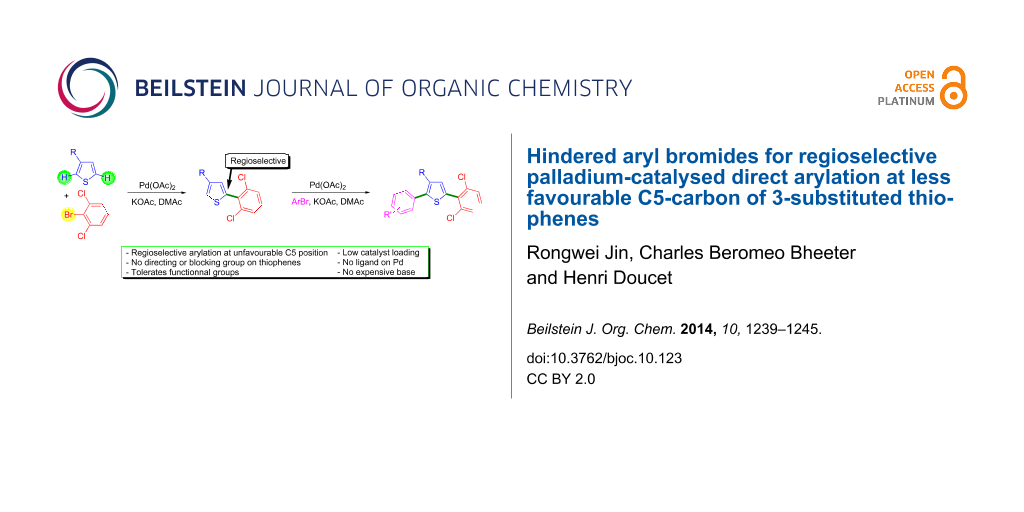
Abstract
The use of the congested aryl bromide 2-bromo-1,3-dichlorobenzene as coupling partner allows to modify the regioselectivity of the arylation of 3-substituted thiophene derivatives in favour of carbon C5. The coupling of this aryl bromide with a variety of 3-substituted thiophenes gave in all cases the desired 5-arylation products in moderate to good yields using only 0.5 mol % of a phosphine-free and air-stable palladium catalyst. Then, from these 5-arylthiophenes, a second palladium-catalysed C–H bond functionalization at C2 of the thiophene ring allows the synthesis of 2,5-diarylthiophenes with two different aryl units.

Graphical Abstract
Introduction
As thiophenes bearing aryl substituents are known to be present in several bioactive molecules and are used as precursors of materials, the regioselective introduction of aryls on thiophenes is an important research area in organic synthesis. The coupling of thiophene derivatives with aryl halides via a C–H bond activation/functionalisation [1-12] provides an environmentally attractive and cost-effective procedure for the preparation of a variety of arylated thiophenes. For such coupling reactions, the major byproduct is a base associated to HX, instead of metallic salts which are produced under the more classical Negishi, Suzuki or Stille cross-coupling reactions. Moreover, direct arylation avoids the preliminary preparation of organometallics reducing the number of steps to prepare these arylthiophenes.
The regioselective arylation via a Pd-catalysed C–H bond activation at carbon C5 of 2-substituted thiophenes has been largely described in recent years [13-22]. On the other hand, the Pd-catalysed direct arylation of 3-substituted thiophenes has attracted much less attention (Scheme 1, top) [23-31]. With such thiophene derivatives, in most cases, either C2-arylated thiophenes or mixtures of C2- and C5-arylated products have been obtained. For example, Sharp et al. described conditions for the regioselective arylation at carbons C2 or C5 of methyl 3-thiophenecarboxylate [23]. The reaction performed with Pd(PPh3)4 as the catalyst in toluene led selectively to the 2-arylated thiophene; whereas, the use of Pd2(dba)3 in NMP afforded a mixture of 2- and 5-arylated thiophenes in a 15:51 ratio. Lemaire and co-workers have reported the C2-arylation of 3-formyl-, 3-cyano- and 3-nitrothiophenes with iodobenzenes [24,25]. Forgione, Bilodeau et al. reported that the reaction of 3-methylthiophene with bromobenzene using Pd[(P(t-Bu)3]2 as the catalyst affords a mixture of 2- and 5-phenylated thiophenes in a 3.3:1 ratio [26]. Fagnou and co-workers reported that the coupling of 3-n-hexylthiophene with 4-bromonitrobenzene also led to a mixture of C2- and C5-arylation products in a 1.3:1 ratio [27]. Then, they blocked one position on the thiophene ring using a chloro-substituent in order to selectively arylate positions C2 or C5. The direct arylation of 3-methoxythiophene, which was studied by Borghese and co-workers afforded regioselectively the C2-arylated thiophenes in moderate to high yields [28]. Finally, in the course of their studies on sp3 C–H bond activation, Baudoin and Pierre recently reported that in the presence of an extremely bulky substituent at C3 of a thiophene derivative, the C5-arylated compounds were selectively obtained in good yields [29]. In summary, due to the presence of two reactive C–H bonds in 3-substituted thiophenes (with position C2 generally slightly more reactive than position C5), the control of the regioselectivity of the palladium-catalysed direct arylation of such thiophene derivatives especially to provide 5-arylthiophenes remains a challenging reaction.
