Abstract
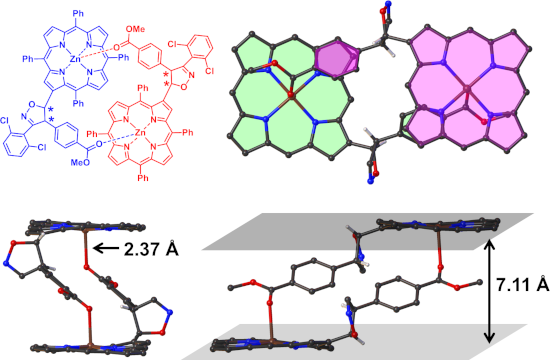
Isoxazoline-linked porphyrins have been synthesized by a regioselective 1,3-dipolar cycloaddition reaction between vinylporphyrin 2 and nitrile oxides. The steric interaction directed the reaction trajectory, in which only the product with a link between the 5-position of the isoxazoline and the β-position of porphyrin was observed. The isoxazoline-porphyrins 3a,b have been characterized by absorption, emission, 1H NMR and mass spectra. Later, the crystal structure of 3a was obtained and confirmed the basic features of the NMR-derived structure. Furthermore, a pair of enantiomers of 3a presented in the crystal, which formed a dimeric complex through intermolecular coordination between the Zn2+ center and the carbonyl group of the second molecule.
Graphical Abstract
Introduction
In nature, porphyrin-type compounds play a prominent role in life [1]. It is well known that certain vital functions, like O2 transport, photosynthesis etc. depend on the action of porphyrin–metal complexes [2-5]. Inspired by the natural porphyrinoids, some man-made porphyrinoids have been designed to mimic the characteristic functions with purposes of utilization in various fields (please see recent reviews [6-14]). In order to achieve application-oriented molecules, lots of modification approaches have been developed [15-24]. Among them, the 1,3-dipolar cycloaddition reaction [25] is an efficient method to fuse five-membered rings on the periphery of the porphyrin framework. Because the periphery double bonds of the porphyrin macrocycle are nice dipolarophiles, and can trap 1,3-dipoles to furnish the chlorin or bacteriochlorin analogues [26-37]. On the other hand, the formed heterocycles are also very important. For example, isoxazoline derivatives are not only significant intermediates in organic synthesis, e.g., masked aldols [38,39], but also have a broad spectrum of interesting bioactivities, e.g., anti-inflammatory [40,41]. In addition, a few porphyrin-based dipoles have been also reported [42]. Recently, a 1,3-dipolar cycloaddition reaction was performed between the vinyl group at methyl pheophorbide A and the in-situ-generated nitrile oxide, which showed inferior selectivity and afforded regio-/stereoisomers [43]. NMR analysis assisted the identification of various isomeric isoxazoline-linked chlorin products. Here, we would like to report an artificial vinylporphyrin 2, which has been designed to control the regioselectivity and reaction trajectory of the 1,3-dipolar cycloaddition reaction by steric hindrance. The obtained novel isoxazoline-substituted porphyrin derivatives 3a,b have been characterized by absorption, emission, NMR and mass spectrometry. In addition, the crystal structure of 3a is discussed.
Results and Discussion
The synthetic route to the β-isoxazoline-substituted porphyrin is depicted in Scheme 1, in which the double bond was furnished by Wittig reaction, followed by 1,3-dipolar cycloaddition reaction with stable dipoles. In detail, tetraphenylporphyrin (TPP) was used as starting material. A Vilsmeier reaction was carried out after insertion of Cu2+ into the cavity of TPP. In the presence of concentrated H2SO4 the Cu2+ was removed to give the 2-formyl derivative TPP-CHO. Subsequently, the formyl group was reduced by NaBH4, accompanied with chlorination by SOCl2, to afford the chloromethyl derivative TPP-CH2Cl. Notably, this intermediate was labile on silica-gel column to give the precursor hydroxymethylporphyrin. Hence recrystallization was employed to purify it. Later, refluxing of the solution of TPP-CH2Cl and PPh3 in toluene gave the phosphonium salt 1 [44]. To introduce the vinyl group at the β-position, a Wittig reaction was performed, in which the porphyrin phosphonium salt 1 reacted with 4-MeCO2-benzaldehyde in the presence of DBU to furnish the double bond, followed by insertion of Zn ions into the porphyrin cavity to give compound 2. Nitrile oxides [45] as one of the most reactive dipoles not only react with various dipolarophiles but also undergo spontaneous dimerization to form furoxans. To avoid or minimize this drawback, the substitutents (e.g., Cl or Me) around the CNO group were introduced to stabilize the nitrile oxides even at room temperature [46,47]. Here, the 2,6-dichlorophenyl- or 2,4,6-trimethylphenyl nitrile oxides were used to react with 2 at 110 °C for 12 hours in toluene. Generally, two products with different positions (C4 or C5) of isoxazoline connected to the β-position of porphyrin should be obtained if the stereochemistry was not considered (Scheme 1) [43]. However, only one fraction was isolated in this reaction besides the unconsumed starting material 2.
