|---|---|---|---|
| 1 | PdCl2 | none | none | 0 |
| 2 | PdCl2 | PPh3 | K3PO4 | <5 |
| 3 | Pd(PPh3)4 | none | K2CO3 | 21 |
| 4 | Pd(PPh3)2Cl2 | none | K2CO3 | 18 |
| 5 | Pd(PPh3)4 | none | K3PO4·H2O | 10 |
| 6 | Pd(PPh3)2Cl2 | PCy3 | K3PO4·H2O | 33 |
| 7 | PdCl2 | PCy3 | K3PO4·H2O | <5 |
| 8 | Pd(OAc)2 | PPh3 | K2CO3 | 51 |
| 9 | Pd(OAc)2 | PCy3 | K3PO4·H2O | 88 |
| 10 | Pd(OAc)2 | PCy3/PPh3c | K3PO4 | 67 |
| 11 | Pd(OAc)2 | PCy3 | K2CO3 | 76 |
| 12 | Pd (PPh3)2Cl2 | PPh3 | K2CO3 | 20 |
aReaction conditions: compound 9 (0. 85 mmol), bromobenzene (13a, 0.77 mmol), Pd cat. (4 mol %), ligand (8 mol %), base (3 equiv), toluene (0.50 mL) and water (0.1 mL). Closed vessel, 80 W of microwave energy, 100 °C, 100 psi of pressure, 10 minutes. bIsolated yields after column chromatography. c4 mol % each of the ligands.
The addition of PPh3 as a ligand and K3PO4 as a base failed to afford the desired coupled product in any substantive yield (Table 1, entry 2). Poor conversion of the starting material and low assay yield of the desired product were observed when more bulky Pd(PPh3)4 as a catalyst was used (Table 1, entries, 3 and 5). The use of Pd(PPh3)2Cl2 as a catalyst in conjunction with PPh3 as a ligand and K2CO3 or K3PO4·H2O as bases also failed to optimise the reaction conditions (Table 1, entries 4, 6 and 12). This was attributed to the decomposition of Pd(PPh3)2Cl2 catalyst to Pd-black. Moderate to good yields were obtained when of Pd(OAc)2/PCy3 or Pd(OAc)2/PPh3 combinations were used (Table 1, entries 8–11). Pd(OAc)2/PCy3 and K3PO4·H2O (Table 1, entry 9) was recognised to be the most effective combination and was thus chosen as optimal reaction conditions for the purpose of this study. With the optimized reaction conditions in hand, the scope and the limitations of the cross-coupling reaction was investigated using bromobenzene (13a) and 4-bromoanisole (13b) as elelctrophilic coupling partners and boronates 9–12 (Scheme 1) as the corresponding nucleophilic coupling partners (Table 2). The cross-coupling reaction of bromobenzene (13a) with boronates 9–12 went smooth affording the coupled products in yields ranging from 68% to 88% (Table 2, entries 1, 3, 5 and 7). The sterically hindered ortho-substituted boronate 11 generally afforded lower yields when compared to other boronate derivatives (Table 2, entries 5 and 6). This was attributed to incomplete conversion of the starting materials possibly due to a steric effect around the boron atom which is consistent with the literature [6,18,19]. The cross-coupling of an electron-rich aromatic system (4-bromoanisole) was generally less efficient compared to bromobenzene (Table 2, entries 2 and 6). This effect was attributed to the deactivation of the carbon–bromine bond as a result of electron-donating substituent (OMe) [20].