![[1860-5397-10-123-i1]](/bjoc/content/inline/1860-5397-10-123-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Regioselectivity of coupling reactions of 3-substituted thiophenes with aryl halides.
Scheme 1: Regioselectivity of coupling reactions of 3-substituted thiophenes with aryl halides.
Our goal was to promote arylation at carbon C5 of a range of 3-substituted thiophenes without the use of a blocking group at carbon C2. To our knowledge, ortho-substituents on aryl bromides have not been employed as directing groups for palladium-catalysed direct arylation of 3-substituted thiophenes. The use of congested aryl bromides for such couplings would certainly modify the regioselectivity in favour of arylation at the less hindered thiophene position. Herein, we wish to report on the influence of such ortho-substituents on aryl bromides on the regioselectivity of the palladium-catalysed direct arylations of 3-substituted thiophenes.
Results and Discussion
We first studied the palladium-catalysed direct arylation of 3-methylthiophene using tert-butyl 2-bromobenzoate as the coupling partner (Scheme 2). Based on previous results [19], DMA was initially chosen as the solvent and KOAc as the base. The reactions were performed at 150 °C under argon in the presence of 0.5 mol % Pd(OAc)2 as the catalyst. However, under these conditions, a poor regioselectivity was observed as the desired C5-arylation product 1b was only obtained in 34% selectivity together with 66% of C2-arylation product 1a. Moreover, a moderate conversion of this aryl bromide was observed and purification by silica gel chromatography afforded a mixture of 1a and 1b. A slightly lower selectivity in favour of the formation of C5-arylation product 2b was observed using 2-bromonitrobenzene as the coupling partner, as a large amount of 2,5-diarylated product 2c was also produced with a 2a:2b:2c ratio of 34:21:45. However, a complete conversion of 2-bromonitrobenzene was observed. 2-Bromoaniline was found to be unreactive and was recovered. It should be noted that no amination reaction of 2-bromoaniline due to self-coupling was observed. From 2-(trifluoromethyl)bromobenzene, a very similar mixture of regioisomers than with 2-bromonitrobenzene was obtained, as 4a, 4b and 4c were formed in a 24:31:45 ratio. The use of 2-bromobenzaldehyde was not successful, as the desired product 5b was only obtained with 10% selectivity. From more hindered 2-bromobenzaldehyde diethyl acetal, the selectivity in favour of desired product 6b was slightly higher (34%), although still not synthetically useful. Next, we employed 2-chlorobromobenzene as the coupling partner. However, again the desired product 7b was only formed with 15% selectivity. It should be noted that with these aryl bromides, in all cases, mixtures of regioisomers a and b were obtained after column chromatography. On the other hand, the use of 2-bromo-1,3-dichlorobenzene allowed to obtain very selectively the desired 5-arylation product 8b. Only traces of the C2-arylated thiophene 8a and a low amount of 2,5-diarylated product 8c were detected by 1H NMR and GC–MS analysis of the crude mixture. Moreover a high conversion of 86% of this aryl bromide was observed using only 0.5 mol % Pd(OAc)2 as the catalyst.
![[1860-5397-10-123-i2]](/bjoc/content/inline/1860-5397-10-123-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Regioselectivity of the arylation of 2-methylthiophene with ortho-substituted aryl bromides.
Scheme 2: Regioselectivity of the arylation of 2-methylthiophene with ortho-substituted aryl bromides.
Then, we studied the scope of the coupling of 2-bromo-1,3-dichlorobenzene, using other 3-substituted thiophene derivatives (Scheme 3, Table 1). Both 3-chlorothiophene and ethyl thiophene-3-carboxylate led to the desired 5-arylation products 9b and 10b with 89% selectivity and in 44% and 65% yields, respectively (Table 1, entries 1 and 2). A slightly higher regioselectivity in favour of the C5-arylation was observed in the presence of 3-acetylthiophene, as 11b was obtained with 92% selectivity (Table 1, entry 3). From ethyl thiophen-3-ylacetate, 12b was formed with 90% selectivity and in 62% yield (Table 1, entry 4). On the other hand, from methyl (E)-3-thiophen-3-ylacrylate, 3-bromothiophene or 3-formylthiophene, mixtures of unidentified products were obtained.
![[1860-5397-10-123-i3]](/bjoc/content/inline/1860-5397-10-123-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Direct arylation of 3-substituted thiophenes with 2-bromo-1,3-dichlorobenzene.
Scheme 3: Direct arylation of 3-substituted thiophenes with 2-bromo-1,3-dichlorobenzene.