![[1860-5397-15-143-i1]](/bjoc/content/inline/1860-5397-15-143-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route of β-isoxazoline linked porphyrin 3.
Scheme 1: Synthetic route of β-isoxazoline linked porphyrin 3.
The structure of 1,3-dipole cycloaddition products 3a,b were firstly characterized by absorption spectroscopy (Figure 1). The two compounds 3a,b showed approximately overlapped traces with typical intense Soret band and two weak Q bands. When compared to the spectrum of the precursor 2, a blue shift of ≈10 nm was observed in those of 3a or 3b, which may result from the loss of partial conjugation after the cycloaddition reaction between the nitrile oxides and C=C double bond. However, the substitution at the β-position in 3a or 3b led to a ≈5 nm red shift when compared with the absorption spectrum of ZnTPP [48]. In addition, the fluorescence spectra of 3a and 3b were comparable with that of ZnTPP, and again shifted to short wavelength (≈14 nm) with respect to that of 2. Later the ESIMS spectrum confirmed the constitution of two compounds, respectively. The pseudo molecular ions were observed at m/z = 1024.1765, demonstrating the molecular formula of C61H39Cl2N5O3Zn for 3a, and m/z = 998.3011, indicating the molecular formula of C64H47N5O3Zn for 3b. Thus, the formal formulas of 3a,b are the sum of porphyrin 2 and the corresponding nitrile oxides.
![[1860-5397-15-143-1]](/bjoc/content/figures/1860-5397-15-143-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Normalized UV–vis and emission spectra of β-isoxazoline porphyrins 3a,b, 2 and ZnTPP in CH2Cl2.
Figure 1: Normalized UV–vis and emission spectra of β-isoxazoline porphyrins 3a,b, 2 and ZnTPP in CH2Cl2.
Subsequently, the analysis of the 1H NMR spectra helped to establish the structure of compound 3a,b (Figure 2). In the 1H NMR spectrum of 2, the signals of the vinyl group were observed at 7.15 ppm (J = 16.0 Hz) and 7.28 ppm (J = 16.0 Hz, Figure 2, top), indicating a trans-configuration of the double bond. The signal of the CO2Me group occurred at 3.93 ppm, while those of the β-pyrrolic protons were in the range of 8.82 ppm to 9.13 ppm.
![[1860-5397-15-143-2]](/bjoc/content/figures/1860-5397-15-143-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The 1H NMR spectra of 2 (top, partially zoomed around 7.26 ppm), 3a (middle, red arrows indicate the high field shift of the protons after 1,3-diploar cycloaddition, while the blue one is the signal of aromatic protons of the dichlorophenyl group) and 3b (bottom, inset is the signal of CH3 around 2 ppm, x marks the signal of H2O, the blue one is the signal of aromatic protons on trimethylphenyl group) in CDCl3 (500 MHz, 25 °C).
Figure 2: The 1H NMR spectra of 2 (top, partially zoomed around 7.26 ppm), 3a (middle, red arrows indicate th...
As consequence of the cycloaddition reaction, the signals of the vinyl group vanished in the spectra of 3a or 3b. Taking the analysis of 3a as example. Two doublets at 5.07 ppm (J = 4.0 Hz) and 6.15 ppm (J = 4.0 Hz) aroused, which should be assigned to the two saturated CHs after dipolar cycloaddition reaction between the vinyl group and dichlorophenyl nitrile oxides. A multiplet was observed around 7.13 ppm and assigned to the aromatic protons of the 2,6-dichlorophenyl group. Meanwhile the aromatic signals of C6H4CO2Me shifted to the higher field when compared to that of 2, owing to the disturbing of the de-shielding effect from the double bond and porphyrin framework. Similar variation was also realized in the spectrum of 3b. Based on these, the cycloaddition reaction should take place on the β-vinyl group but not the double bond on the porphyrin framework, indicating the high activity of the β-vinyl group over the double bond on the macrocycle. However, at which position of the newly formed isoxazoline is linked to the porphyrin is still elusive.