Table 2: Palladium-catalysed cross-coupling of boronate 9–12 with bromobenzene and 4-bromoanisole.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-109-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Boronate | ArBr | Product | Yieldsb |
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-109-i4.svg?max-width=637&scale=1.0)
9 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-109-i5.svg?max-width=637&scale=1.0)
13a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-109-i6.svg?max-width=637&scale=1.0)
14 |
88 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-109-i7.svg?max-width=637&scale=1.0)
9 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-109-i8.svg?max-width=637&scale=1.0)
13b |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-109-i9.svg?max-width=637&scale=1.0)
15 |
62 |
| 3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-109-i10.svg?max-width=637&scale=1.0)
10 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-109-i11.svg?max-width=637&scale=1.0)
13a |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-109-i12.svg?max-width=637&scale=1.0)
16 |
85 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-109-i13.svg?max-width=637&scale=1.0)
10 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-109-i14.svg?max-width=637&scale=1.0)
13b |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-109-i15.svg?max-width=637&scale=1.0)
17 |
68 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-109-i16.svg?max-width=637&scale=1.0)
11 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-109-i17.svg?max-width=637&scale=1.0)
13a |
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-109-i18.svg?max-width=637&scale=1.0)
18 |
68 |
| 6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-109-i19.svg?max-width=637&scale=1.0)
11 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-109-i20.svg?max-width=637&scale=1.0)
13b |
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-109-i21.svg?max-width=637&scale=1.0)
19 |
65 |
| 7 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-109-i22.svg?max-width=637&scale=1.0)
12 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-109-i23.svg?max-width=637&scale=1.0)
13a |
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-109-i24.svg?max-width=637&scale=1.0)
20 |
72 |
| 8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-109-i25.svg?max-width=637&scale=1.0)
12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-10-109-i26.svg?max-width=637&scale=1.0)
13b |
![[Graphic 26]](/bjoc/content/inline/1860-5397-10-109-i27.svg?max-width=637&scale=1.0)
21 |
76 |
aReaction conditions: Boronate 9–12 (1.1 eqiuv), 13a and 13b (1.0 equiv), Pd(OAc)2 (4.0 mol %), PCy3, (8.0 mol %), K2PO4·H2O (3.0 equiv), toluene (0.50 mL) and H2O (0.10 mL), 80 W of microwave energy, 100 °C, 100 psi, 10 minutes. bYields of isolated products after a flash column chromatography.
Encouraged by our results (Table 2), we then turned our attention to investigate the reactivity of activated as well as conjugated electrophiles in our catalytic system (Table 3). Unlike bromobenzene and 4-bromoanisole, the cross-coupling reaction of electron-deficient electrophiles (4-bromoacetophenone (13d) and 4-bromonitrobenzene (13c)) furnished the desired coupled products in excellent yields ranging from 85 to 96% (Table 3) [6,21]. These observations are consistent with the literature and are attributed to the activation of the carbon–bromine bonds due to the electron-withdrawing functional groups. The presence of an electron-withdrawing group induces oxidative addition of the carbon–bromine bond to the metal centre (catalyst) compared to the corresponding electron-neutral and electron-rich functionalities [22]. The cross-coupling reaction of boronate 9 with both substrates 13c and 13d afforded the desired products, as expected, in high yields (Table 3, entries 1 and 2). We noticed that steric hinderance on the boronate 10 did not have any negative impact on the cross-coupling reaction investigated herein. Sterically hindered boronate 10 was smoothly coupled with 4-bromonitrobenzene (13c) and 4-bromoacetophenone (13d) providing coupled products 24 and 25 in 96 and 94% yields, respectively (Table 3, entries 3 and 4). Although it is known that ortho-substituted boron counterparts usually suffer from facile protodeborination providing coupled products in low yields [5], ortho-substituted boronate 11 afforded the desired products 26 and 27 in 85 and 86% yields, respectively (Table 3, entries 5 and 6). The cross-coupling reaction of boronate 12, with each of the substrates (13c and 13d), furnished the desired products in 91 and 86% yields (Table 3, entries 7 and 8). The reaction was more sensitive towards increasing steric hindrance on the electrophilic counterpart (13e) only affording the coupled products in moderate yields (Table 3, entries 9–11).