Table 1: Direct arylation of 3-substituted thiophenes with 2-bromo-1,3-dichlorobenzene (Scheme 3).a
| Entry | Heteroarene | Major product | Ratio a:b:c | Yieldb of regioisomer b (%) |
|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-123-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-123-i6.svg?max-width=637&scale=1.0)
9b |
3:89:8 | 44 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-123-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-123-i8.svg?max-width=637&scale=1.0)
10b |
3:89:8 | 65 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-123-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-123-i10.svg?max-width=637&scale=1.0)
11b |
0:92:8 | 57 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-123-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-123-i12.svg?max-width=637&scale=1.0)
12b |
2:90:8 | 62 |
aConditions: Pd(OAc)2 (0.5 mol %), aryl bromide (1 mmol), thiophene derivative (2 mmol), KOAc (2 mmol), DMA (3 mL), 150 °C, 20 h; bisolated yields of regioisomers 9b–12b.
The reactivity for arylation at carbon C2 of the previously prepared 2-(2,6-dichlorophenyl)-4-methylthiophene (8b), via a second Pd-catalysed C–H bond activation, was also investigated (Scheme 4). 4-Bromobenzonitrile and 8b in the presence of 0.5 mol % Pd(OAc)2 and KOAc as the base gave the desired coupling product 13 in 82% yield. A similar reactivity was observed with 4-bromobenzaldehyde and 3-bromoacetophenone affording 14 and 15 in 80% and 85% yields, respectively. Finally, more congested 2-bromobenzonitrile was reacted with 8b to provide 16 in 88% yield. These regioselective sequential arylations of a 3-substituted thiophene offer a simple access to a variety of 2,5-diarylthiophenes with two different aryl units.
![[1860-5397-10-123-i4]](/bjoc/content/inline/1860-5397-10-123-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Pd-catalysed C2-arylation of 8b with aryl bromides.
Scheme 4: Pd-catalysed C2-arylation of 8b with aryl bromides.
Conclusion
In summary, we have demonstrated that the use of the congested coupling partner 2-bromo-1,3-dichlorobenzene allows to direct the arylation to the unfavourable C5 position of 3-substituted thiophenes. These less favoured regioisomers can be selectively obtained in moderate to good yields using a range of 3-substituted thiophenes, as chloro, ester, acetyl or ethyl acetate substituents are tolerated. Moreover, the sequential catalytic C5 and C2 arylations, allow the preparation of 2,5-diarylthiophenes with two different aryl units in two steps. The major byproduct of these couplings is KBr/AcOH instead of metallic salts as with more classical coupling procedures. For these reasons, this process gives an economically viable access to C5-arylated 3-substituted heteroaromatics.
Experimental
General
All reactions were perfomed in Schlenk tubes under argon. DMA analytical grade was not distilled before use. Commercial aryl bromide derivatives were used without purification. 1H NMR (400 MHz), 13C NMR (100 MHz) spectra were recorded in CDCl3 solutions on Bruker GPX (400 MHz). GC was performed on a Shimadzu GC-2014 and GC–MS was performed on a Shimadzu QP-2010S. Chemical shifts are reported in ppm relative to CDCl3 (1H: 7.26 and 13C: 77.0). Flash chromatography was performed on silica gel (230–400 mesh).
General procedure for direct arylations
The aryl bromide (1 mmol), thiophene derivative (2 mmol), KOAc (2 mmol), Pd(OAc)2 (0.005 mmol, 1.1 mg) and DMA (3 mL) were introduced in a Schlenk tube under argon equipped with a magnetic stirring bar. The Schlenk tube was placed in an oil bath pre-heated at 150 °C, and the reaction mixture was allowed to stir for 20 h. After cooling, the crude reaction mixture was analysed by gas chromatography and 1H NMR to determine the conversion of the aryl bromide and the regioselectivity of the arylation. The solvent was removed by heating under vacuum, then the residue was charged onto a silica gel column.
tert-Butyl 2-(4-methylthiophen-2-yl)benzoate (1b): After column chromatography (pentane/diethyl ether 98:2), a mixture of products 1a and 1b was obtained in 24% (0.066 g) yield. 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 1H), 2.20 (s, 3H). 1a was also observed: 1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 5.0 Hz, 1H), 6.81 (d, J = 5.0 Hz, 1H), 1.97 (s, 3H).
4-Methyl-2-(2-nitrophenyl)thiophene (2b): After column chromatography (pentane/diethyl ether 95:5), a mixture of products 2a and 2b was obtained in 60% (0.131 g) yield. 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 8.0 Hz, 1H), 6.91 (s, 1H), 2.19 (s, 3H). 2a was also observed: 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 5.0 Hz, 1H), 6.83 (d, J = 5.0 Hz, 1H), 1.99 (s, 3H).
4-Methyl-2-(2-trifluoromethylphenyl)thiophene (4b): After column chromatography (pentane/diethyl ether 90:10), a mixture of products 4a, 4b and 4c was obtained in 66% (0.160 g) yield. 1H NMR (400 MHz, CDCl3) δ 6.88 (s, 1H), 6.83 (s, 1H), 1.95 (s, 3H). 4a was also observed: 1H NMR (400 MHz, CDCl3) δ 7.20 (d, J = 5.1 Hz, 1H), 6.81 (d, J = 5.1 Hz, 1H). 4c was also observed: 1H NMR (400 MHz, CDCl3) δ 7.75–7.65 (m, 2H), 7.55–7.35 (m, 6H), 6.82 (s, 1H), 1.95 (s, 3H).