Later, a single crystal of 3a was obtained by diffusion of n-hexane to the solution of 3a in CHCl3 at 23 °C. The structure of 3a was unambiguously established by single-crystal X-ray diffraction analysis. Compound 3a crystallized in the monoclinic with space group C2/c. Figure 3 shows the dimeric structure of 3a (front view and side view). Notably, only the 5-position of isoxazoline linking to porphyrin was observed. As expected, the steric clash of the substituent on nitrile oxide and porphyrin framework seems to confine the reaction trajectory and direct nitrile oxide to approach the double bond from outside the macrocycle. In addition, a pair of enantiomers is revealed which formed a dimeric fashion with the help of the fifth coordination to the Zn2+ by the carbonyl group from the other molecule. The distance of Zn–O is found to be 2.37 Å, which is longer than the distance when MeOH coordinated to the Zn2+ ions [49]. This may attribute to the larger volume of the C6H4CO2Me group than MeOH when closing to the Zn2+ center. In the dimer structure, two planes defined by porphyrin macrocycles are parallel with a distance of 7.11 Å. The bulky substituent group around the β-position, in conjunction with the presence of four benzene rings attached to the central porphyrin core, twist the overall geometry of the molecule. For instance, the porphyrin core is slightly distorted with deviation of 0.14 Å. The dihedral angles ranges from 2.6° to 8.7° for the four pyrrole rings with respect to the porphyrin mean plane, which are more twisted than that in normal Zn-porphyrin [49]. The lengths of Zn–N bonds range from 2.041 Å to 2.073 Å. The new formed five-membered isoxazoline ring is almost vertical (with a dihedral angle of 82.7°) to the mean plane of the porphyrin core. While the dihedral angle between the plane defined by macrocycle and the methoxycarbonylbenzene ring was found to be 40.5°.
![[1860-5397-15-143-3]](/bjoc/content/figures/1860-5397-15-143-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The representative dimeric structure of 3a according to the crystal structure (a) and enantiomeric dimer structure in crystal (b–d, the phenyl group at meso-positions and 2,6-dichlorophenyl group were omitted for the sake of clarity). The distances of Zn–O (c) and distance between the two mean planes of porphyrins (d) has been labeled.
Figure 3: The representative dimeric structure of 3a according to the crystal structure (a) and enantiomeric ...
Conclusion
In summary, two novel isoxazoline-substituted porphyrin derivatives 3a,b have been synthesized via the regiospecific and steric-oriented 1,3-dipolar cycloaddition reaction of vinylporphyrin 2 and two nitrile oxides, respectively. Only the product with a link between the 5-position of the isoxazoline and the β-position of porphyrin was obtained, directed by steric hindrance when the two components are approaching to each other. Owing to the interruption of the double bond after the cycloaddition reaction, the absorption spectrum shifted to shorter wavelengths. The isoxazoline-modified porphyrins have been identified by absorption, emission, 1H NMR and mass spectrometry, and 3a was further characterized by X-ray analysis. In the crystal, a pair of enantiomers of 3a is present, which assembled to a dimeric structure with the fifth chelation of a Zn2+ ion by the carbonyl group of the other molecule. The self-dimerization property of 3a may be utilized in supramolecular chemistry [50-53].
Experimental
1: Porphyrin phosphonium salt 1 was prepared according to the literature [44], the yield was 51%. 1H NMR (500 MHz, CDCl3) δ –2.77 (s, 2H, inner NH), 5.19–5.22 (d, J = 15.0 Hz, 2H, CH2P), 7.10–7.14 (m, 6H), 7.27–7.30 (m, 5H), 7.39 (d, J = 10.0 Hz, 2H), 7.40–7.89 (m, 18H), 8.17–8.19 (m, 4H), 8.31 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.46 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.76–8.86 (m, 5H, β-pyrrolic H).