Table 3: Palladium-catalysed cross-coupling of boronate 9–12 with 4-bromonitrobenzene, 4-bromoacetophenone and 9-bromoantracene.a
| Entry | Boronate | ArBr | Product | Yields (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-10-109-i28.svg?max-width=637&scale=1.0)
9 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-10-109-i29.svg?max-width=637&scale=1.0)
13c |
![[Graphic 29]](/bjoc/content/inline/1860-5397-10-109-i30.svg?max-width=637&scale=1.0)
22 |
91 |
| 2 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-10-109-i31.svg?max-width=637&scale=1.0)
9 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-10-109-i32.svg?max-width=637&scale=1.0)
13d |
![[Graphic 32]](/bjoc/content/inline/1860-5397-10-109-i33.svg?max-width=637&scale=1.0)
23 |
90 |
| 3 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-10-109-i34.svg?max-width=637&scale=1.0)
10 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-10-109-i35.svg?max-width=637&scale=1.0)
13c |
![[Graphic 35]](/bjoc/content/inline/1860-5397-10-109-i36.svg?max-width=637&scale=1.0)
24 |
96 |
| 4 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-10-109-i37.svg?max-width=637&scale=1.0)
10 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-10-109-i38.svg?max-width=637&scale=1.0)
13d |
![[Graphic 38]](/bjoc/content/inline/1860-5397-10-109-i39.svg?max-width=637&scale=1.0)
25 |
94 |
| 5 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-10-109-i40.svg?max-width=637&scale=1.0)
11 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-10-109-i41.svg?max-width=637&scale=1.0)
13c |
![[Graphic 41]](/bjoc/content/inline/1860-5397-10-109-i42.svg?max-width=637&scale=1.0)
26 |
85 |
| 6 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-10-109-i43.svg?max-width=637&scale=1.0)
11 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-10-109-i44.svg?max-width=637&scale=1.0)
13d |
![[Graphic 44]](/bjoc/content/inline/1860-5397-10-109-i45.svg?max-width=637&scale=1.0)
27 |
86 |
| 7 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-10-109-i46.svg?max-width=637&scale=1.0)
12 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-10-109-i47.svg?max-width=637&scale=1.0)
13c |
![[Graphic 47]](/bjoc/content/inline/1860-5397-10-109-i48.svg?max-width=637&scale=1.0)
28 |
91 |
| 8 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-10-109-i49.svg?max-width=637&scale=1.0)
12 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-10-109-i50.svg?max-width=637&scale=1.0)
13d |
![[Graphic 50]](/bjoc/content/inline/1860-5397-10-109-i51.svg?max-width=637&scale=1.0)
29 |
86 |
| 9 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-10-109-i52.svg?max-width=637&scale=1.0)
9 |
![[Graphic 52]](/bjoc/content/inline/1860-5397-10-109-i53.svg?max-width=637&scale=1.0)
13e |
![[Graphic 53]](/bjoc/content/inline/1860-5397-10-109-i54.svg?max-width=637&scale=1.0)
30 |
75 |
| 10 |
![[Graphic 54]](/bjoc/content/inline/1860-5397-10-109-i55.svg?max-width=637&scale=1.0)
11 |
![[Graphic 55]](/bjoc/content/inline/1860-5397-10-109-i56.svg?max-width=637&scale=1.0)
13e |
![[Graphic 56]](/bjoc/content/inline/1860-5397-10-109-i57.svg?max-width=637&scale=1.0)
31 |
72 |
| 11 |
![[Graphic 57]](/bjoc/content/inline/1860-5397-10-109-i58.svg?max-width=637&scale=1.0)
12 |
![[Graphic 58]](/bjoc/content/inline/1860-5397-10-109-i59.svg?max-width=637&scale=1.0)
13e |
![[Graphic 59]](/bjoc/content/inline/1860-5397-10-109-i60.svg?max-width=637&scale=1.0)
32 |
69 |
aReaction conditions: boronate 9–12 (1.1 eqiuv), 13c–e (1.0 equiv), Pd(OAc)2 (4.0 mol %), PCy3, (8.0 mol %), K2PO4·H2O (3.0 equiv), toluene (0.50 mL) and H2O (0.10 mL), 80 W of microwave energy, 100 °C, 100 psi, 10 minutes. bYields of isolated products after a flash column chromatography.