2-(4-Methylthiophen-2-yl)benzaldehyde (5b): After column chromatography (pentane/diethyl ether 95:5), a mixture of products 5a and 5b was obtained in 51% (0.103 g) yield. 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 7.92 (d, J = 7.8 Hz, 1H), 7.60–7.35 (m, 3H), 6.97 (s, 1H), 6.81 (s, 1H), 2.25 (s, 3H). 5a was also observed: 1H NMR (400 MHz, CDCl3) δ 9.85 (s, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 7.36 (d, J = 7.6 Hz, 1H), 7.27 (d, J = 5.0 Hz, 1H), 6.89 (d, J = 5.0 Hz, 1H), 2.02 (s, 3H).
2-(2-Chlorophenyl)-4-methylthiophene (7b): After column chromatography (pentane/diethyl ether 95:5), a mixture of products 7a and 7b was obtained in 64% (0.133 g) yield. 1H NMR (400 MHz, CDCl3) δ 7.11 (s, 1H), 6.90 (s, 1H), 2.24 (s, 3H). 7a was also observed: 1H NMR (400 MHz, CDCl3) δ 6.87 (d, J = 5.0 Hz, 1H), 2.04 (s, 3H).
2-(2,6-Dichlorophenyl)-4-methylthiophene (8b): From 2-bromo-1,3-dichlorobenzene (0.226 g, 1 mmol) and 3-methylthiophene (0.196 g, 2 mmol), 8b was obtained in 63% (0.153 g) yield as an oil after column chromatography (pentane). 1H NMR (400 MHz, CDCl3) δ 7.31 (d, J = 7.5 Hz, 2H), 7.15 (t, J = 7.5 Hz, 1H), 6.98 (s, 1H), 6.75 (s, 1H), 2.25 (s, 3H); 13C NMR (50 MHz, CDCl3) δ 137.4, 136.6, 136.4, 133.1, 130.8, 129.7, 128.0, 122.1, 15.8; Anal. calcd for C11H8Cl2S (243.15): C, 54.34; H, 3.32; found: C, 54.19; H, 3.17. Traces of 8a and 8c were also detected by GC–MS analysis of the crude mixture.
4-Chloro-2-(2,6-dichlorophenyl)thiophene (9b): From 2-bromo-1,3-dichlorobenzene (0.226 g, 1 mmol) and 3-chlorothiophene (0.237 g, 2 mmol), 9b was obtained in 44% (0.116 g) yield as an oil after column chromatography (pentane). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 7.5 Hz, 2H), 7.23–7.17 (m, 2H), 6.83 (d, J = 1.2 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 137.4, 136.5, 131.7, 130.4, 128.8, 128.2, 124.9, 121.4; Anal. calcd for C10H5Cl3S (263.57): C, 45.57; H, 1.91; found: C, 45.67; H, 1.90. Traces of 9a and 9c were also detected by GC–MS analysis of the crude mixture.
Ethyl 5-(2,6-dichlorophenyl)thiophene-3-carboxylate (10b): From 2-bromo-1,3-dichlorobenzene (0.226 g, 1 mmol) and ethyl thiophene-3-carboxylate (0.312 g, 2 mmol), 10b was obtained in 65% (0.196 g) yield as an oil after column chromatography (pentane/diethyl ether 98:2). 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 7.37 (d, J = 7.5 Hz, 2H), 7.34 (s, 1H), 7.22 (t, J = 7.5 Hz, 1H), 4.29 (q, J = 7.5 Hz, 2H), 1.31 (t, J = 7.5 Hz, 3H); 13C NMR (50 MHz, CDCl3) δ 162.6, 137.3, 136.6, 133.8, 133.7, 131.8, 130.3, 129.3, 128.2, 60.8, 14.3; Anal. calcd for C13H10Cl2O2S (301.19): C, 51.84; H, 3.35; found: C, 51.99; H, 3.17. Traces of 10a and 10c were also detected by GC–MS analysis of the crude mixture.
1-[5-(2,6-Dichlorophenyl)thiophen-3-yl]ethanone (11b): From 2-bromo-1,3-dichlorobenzene (0.226 g, 1 mmol) and 1-thiophen-3-ylethanone (0.252 g, 2 mmol), 11b was obtained in 57% (0.154 g) yield as an oil after column chromatography (pentane/diethyl ether 95:5). 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.37 (d, J = 7.5 Hz, 2H), 7.34 (s, 1H), 7.22 (t, J = 7.5 Hz, 1H), 2.49 (s, 3H); 13C NMR (50 MHz, CDCl3) δ 191.0, 141.3, 136.7, 135.4, 132.5, 130.6, 129.4, 127.4, 127.2, 26.4; Anal. calcd (%) for C12H8Cl2OS (271.16): C, 53.15; H, 2.97; found: C, 53.31; H, 3.07. Traces of 11c were also detected by GC–MS analysis of the crude mixture.