2: To the solution of compound 1 (0.10 g, 0.11 mmol) and 4-methoxycarbonylbenzaldehyde (18 mg, 0.11 mmol) in CH2Cl2 (20 mL) was added DBU (83 mg, 0.55 mmol, 5.0 equiv vs 1). The reaction mixture was stirred for 8 hours. H2O (15 mL) was added to quench the reaction. The organic phase was separated and dried with MgSO4, followed by filtration and concentration to obtain a dark red residue. The product was isolated as first fraction by silica gel column chromatography with an eluent consisting of CH2Cl2/petrol ether 1:1 (v:v). Finally, the free-base porphyrin 2-H2 was obtained in a yield of 68 mg (82%). The obtained free-base porphyrin 2-H2 (50 mg, 0.065 mmol) was dissolved in CHCl3 (20 mL). Then the solution of Zn(OAc)2·2H2O (28.5 mg, 0.13 mmol, 2 equiv) in MeOH (6 mL). After 2 hours, TLC analysis showed the completion of the reaction. H2O was added to quench the reaction and the organic phase was collected. Then the organic phase was dried by MgSO4, followed by filtration and concentration under reduced pressure. The raw product was purified by silica gel column chromatography with eluent of CH2Cl2/petrol ether 3:1 (v:v). Finally, compound 2 was obtained in a yield of 92% (50.2 mg). 1H NMR (500 MHz, CDCl3) δ 3.93 (s, 3H, CO2CH3), 7.15 (d, J = 16.0 Hz, 1H, CH=CH), 7.28 (d, J = 16.0 Hz, 1H, CH=CH), 7.30 (d, J = 8.0 Hz, 2H), 7.75–7.85 (m, 12H), 7.97 (d, J = 8.0 Hz, 2H), 8.18–8.22 (m, 6H), 8.24–8.26 (m, 2H), 8.83 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.90 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.91–8.92 (m, 3H, β-pyrrolic H), 8.94 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 9.13 (s, 1H, β-pyrrolic H); ESIMS m/z: [M + H]+ calcd for C54H39N4O2ZnF, 837.2; found, 837.1; UV–vis (CH2Cl2) λmax: 432, 561, 597 nm.
3a: A solution of 2 (100 mg, 0.12 mmol) and nitrile oxide (0.6 mmol, 5 equiv) in dry toluene (20 mL) was heated to reflux for 6 hours under N2. Subsequently, addition of two portions of 2,6-dichlorophenyl nitrile oxide [45] (0.3 mmol) were added to the reaction mixture every 3 hours. The reaction mixture was concentrated to dryness under reduced pressure and the product was isolated by silica gel column chromatography with eluent of DCM/ethyl acetate 50:1 (v/v). The second purple fraction was collected. Finally, 3a was obtained in a yield of 48% (59 mg) as purple powder. 1H NMR (500 MHz, CDCl3) δ 3.95 (s, 3H, CO2CH3), 5.07 (d, J = 4.0 Hz, 1H, CH), 6.15 (d, J = 4.0 Hz, 1H, CH), 6.79 (t, J = 6.0 Hz, 1H), 7.05 (d, J = 8.0 Hz, 2H), 7.12–7.15 (m, 3H), 7.44 (t, J = 6.0 Hz, 1H), 7.67 (t, J = 6.0 Hz, 1H), 7.76–7.77 (m, 10H), 7.91 (d, J = 8.0 Hz, 2H), 8.17–8.22 (m, 7H), 8.63 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.85 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.92–8.95 (m, 4H, β-pyrrolic H), 9.30 (s, 1H, β-pyrrolic H). ESIMS m/z: [M + H]+ calcd for C61H40Cl2N5O3Zn, 1024.1794; found, 1024.1765; UV–vis (CH2Cl2) λmax: 403, 424, 552, 592 nm. Some crystals of C61H39Cl2N5O3Zn were recrystallized from CHCl3/n-hexane. A suitable crystal was selected and mounted on a Rigaku Saturn CCD area detector diffractometer. The crystal was kept at 113 K during data collection. Using Olex2 [54], the structure was solved with the XS [55] structure solution program using Direct Methods and refined with the olex2.refine [56] refinement package using Gauss–Newton minimisation. Crystal data for C61H39Cl2N5O3Zn (M = 1026.32 g/mol): monoclinic, space group C2/c (no. 15), a = 28.166(6) Å, b = 17.330(4) Å, c = 26.363(5) Å, β = 117.96(3)°, V = 11366(5) Å3, Z = 8, T = 113 K, μ(Mo Kα) = 0.573 mm−1, Dcalc = 1.1995 g/cm3, 40330 reflections measured (4.7° ≤ 2Θ ≤ 50.04°), 9924 unique (Rint = 0.0578, Rsigma = 0.0601) which were used in all calculations. The final R1 was 0.0861 (I>=2u(I)) and wR2 was 0.2556 (all data). The CCDC number of the crystal of 3a is 1910675, which can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
3b: Through the same procedure as that for 3a, besides using 2,4,6-trimethylphenyl nitrile oxide [45], compound 3b was obtained in a yield of 30% (36 mg). 1H NMR (500 MHz, CDCl3) δ 1.40 (s, 6H, CH3), 2.09 (s, 3H, CH3), 3.98 (s, 3H, CO2CH3), 4.42 (s, 1H, C-H), 6.29 (s, 1H, CH), 6.52 (s, 2H), 6.62 (t, J = 6.0 Hz, 1H), 6.95 (d, J = 7.5 Hz, 2H), 7.55 (t, J = 6.0 Hz, 1H), 7.69–7.77 (m, 10H), 7.82 (d, J = 7.0 Hz, 2H), 7.91(d, J = 7.5 Hz, 2H), 8.00 (d, J = 7.0 Hz, 1H), 8.56 (d, J = 7.0 Hz, 1H), 8.16–8.22 (m, 5H), 8.56 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.84 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 8.90–8.92 (m, 3H, β-pyrrolic H), 8.95 (d, J = 4.0 Hz, 1H, β-pyrrolic H), 9.19 (s, 1H, β-pyrrolic H); ESIMS m/z: [M + H]+ calcd for C64H48N5O3Zn, 998.3043; found, 998.3011; UV–vis (CH2Cl2) λmax: 402, 424, 552, 595 nm.