Conclusion
Although arylboronic acids, arylboronate esters and potassium aryltrifluoroborate salts are powerful coupling partners in the Suzuki–Miyaura cross-coupling realm, extending the scope of organoboron compounds that can participate effectively as coupling partners in the cross-coupling reaction is still necessary. We have synthesised a range of 2-aryl-1,3-dihydro-1H-benzo[d]1,3,2-diazaborole compounds and developed their first Pd-catalysed Suzuki–Miyaura cross-coupling reaction with a range of aryl bromides bearing electron-rich, electron-neutral and electron-deficient functionalities using cost-effective and commercially available combination of Pd(OAc)2/PCy3 as a catalyst and K3PO4·H2O as a base. The catalytic system appeared versatile and general, tolerating a large range of functional groups such as NO2, OMe, COMe and diazaborolyl whilst furnishing the coupled product with isolated yields of up to 96% in only 10 minutes.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and copies of 1H and 13C NMR spectra of all synthesised compounds. | ||
| Format: PDF | Size: 1.0 MB | Download |
References
-
Hall, D. G., Ed. Boronic Acids; Wiley-VCH: Weinheim, Germany, 2005; pp 1–35.
Return to citation in text: [1] -
Darses, S.; Genet, J.-P. Chem. Rev. 2008, 108, 288–325. doi:10.1021/cr0509758
Return to citation in text: [1] -
Chow, W. K.; Yuen, Y. O.; So, C. M.; Wong, W. T.; Kwong, F. Y. J. Org. Chem. 2012, 77, 3543–3548. doi:10.1021/jo202472k
Return to citation in text: [1] -
Rosen, B. M.; Huang, C.; Percec, V. Org. Lett. 2008, 10, 2597–2600. doi:10.1021/ol800832n
Return to citation in text: [1] -
Molander, G. A.; Beaumard, F. Org. Lett. 2010, 12, 4022–4025. doi:10.1021/ol101592r
Return to citation in text: [1] [2] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] [2] [3] -
Dreher, S. D.; Lim, S.-E.; Sandrock, D. L.; Molander, G. A. J. Org. Chem. 2009, 74, 3626–3631. doi:10.1021/jo900152n
Return to citation in text: [1] -
Weber, L.; Halama, J.; Böhling, L.; Chrostowska, A.; Dargelos, A.; Stammler, H.-G.; Neumann, B. Eur. J. Inorg. Chem. 2011, 3091–3101. doi:10.1002/ejic.201100123
Return to citation in text: [1] -
Entwistle, C. D.; Marder, T. B. Chem. Mater. 2004, 16, 4574–4585. doi:10.1021/cm0495717
Return to citation in text: [1] -
Weber, L.; Werner, V.; Fox, M. A.; Marder, T. B.; Schwedler, S.; Brockhinke, A.; Stammler, H.-G.; Neumann, B. Dalton Trans. 2009, 1339–1351. doi:10.1039/b815931a
Return to citation in text: [1] -
Wade, C. R.; Broomsgrove, A. E. J.; Aldridge, S.; Gabbaï, F. P. Chem. Rev. 2010, 110, 3958–3984. doi:10.1021/cr900401a
Return to citation in text: [1] [2] -
Weber, L.; Halama, J.; Werner, V.; Hanke, K.; Böhling, L.; Chrostowska, A.; Dargelos, A.; Maciejczyk, M.; Raza, A.-L.; Stammler, H.-G.; Neumann, B. Eur. J. Inorg. Chem. 2010, 5416–5425. doi:10.1002/ejic.201000665
Return to citation in text: [1] -
Slabber, C. A.; Grimmer, C. D.; Robinson, R. S. J. Organomet. Chem. 2013, 723, 122–128. doi:10.1016/j.jorganchem.2012.09.018
Return to citation in text: [1] -
Hadebe, S. W.; Sithebe, S.; Robinson, R. S. Tetrahedron 2011, 67, 4277–4282. doi:10.1016/j.tet.2011.03.095
Return to citation in text: [1] [2] -
Noguchi, H.; Hojo, K.; Suginome, M. J. Am. Chem. Soc. 2007, 129, 758–759. doi:10.1021/ja067975p
Return to citation in text: [1] -
Gills, P. E.; Burke, D. M. J. Am. Chem. Soc. 2007, 129, 6716–6717. doi:10.1021/ja0716204
Return to citation in text: [1] -
Sithebe, S. Synthetic, Photophysical, Studies of Alkenyl/benzo-1,3,2-diazaborolane Compounds and their Palladium-Catalyzed Cross-Coupling Reactions. Ph.D. Thesis, University of KwaZulu Natal, South Africa, 2013; pp 1–227.