Ethyl [5-(2,6-dichlorophenyl)thiophen-3-yl]acetate (12b): From 2-bromo-1,3-dichlorobenzene (0.226 g, 1 mmol) and ethyl thiophen-3-ylacetate (0.340 g, 2 mmol), 12b was obtained in 62% (0.195 g) yield as an oil after column chromatography (pentane/diethyl ether 95:5). 1H NMR (400 MHz, CDCl3) δ 7.32 (d, J = 7.5 Hz, 2H), 7.22 (s, 1H), 7.16 (t, J = 7.5 Hz, 1H), 6.89 (s, 1H), 4.12 (q, J = 7.5 Hz, 2H), 3.60 (s, 2H), 1.19 (t, J = 7.5 Hz, 3H); 13C NMR (50 MHz, CDCl3) δ 170.9, 136.7, 136.6, 133.5, 132.8, 130.2, 129.9, 128.1, 124.2, 60.9, 36.2, 14.2; Anal. calcd for C14H12Cl2O2S (315.22): C, 53.34; H, 3.84; found: C, 53.21; H, 3.70. Traces of 12a and 12c were also detected by GC–MS analysis of the crude mixture.
4-[5-(2,6-Dichlorophenyl)-3-methylthiophen-2-yl]benzonitrile (13): From 4-bromobenzonitrile (0.182 g, 1 mmol) and 2-(2,6-dichlorophenyl)-4-methylthiophene (8b, 0.486 g, 2 mmol), 13 was obtained in 82% (0.282 g) yield as an oil after column chromatography (pentane/diethyl ether 95:5). 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 7.5 Hz, 2H), 7.34 (d, J = 7.5 Hz, 2H), 7.19 (t, J = 7.5 Hz, 1H), 6.80 (s, 1H), 2.33 (s, 3H); 13C NMR (50 MHz, CDCl3) δ 139.1, 137.2, 136.4, 136.3, 134.7, 133.2, 132.3, 132.2, 130.0, 129.2, 128.2, 118.8, 110.6, 15.8; Anal. calcd for C18H11Cl2NS (344.26): C, 62.80; H, 3.22; found: C, 63.04; H, 3.17.
4-(5-(2,6-Dichlorophenyl)-3-methylthiophen-2-yl)benzaldehyde (14): From 4-bromobenzaldehyde (0.185 g, 1 mmol) and 2-(2,6-dichlorophenyl)-4-methylthiophene (8b, 0.486 g, 2 mmol), 16 was obtained in 80% (0.278 g) yield as an oil after column chromatography (pentane/diethyl ether 85:15). 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.86 (d, J = 7.5 Hz, 2H), 7.63 (d, J = 7.5 Hz, 2H), 7.34 (d, J = 7.5 Hz, 2H), 7.19 (t, J = 7.5 Hz, 1H), 6.81 (s, 1H), 2.36 (s, 3H); 13C NMR (50 MHz, CDCl3) δ 191.6, 140.6, 137.9, 136.4, 136.1, 134.8, 134.6, 133.2, 132.3, 130.0, 129.1, 128.2, 15.5; Anal. calcd for C18H12Cl2OS (347.26): C, 62.26; H, 3.48; found: C, 62.09; H, 3.50.
1-{3-[5-(2,6-Dichlorophenyl)-3-methylthiophen-2-yl]phenyl}ethanone (15): From 3-bromoacetophenone (0.199 g, 1 mmol) and 2-(2,6-dichlorophenyl)-4-methylthiophene (8b, 0.486 g, 2 mmol), 15 was obtained in 85% (0.307 g) yield as a white solid after column chromatography pentane/diethyl ether 85:15). 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1H), 7.84 (d, J = 7.5 Hz, 1H), 7.65 (d, J = 7.5 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.34 (d, J = 7.5 Hz, 2H), 7.17 (t, J = 7.5 Hz, 1H), 6.79 (s, 1H), 2.58 (s, 3H), 2.31 (s, 3H); 13C NMR (50 MHz, CDCl3) δ 196.8, 137.1, 136.4, 135.5, 134.0, 133.9, 132.5, 132.4, 131.7, 131.6, 128.8, 127.9, 127.8, 127.1, 126.1, 25.7, 14.1; Anal. calcd for C19H14Cl2OS (361.29): C, 63.19; H, 3.91; found: C, 63.04; H, 3.99.