ZnTPP: The Zn-complex of TPP was prepared according to the literature [48], the yield was 89%. 1H NMR (500 MHz, CDCl3) δ 7.73–7.80 (m, 12H), 8.23 (dd, J = 7.5/1.4 Hz, 8H), 8.95 (s, 8H, β-pyrrolic H).
Supporting Information
| Supporting Information File 1: X-ray data of compound 3a. | ||
| Format: CIF | Size: 719.2 KB | Download |
References
-
Battersby, A. R. Nat. Prod. Rep. 2000, 17, 507–526. doi:10.1039/b002635m
Return to citation in text: [1] -
Kraeutler, B. Chimia 1987, 41, 277–292. doi:10.2524/jtappij.41.3_292
Return to citation in text: [1] -
Eschenmoser, A. Angew. Chem., Int. Ed. Engl. 1988, 27, 5–39. doi:10.1002/anie.198800051
Return to citation in text: [1] -
Severance, S.; Hamza, I. Chem. Rev. 2009, 109, 4596–4616. doi:10.1021/cr9001116
Return to citation in text: [1] -
Maxwell, K.; Johnson, G. N. J. Exp. Bot. 2000, 51, 659–668. doi:10.1093/jxb/51.345.659
Return to citation in text: [1] -
Ding, Y.; Zhu, W.-H.; Xie, Y. Chem. Rev. 2017, 117, 2203–2256. doi:10.1021/acs.chemrev.6b00021
Return to citation in text: [1] -
Zhang, W.; Lai, W.; Cao, R. Chem. Rev. 2017, 117, 3717–3797. doi:10.1021/acs.chemrev.6b00299
Return to citation in text: [1] -
Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; Di Natale, C. Chem. Rev. 2017, 117, 2517–2583. doi:10.1021/acs.chemrev.6b00361
Return to citation in text: [1] -
Baglia, R. A.; Zaragoza, J. P. T.; Goldberg, D. P. Chem. Rev. 2017, 117, 13320–13352. doi:10.1021/acs.chemrev.7b00180
Return to citation in text: [1] -
Lu, H.; Kobayashi, N. Chem. Rev. 2016, 116, 6184–6261. doi:10.1021/acs.chemrev.5b00588
Return to citation in text: [1] -
Rajora, M. A.; Lou, J. W. H.; Zheng, G. Chem. Soc. Rev. 2017, 46, 6433–6469. doi:10.1039/c7cs00525c
Return to citation in text: [1] -
Thompson, S. J.; Brennan, M. R.; Lee, S. Y.; Dong, G. Chem. Soc. Rev. 2018, 47, 929–981. doi:10.1039/c7cs00582b
Return to citation in text: [1] -
Tanaka, T.; Osuka, A. Chem. Soc. Rev. 2015, 44, 943–969. doi:10.1039/c3cs60443h
Return to citation in text: [1] -
Li, L.-L.; Diau, E. W.-G. Chem. Soc. Rev. 2013, 42, 291–304. doi:10.1039/c2cs35257e
Return to citation in text: [1] -
Szyszko, B.; Latos-Grażyński, L. Chem. Soc. Rev. 2015, 44, 3588–3616. doi:10.1039/c4cs00398e
Return to citation in text: [1] -
Chatterjee, T.; Shetti, V. S.; Sharma, R.; Ravikanth, M. Chem. Rev. 2017, 117, 3254–3328. doi:10.1021/acs.chemrev.6b00496
Return to citation in text: [1] -
Hiroto, S.; Miyake, Y.; Shinokubo, H. Chem. Rev. 2017, 117, 2910–3043. doi:10.1021/acs.chemrev.6b00427
Return to citation in text: [1] -
Lash, T. D. Chem. Rev. 2017, 117, 2313–2446. doi:10.1021/acs.chemrev.6b00326
Return to citation in text: [1] -
Matano, Y. Chem. Rev. 2017, 117, 3138–3191. doi:10.1021/acs.chemrev.6b00460
Return to citation in text: [1] -
Sarma, T.; Panda, P. K. Chem. Rev. 2017, 117, 2785–2838. doi:10.1021/acs.chemrev.6b00411
Return to citation in text: [1] -
Anguera, G.; Sánchez-García, D. Chem. Rev. 2017, 117, 2481–2516. doi:10.1021/acs.chemrev.6b00345