Return to citation in text: [1] [2] -
Xu, L.; Li, B.-J.; Wu, Z.-H.; Lu, Y.-X.; Guan, T.-B.; Wang, B.-Q.; Zhao, K.-Q.; Shi, Z.-J. Org. Lett. 2010, 12, 884–887. doi:10.1021/ol9029534
Return to citation in text: [1] -
Khedkar, M. V.; Tambede, P. J.; Qureshi, Z. S.; Bhanage, B. M. Eur. J. Org. Chem. 2010, 6981–6986. doi:10.1002/ejoc.201001134
Return to citation in text: [1] -
de Luna Martins, D.; Alvarez, H. M.; Aguiar, L. C. S. Tetrahedron Lett. 2010, 51, 6814–6817. doi:10.1016/j.tetlet.2010.09.145
Return to citation in text: [1] -
Lee, C.-C.; Ke, W.-C.; Chan, K.-T.; Lai, C.-L.; Hu, C.-H.; Lee, H. M. Chem.–Eur. J. 2007, 13, 582–591. doi:10.1002/chem.200600502
Return to citation in text: [1] -
Kaur, H.; Shah, D.; Pal, U. Catal. Commun. 2011, 12, 1384–1388. doi:10.1016/j.catcom.2011.05.012
Return to citation in text: [1]
| 1. | Hall, D. G., Ed. Boronic Acids; Wiley-VCH: Weinheim, Germany, 2005; pp 1–35. |
| 2. | Darses, S.; Genet, J.-P. Chem. Rev. 2008, 108, 288–325. doi:10.1021/cr0509758 |
| 3. | Chow, W. K.; Yuen, Y. O.; So, C. M.; Wong, W. T.; Kwong, F. Y. J. Org. Chem. 2012, 77, 3543–3548. doi:10.1021/jo202472k |
| 4. | Rosen, B. M.; Huang, C.; Percec, V. Org. Lett. 2008, 10, 2597–2600. doi:10.1021/ol800832n |
| 5. | Molander, G. A.; Beaumard, F. Org. Lett. 2010, 12, 4022–4025. doi:10.1021/ol101592r |
| 11. | Wade, C. R.; Broomsgrove, A. E. J.; Aldridge, S.; Gabbaï, F. P. Chem. Rev. 2010, 110, 3958–3984. doi:10.1021/cr900401a |
| 22. | Kaur, H.; Shah, D.; Pal, U. Catal. Commun. 2011, 12, 1384–1388. doi:10.1016/j.catcom.2011.05.012 |
| 8. | Weber, L.; Halama, J.; Böhling, L.; Chrostowska, A.; Dargelos, A.; Stammler, H.-G.; Neumann, B. Eur. J. Inorg. Chem. 2011, 3091–3101. doi:10.1002/ejic.201100123 |
| 9. | Entwistle, C. D.; Marder, T. B. Chem. Mater. 2004, 16, 4574–4585. doi:10.1021/cm0495717 |
| 10. | Weber, L.; Werner, V.; Fox, M. A.; Marder, T. B.; Schwedler, S.; Brockhinke, A.; Stammler, H.-G.; Neumann, B. Dalton Trans. 2009, 1339–1351. doi:10.1039/b815931a |
| 11. | Wade, C. R.; Broomsgrove, A. E. J.; Aldridge, S.; Gabbaï, F. P. Chem. Rev. 2010, 110, 3958–3984. doi:10.1021/cr900401a |
| 5. | Molander, G. A.; Beaumard, F. Org. Lett. 2010, 12, 4022–4025. doi:10.1021/ol101592r |