2-[5-(2,6-Dichlorophenyl)-3-methylthiophen-2-yl]benzonitrile (16): From 2-bromobenzonitrile (0.182 g, 1 mmol) and 2-(2,6-dichlorophenyl)-4-methylthiophene (8b, 0.486 g, 2 mmol), 16 was obtained in 88% (0.303 g) yield as an oil after column chromatography (pentane/diethyl ether 95:5). 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.8 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.40 (d, J = 7.8 Hz, 2H), 7.24 (t, J = 7.8 Hz, 1H), 6.88 (s, 1H), 2.27 (s, 3H); 13C NMR (50 MHz, CDCl3) δ 136.9, 135.7, 135.6, 135.1, 133.5, 132.4, 131.4, 131.3, 130.9, 130.8, 128.9, 127.2, 127.1, 117.0, 112.8, 13.9; Anal. calcd for C18H11Cl2NS (344.26): C, 62.80; H, 3.22; found: C, 63.01; H, 3.40.
References
-
Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269. doi:10.1016/j.tet.2009.10.015
Return to citation in text: [1] -
Satoh, T.; Miura, M. Chem. Lett. 2007, 36, 200. doi:10.1246/cl.2007.200
Return to citation in text: [1] -
Li, B.-J.; Yang, S.-D.; Shi, Z.-J. Synlett 2008, 949. doi:10.1055/s-2008-1042907
Return to citation in text: [1] -
Ackermann, L.; Vincente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792. doi:10.1002/anie.200902996
Return to citation in text: [1] -
Fischmeister, C.; Doucet, H. Green Chem. 2011, 13, 741. doi:10.1039/c0gc00885k
Return to citation in text: [1] -
Ackermann, L. Chem. Rev. 2011, 111, 1315. doi:10.1021/cr100412j
Return to citation in text: [1] -
Kuhl, N.; Hopkinson, M. N.; Wencel-Delord, J.; Glorius, F. Angew. Chem., Int. Ed. 2012, 51, 10236. doi:10.1002/anie.201203269
Return to citation in text: [1] -
Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936. doi:10.1021/ar300014f
Return to citation in text: [1] -
Wencel-Delord, J.; Glorius, F. Nat. Chem. 2013, 5, 369. doi:10.1038/nchem.1607
Return to citation in text: [1] -
Akita, Y.; Inoue, A.; Yamamoto, K.; Ohta, A.; Kurihara, T.; Shimizu, M. Heterocycles 1985, 23, 2327. doi:10.3987/R-1985-09-2327
Return to citation in text: [1] -
Ohta, A.; Akita, Y.; Ohkuwa, T.; Chiba, M.; Fukunaga, R.; Miyafuji, A.; Nakata, T.; Tani, N.; Aoyagi, Y. Heterocycles 1990, 31, 1951. doi:10.3987/COM-90-5467
Return to citation in text: [1] -
Aoyagi, Y.; Inoue, A.; Koizumi, I.; Hashimoto, R.; Tokunaga, K.; Gohma, K.; Komatsu, J.; Sekine, K.; Miyafuji, A.; Kunoh, J.; Honma, R.; Akita, Y.; Ohta, A. Heterocycles 1992, 33, 257. doi:10.3987/COM-91-S29
Return to citation in text: [1] -
Okazawa, T.; Satoh, T.; Miura, M.; Nomura, M. J. Am. Chem. Soc. 2002, 124, 5286. doi:10.1021/ja0259279
Return to citation in text: [1] -
Masui, K.; Ikegami, H.; Mori, A. J. Am. Chem. Soc. 2004, 126, 5074. doi:10.1021/ja031855p
Return to citation in text: [1] -
Masui, K.; Mori, A.; Okano, K.; Takamura, K.; Kinoshita, M.; Ikeda, T. Org. Lett. 2004, 6, 2011. doi:10.1021/ol049386z
Return to citation in text: [1] -
Amaladass, P.; Clement, J. A.; Mohanakrishnan, A. K. Tetrahedron 2007, 63, 10363. doi:10.1016/j.tet.2007.07.037
Return to citation in text: [1] -
Turner, G. L.; Morris, J. A.; Greaney, M. F. Angew. Chem., Int. Ed. 2007, 46, 7996. doi:10.1002/anie.200702141
Return to citation in text: [1] -
Arai, N.; Miyaoku, T.; Teruya, S.; Mori, A. Tetrahedron Lett. 2008, 49, 1000. doi:10.1016/j.tetlet.2007.12.010
Return to citation in text: [1] -
Roger, J.; Požgan, F.; Doucet, H. Green Chem. 2009, 11, 425. doi:10.1039/b819912d
Return to citation in text: [1] [2] -
Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. J. Org. Chem. 2009, 74, 1826. doi:10.1021/jo8026565
Return to citation in text: [1] -
Zinovyeva, V. A.; Vorotyntsev, M. A.; Bezverkhyy, I.; Chaumont, D.; Hierso, J.-C. Adv. Funct. Mater. 2011, 21, 1064. doi:10.1002/adfm.201001912
Return to citation in text: [1] -
Beydoun, K.; Roger, J.; Boixel, J.; Le Bozec, H.; Guerchais, V.; Doucet, H. Chem. Commun. 2012, 48, 11951. doi:10.1039/c2cc37046h
Return to citation in text: [1] -
Glover, B.; Harvey, K. A.; Liu, B.; Sharp, M. J.; Tymoschenko, M. F. Org. Lett. 2003, 5, 301. doi:10.1021/ol027266q
Return to citation in text: [1] [2] -
Lavenot, L.; Gozzi, C.; Ilg, K.; Orlova, I.; Penalva, V.; Lemaire, M. J. Organomet. Chem. 1998, 567, 49. doi:10.1016/S0022-328X(98)00667-6
Return to citation in text: [1] [2] -
Fournier dit Chabert, J.; Marquez, B.; Neville, L.; Joucla, L.; Broussous, S.; Bouhours, P.; David, E.; Pellet-Rostaing, S.; Marquet, B.; Moreau, N.; Lemaire, M. Bioorg. Med. Chem. 2007, 15, 4482. doi:10.1016/j.bmc.2007.04.023
Return to citation in text: [1] [2] -
Forgione, P.; Brochu, M.-C.; St-Onge, M.; Thesen, K. H.; Bailey, M. D.; Bilodeau, F. J. Am. Chem. Soc. 2006, 128, 11350. doi:10.1021/ja063511f
Return to citation in text: [1] [2] -
Liégault, B.; Petrov, I.; Gorlesky, S. I.; Fagnou, K. J. Org. Chem. 2010, 75, 1047. doi:10.1021/jo902515z
Return to citation in text: [1] [2] -
Borghese, A.; Geldhof, G.; Antoine, L. Tetrahedron Lett. 2006, 47, 9249. doi:10.1016/j.tetlet.2006.10.130
Return to citation in text: [1] [2] -
Pierre, C.; Baudoin, O. Tetrahedron 2013, 69, 4473. doi:10.1016/j.tet.2012.11.060
Return to citation in text: [1] [2] -
René, O.; Fagnou, K. Org. Lett. 2010, 12, 2116. doi:10.1021/ol1006136
Return to citation in text: [1] -
Dong, J. J.; Roy, D.; Roy, R. J.; Ionita, M.; Doucet, H. Synthesis 2011, 3530. doi:10.1055/s-0030-1260213
Return to citation in text: [1]
| 1. | Bellina, F.; Rossi, R. Tetrahedron 2009, 65, 10269. doi:10.1016/j.tet.2009.10.015 |
| 2. | Satoh, T.; Miura, M. Chem. Lett. 2007, 36, 200. doi:10.1246/cl.2007.200 |
| 3. | Li, B.-J.; Yang, S.-D.; Shi, Z.-J. Synlett 2008, 949. doi:10.1055/s-2008-1042907 |
| 4. | Ackermann, L.; Vincente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792. doi:10.1002/anie.200902996 |
| 5. | Fischmeister, C.; Doucet, H. Green Chem. 2011, 13, 741. doi:10.1039/c0gc00885k |
| 6. | Ackermann, L. Chem. Rev. 2011, 111, 1315. doi:10.1021/cr100412j |
| 7. | Kuhl, N.; Hopkinson, M. N.; Wencel-Delord, J.; Glorius, F. Angew. Chem., Int. Ed. 2012, 51, 10236. doi:10.1002/anie.201203269 |
| 8. | Neufeldt, S. R.; Sanford, M. S. Acc. Chem. Res. 2012, 45, 936. doi:10.1021/ar300014f |
| 9. | Wencel-Delord, J.; Glorius, F. Nat. Chem. 2013, 5, 369. doi:10.1038/nchem.1607 |
| 10. | Akita, Y.; Inoue, A.; Yamamoto, K.; Ohta, A.; Kurihara, T.; Shimizu, M. Heterocycles 1985, 23, 2327. doi:10.3987/R-1985-09-2327 |
| 11. | Ohta, A.; Akita, Y.; Ohkuwa, T.; Chiba, M.; Fukunaga, R.; Miyafuji, A.; Nakata, T.; Tani, N.; Aoyagi, Y. Heterocycles 1990, 31, 1951. doi:10.3987/COM-90-5467 |
| 12. | Aoyagi, Y.; Inoue, A.; Koizumi, I.; Hashimoto, R.; Tokunaga, K.; Gohma, K.; Komatsu, J.; Sekine, K.; Miyafuji, A.; Kunoh, J.; Honma, R.; Akita, Y.; Ohta, A. Heterocycles 1992, 33, 257. doi:10.3987/COM-91-S29 |
| 24. | Lavenot, L.; Gozzi, C.; Ilg, K.; Orlova, I.; Penalva, V.; Lemaire, M. J. Organomet. Chem. 1998, 567, 49. doi:10.1016/S0022-328X(98)00667-6 |