Return to citation in text: [1] -
Tanaka, T.; Osuka, A. Chem. Rev. 2017, 117, 2584–2640. doi:10.1021/acs.chemrev.6b00371
Return to citation in text: [1] -
Taniguchi, M.; Lindsey, J. S. Chem. Rev. 2017, 117, 344–535. doi:10.1021/acs.chemrev.5b00696
Return to citation in text: [1] -
Li, Q.; Li, C.; Kim, J.; Ishida, M.; Li, X.; Gu, T.; Liang, X.; Zhu, W.; Ågren, H.; Kim, D.; Furuta, H.; Xie, Y. J. Am. Chem. Soc. 2019, 141, 5294–5302. doi:10.1021/jacs.8b13148
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651
Return to citation in text: [1] -
Gałȩzowski, M.; Gryko, D. T. J. Org. Chem. 2006, 71, 5942–5950. doi:10.1021/jo060545x
Return to citation in text: [1] -
Liu, X.; Feng, Y.; Hu, X.; Li, X. Synthesis 2005, 3632–3638. doi:10.1055/s-2005-918415
Return to citation in text: [1] -
Liu, X.; Feng, Y.; Chen, X.; Li, F.; Li, X. Synlett 2005, 1030–1032. doi:10.1055/s-2005-864811
Return to citation in text: [1] -
Li, X.; Zhuang, J.; Li, Y.; Liu, H.; Wang, S.; Zhu, D. Tetrahedron Lett. 2005, 46, 1555–1559. doi:10.1016/j.tetlet.2004.12.138
Return to citation in text: [1] -
Almeida, J.; Aguiar, A.; Leite, A.; Silva, A. M. N.; Cunha-Silva, L.; de Castro, B.; Rangel, M.; Barone, G.; Tomé, A. C.; Silva, A. M. G. Org. Chem. Front. 2017, 4, 534–544. doi:10.1039/c6qo00771f
Return to citation in text: [1] -
Wyrębek, P.; Mikus, A.; Ostrowski, S. Heterocycles 2012, 85, 57–64. doi:10.3987/com-11-12347
Return to citation in text: [1] -
Tomé, A. C.; Neves, M. G. P. M. S.; Cavaleiro, J. A. S. J. Porphyrins Phthalocyanines 2009, 13, 408–414. doi:10.1142/s1088424609000619
Return to citation in text: [1] -
Ostrowski, S.; Wyrebek, P.; Mikus, A. Heterocycles 2006, 68, 885–888. doi:10.3987/com-06-10691
Return to citation in text: [1] -
Silva, A. M. G.; Tomé, A. C.; Neves, M. G. P. M. S.; Silva, A. M. S.; Cavaleiro, J. A. S.; Perrone, D.; Dondoni, A. Tetrahedron Lett. 2002, 43, 603–605. doi:10.1016/s0040-4039(01)02243-2
Return to citation in text: [1] -
Silva, A. M. G.; Tomé,, A. C.; Neves, M. G. P. M. S.; Silva, A. M. S.; Cavaleiro, J. A. S. J. Org. Chem. 2005, 70, 2306–2314. doi:10.1021/jo048349i
Return to citation in text: [1] -
Séverac, M.; Pleux, L. L.; Scarpaci, A.; Blart, E.; Odobel, F. Tetrahedron Lett. 2007, 48, 6518–6522. doi:10.1016/j.tetlet.2007.07.049
Return to citation in text: [1] -
Flemming, J.; Dolphin, D. Tetrahedron Lett. 2002, 43, 7281–7283. doi:10.1016/s0040-4039(02)01619-2
Return to citation in text: [1] -
Jäger, V.; Grund, H.; Buß, V.; Schwab, W.; Müller, I.; Schohe, R.; Franz, R.; Ehrler, R. Bull. Soc. Chim. Belg. 1983, 92, 1039–1054. doi:10.1002/bscb.19830921113
Return to citation in text: [1] -
Evans, D. A.; Ripin, D. H. B.; Halstead, D. P.; Campos, K. R. J. Am. Chem. Soc. 1999, 121, 6816–6826. doi:10.1021/ja990789h
Return to citation in text: [1] -
Habeeb, A. G.; Praveen Rao, P. N.; Knaus, E. E. J. Med. Chem. 2001, 44, 2921–2927. doi:10.1021/jm0101287
Return to citation in text: [1] -