| 7. | Dreher, S. D.; Lim, S.-E.; Sandrock, D. L.; Molander, G. A. J. Org. Chem. 2009, 74, 3626–3631. doi:10.1021/jo900152n |
| 20. | de Luna Martins, D.; Alvarez, H. M.; Aguiar, L. C. S. Tetrahedron Lett. 2010, 51, 6814–6817. doi:10.1016/j.tetlet.2010.09.145 |
| 6. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 6. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 21. | Lee, C.-C.; Ke, W.-C.; Chan, K.-T.; Lai, C.-L.; Hu, C.-H.; Lee, H. M. Chem.–Eur. J. 2007, 13, 582–591. doi:10.1002/chem.200600502 |
| 15. | Noguchi, H.; Hojo, K.; Suginome, M. J. Am. Chem. Soc. 2007, 129, 758–759. doi:10.1021/ja067975p |
| 16. | Gills, P. E.; Burke, D. M. J. Am. Chem. Soc. 2007, 129, 6716–6717. doi:10.1021/ja0716204 |
| 17. | Sithebe, S. Synthetic, Photophysical, Studies of Alkenyl/benzo-1,3,2-diazaborolane Compounds and their Palladium-Catalyzed Cross-Coupling Reactions. Ph.D. Thesis, University of KwaZulu Natal, South Africa, 2013; pp 1–227. |
| 14. | Hadebe, S. W.; Sithebe, S.; Robinson, R. S. Tetrahedron 2011, 67, 4277–4282. doi:10.1016/j.tet.2011.03.095 |
| 6. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 18. | Xu, L.; Li, B.-J.; Wu, Z.-H.; Lu, Y.-X.; Guan, T.-B.; Wang, B.-Q.; Zhao, K.-Q.; Shi, Z.-J. Org. Lett. 2010, 12, 884–887. doi:10.1021/ol9029534 |
| 19. | Khedkar, M. V.; Tambede, P. J.; Qureshi, Z. S.; Bhanage, B. M. Eur. J. Org. Chem. 2010, 6981–6986. doi:10.1002/ejoc.201001134 |
| 13. | Slabber, C. A.; Grimmer, C. D.; Robinson, R. S. J. Organomet. Chem. 2013, 723, 122–128. doi:10.1016/j.jorganchem.2012.09.018 |
| 14. | Hadebe, S. W.; Sithebe, S.; Robinson, R. S. Tetrahedron 2011, 67, 4277–4282. doi:10.1016/j.tet.2011.03.095 |
| 12. | Weber, L.; Halama, J.; Werner, V.; Hanke, K.; Böhling, L.; Chrostowska, A.; Dargelos, A.; Maciejczyk, M.; Raza, A.-L.; Stammler, H.-G.; Neumann, B. Eur. J. Inorg. Chem. 2010, 5416–5425. doi:10.1002/ejic.201000665 |
| 17. | Sithebe, S. Synthetic, Photophysical, Studies of Alkenyl/benzo-1,3,2-diazaborolane Compounds and their Palladium-Catalyzed Cross-Coupling Reactions. Ph.D. Thesis, University of KwaZulu Natal, South Africa, 2013; pp 1–227. |
© 2014 Sithebe and Robinson; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)