| 25. | Fournier dit Chabert, J.; Marquez, B.; Neville, L.; Joucla, L.; Broussous, S.; Bouhours, P.; David, E.; Pellet-Rostaing, S.; Marquet, B.; Moreau, N.; Lemaire, M. Bioorg. Med. Chem. 2007, 15, 4482. doi:10.1016/j.bmc.2007.04.023 |
| 23. | Glover, B.; Harvey, K. A.; Liu, B.; Sharp, M. J.; Tymoschenko, M. F. Org. Lett. 2003, 5, 301. doi:10.1021/ol027266q |
| 23. | Glover, B.; Harvey, K. A.; Liu, B.; Sharp, M. J.; Tymoschenko, M. F. Org. Lett. 2003, 5, 301. doi:10.1021/ol027266q |
| 24. | Lavenot, L.; Gozzi, C.; Ilg, K.; Orlova, I.; Penalva, V.; Lemaire, M. J. Organomet. Chem. 1998, 567, 49. doi:10.1016/S0022-328X(98)00667-6 |
| 25. | Fournier dit Chabert, J.; Marquez, B.; Neville, L.; Joucla, L.; Broussous, S.; Bouhours, P.; David, E.; Pellet-Rostaing, S.; Marquet, B.; Moreau, N.; Lemaire, M. Bioorg. Med. Chem. 2007, 15, 4482. doi:10.1016/j.bmc.2007.04.023 |
| 26. | Forgione, P.; Brochu, M.-C.; St-Onge, M.; Thesen, K. H.; Bailey, M. D.; Bilodeau, F. J. Am. Chem. Soc. 2006, 128, 11350. doi:10.1021/ja063511f |
| 27. | Liégault, B.; Petrov, I.; Gorlesky, S. I.; Fagnou, K. J. Org. Chem. 2010, 75, 1047. doi:10.1021/jo902515z |
| 28. | Borghese, A.; Geldhof, G.; Antoine, L. Tetrahedron Lett. 2006, 47, 9249. doi:10.1016/j.tetlet.2006.10.130 |
| 29. | Pierre, C.; Baudoin, O. Tetrahedron 2013, 69, 4473. doi:10.1016/j.tet.2012.11.060 |
| 30. | René, O.; Fagnou, K. Org. Lett. 2010, 12, 2116. doi:10.1021/ol1006136 |
| 31. | Dong, J. J.; Roy, D.; Roy, R. J.; Ionita, M.; Doucet, H. Synthesis 2011, 3530. doi:10.1055/s-0030-1260213 |
| 13. | Okazawa, T.; Satoh, T.; Miura, M.; Nomura, M. J. Am. Chem. Soc. 2002, 124, 5286. doi:10.1021/ja0259279 |
| 14. | Masui, K.; Ikegami, H.; Mori, A. J. Am. Chem. Soc. 2004, 126, 5074. doi:10.1021/ja031855p |
| 15. | Masui, K.; Mori, A.; Okano, K.; Takamura, K.; Kinoshita, M.; Ikeda, T. Org. Lett. 2004, 6, 2011. doi:10.1021/ol049386z |
| 16. | Amaladass, P.; Clement, J. A.; Mohanakrishnan, A. K. Tetrahedron 2007, 63, 10363. doi:10.1016/j.tet.2007.07.037 |
| 17. | Turner, G. L.; Morris, J. A.; Greaney, M. F. Angew. Chem., Int. Ed. 2007, 46, 7996. doi:10.1002/anie.200702141 |
| 18. | Arai, N.; Miyaoku, T.; Teruya, S.; Mori, A. Tetrahedron Lett. 2008, 49, 1000. doi:10.1016/j.tetlet.2007.12.010 |
| 19. | Roger, J.; Požgan, F.; Doucet, H. Green Chem. 2009, 11, 425. doi:10.1039/b819912d |
| 20. | Liégault, B.; Lapointe, D.; Caron, L.; Vlassova, A.; Fagnou, K. J. Org. Chem. 2009, 74, 1826. doi:10.1021/jo8026565 |
| 21. | Zinovyeva, V. A.; Vorotyntsev, M. A.; Bezverkhyy, I.; Chaumont, D.; Hierso, J.-C. Adv. Funct. Mater. 2011, 21, 1064. doi:10.1002/adfm.201001912 |
| 22. | Beydoun, K.; Roger, J.; Boixel, J.; Le Bozec, H.; Guerchais, V.; Doucet, H. Chem. Commun. 2012, 48, 11951. doi:10.1039/c2cc37046h |
| 29. | Pierre, C.; Baudoin, O. Tetrahedron 2013, 69, 4473. doi:10.1016/j.tet.2012.11.060 |
| 28. | Borghese, A.; Geldhof, G.; Antoine, L. Tetrahedron Lett. 2006, 47, 9249. doi:10.1016/j.tetlet.2006.10.130 |
| 27. | Liégault, B.; Petrov, I.; Gorlesky, S. I.; Fagnou, K. J. Org. Chem. 2010, 75, 1047. doi:10.1021/jo902515z |
| 26. | Forgione, P.; Brochu, M.-C.; St-Onge, M.; Thesen, K. H.; Bailey, M. D.; Bilodeau, F. J. Am. Chem. Soc. 2006, 128, 11350. doi:10.1021/ja063511f |
| 19. | Roger, J.; Požgan, F.; Doucet, H. Green Chem. 2009, 11, 425. doi:10.1039/b819912d |
© 2014 Jin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)