Habeeb, A. G.; Rao, P. N. P.; Knaus, E. E. Drug Dev. Res. 2000, 51, 273–286. doi:10.1002/ddr.9
Return to citation in text: [1] -
da Silva, A. F. F.; Barata, J. F. B.; Silva, A. M. G.; Neves, M. G. P. M. S.; Tomé, A. C.; Silva, A. M. S.; Cavaleiro, J. A. S. Tetrahedron Lett. 2015, 56, 2878–2881. doi:10.1016/j.tetlet.2015.04.066
Return to citation in text: [1] -
Wang, J.-J.; Li, J.-Z.; Li, Y.-W.; Jakus, J.; Shim, Y. K. J. Porphyrins Phthalocyanines 2010, 14, 859–865. doi:10.1142/s1088424610002690
Return to citation in text: [1] [2] -
Bonfantini, E. E.; Officer, D. L. Tetrahedron Lett. 1993, 34, 8531–8534. doi:10.1016/s0040-4039(00)61377-1
Return to citation in text: [1] [2] -
Grundmann, C. Angew. Chem., Int. Ed. Engl. 1963, 2, 260–261. doi:10.1002/anie.196302601
Return to citation in text: [1] [2] [3] -
Grundmann, C.; Dean, J. M. J. Org. Chem. 1965, 30, 2809–2812. doi:10.1021/jo01019a074
Return to citation in text: [1] -
Grundmann, C.; Dean, J. M. Angew. Chem., Int. Ed. Engl. 1964, 3, 585–586. doi:10.1002/anie.196405852
Return to citation in text: [1] -
Vail, S. A.; Schuster, D. I.; Guldi, D. M.; Isosomppi, M.; Tkachenko, N.; Lemmetyinen, H.; Palkar, A.; Echegoyen, L.; Chen, X.; Zhang, J. Z. H. J. Phys. Chem. B 2006, 110, 14155–14166. doi:10.1021/jp061844t
Return to citation in text: [1] [2] -
Banala, S.; Wurst, K.; Kräutler, B. J. Porphyrins Phthalocyanines 2014, 18, 115–122. doi:10.1142/s1088424613501204
Return to citation in text: [1] [2] -
Zhang, W.; Li, G.; Xu, L.; Zhuo, Y.; Wan, W.; Yan, N.; He, G. Chem. Sci. 2018, 9, 4444–4450. doi:10.1039/c8sc00688a
Return to citation in text: [1] -
Jiang, T.; Wang, X.; Wang, J.; Hu, G.; Ma, X. ACS Appl. Mater. Interfaces 2019, 11, 14399–14407. doi:10.1021/acsami.9b03112
Return to citation in text: [1] -
Xu, C.; Xu, L.; Ma, X. Chin. Chem. Lett. 2018, 29, 970–972. doi:10.1016/j.cclet.2017.11.045
Return to citation in text: [1] -
Li, D.; Wang, J.; Ma, X. Adv. Opt. Mater. 2018, 6, 1800273. doi:10.1002/adom.201800273
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1] -
Bourhis, L. J.; Dolomanov, O. V.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 59–75. doi:10.1107/s2053273314022207
Return to citation in text: [1]
| 25. | Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 48. | Vail, S. A.; Schuster, D. I.; Guldi, D. M.; Isosomppi, M.; Tkachenko, N.; Lemmetyinen, H.; Palkar, A.; Echegoyen, L.; Chen, X.; Zhang, J. Z. H. J. Phys. Chem. B 2006, 110, 14155–14166. doi:10.1021/jp061844t |
| 15. | Szyszko, B.; Latos-Grażyński, L. Chem. Soc. Rev. 2015, 44, 3588–3616. doi:10.1039/c4cs00398e |
| 16. | Chatterjee, T.; Shetti, V. S.; Sharma, R.; Ravikanth, M. Chem. Rev. 2017, 117, 3254–3328. doi:10.1021/acs.chemrev.6b00496 |
| 17. | Hiroto, S.; Miyake, Y.; Shinokubo, H. Chem. Rev. 2017, 117, 2910–3043. doi:10.1021/acs.chemrev.6b00427 |
| 18. | Lash, T. D. Chem. Rev. 2017, 117, 2313–2446. doi:10.1021/acs.chemrev.6b00326 |
| 19. | Matano, Y. Chem. Rev. 2017, 117, 3138–3191. doi:10.1021/acs.chemrev.6b00460 |
| 20. | Sarma, T.; Panda, P. K. Chem. Rev. 2017, 117, 2785–2838. doi:10.1021/acs.chemrev.6b00411 |
| 21. | Anguera, G.; Sánchez-García, D. Chem. Rev. 2017, 117, 2481–2516. doi:10.1021/acs.chemrev.6b00345 |
| 22. | Tanaka, T.; Osuka, A. Chem. Rev. 2017, 117, 2584–2640. doi:10.1021/acs.chemrev.6b00371 |
| 23. | Taniguchi, M.; Lindsey, J. S. Chem. Rev. 2017, 117, 344–535. doi:10.1021/acs.chemrev.5b00696 |
| 24. | Li, Q.; Li, C.; Kim, J.; Ishida, M.; Li, X.; Gu, T.; Liang, X.; Zhu, W.; Ågren, H.; Kim, D.; Furuta, H.; Xie, Y. J. Am. Chem. Soc. 2019, 141, 5294–5302. doi:10.1021/jacs.8b13148 |
| 49. | Banala, S.; Wurst, K.; Kräutler, B. J. Porphyrins Phthalocyanines 2014, 18, 115–122. doi:10.1142/s1088424613501204 |
| 6. | Ding, Y.; Zhu, W.-H.; Xie, Y. Chem. Rev. 2017, 117, 2203–2256. doi:10.1021/acs.chemrev.6b00021 |
| 7. | Zhang, W.; Lai, W.; Cao, R. Chem. Rev. 2017, 117, 3717–3797. doi:10.1021/acs.chemrev.6b00299 |
| 8. | Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; Di Natale, C. Chem. Rev. 2017, 117, 2517–2583. doi:10.1021/acs.chemrev.6b00361 |
| 9. | Baglia, R. A.; Zaragoza, J. P. T.; Goldberg, D. P. Chem. Rev. 2017, 117, 13320–13352. doi:10.1021/acs.chemrev.7b00180 |
| 10. | Lu, H.; Kobayashi, N. Chem. Rev. 2016, 116, 6184–6261. doi:10.1021/acs.chemrev.5b00588 |
| 11. | Rajora, M. A.; Lou, J. W. H.; Zheng, G. Chem. Soc. Rev. 2017, 46, 6433–6469. doi:10.1039/c7cs00525c |
| 12. | Thompson, S. J.; Brennan, M. R.; Lee, S. Y.; Dong, G. Chem. Soc. Rev. 2018, 47, 929–981. doi:10.1039/c7cs00582b |
| 13. | Tanaka, T.; Osuka, A. Chem. Soc. Rev. 2015, 44, 943–969. doi:10.1039/c3cs60443h |
| 14. | Li, L.-L.; Diau, E. W.-G. Chem. Soc. Rev. 2013, 42, 291–304. doi:10.1039/c2cs35257e |
| 46. | Grundmann, C.; Dean, J. M. J. Org. Chem. 1965, 30, 2809–2812. doi:10.1021/jo01019a074 |
| 47. | Grundmann, C.; Dean, J. M. Angew. Chem., Int. Ed. Engl. 1964, 3, 585–586. doi:10.1002/anie.196405852 |
| 2. | Kraeutler, B. Chimia 1987, 41, 277–292. doi:10.2524/jtappij.41.3_292 |
| 3. | Eschenmoser, A. Angew. Chem., Int. Ed. Engl. 1988, 27, 5–39. doi:10.1002/anie.198800051 |
| 4. | Severance, S.; Hamza, I. Chem. Rev. 2009, 109, 4596–4616. doi:10.1021/cr9001116 |
| 5. | Maxwell, K.; Johnson, G. N. J. Exp. Bot. 2000, 51, 659–668. doi:10.1093/jxb/51.345.659 |
| 43. | Wang, J.-J.; Li, J.-Z.; Li, Y.-W.; Jakus, J.; Shim, Y. K. J. Porphyrins Phthalocyanines 2010, 14, 859–865. doi:10.1142/s1088424610002690 |
| 42. | da Silva, A. F. F.; Barata, J. F. B.; Silva, A. M. G.; Neves, M. G. P. M. S.; Tomé, A. C.; Silva, A. M. S.; Cavaleiro, J. A. S. Tetrahedron Lett. 2015, 56, 2878–2881. doi:10.1016/j.tetlet.2015.04.066 |
| 44. | Bonfantini, E. E.; Officer, D. L. Tetrahedron Lett. 1993, 34, 8531–8534. doi:10.1016/s0040-4039(00)61377-1 |
| 40. | Habeeb, A. G.; Praveen Rao, P. N.; Knaus, E. E. J. Med. Chem. 2001, 44, 2921–2927. doi:10.1021/jm0101287 |
| 41. | Habeeb, A. G.; Rao, P. N. P.; Knaus, E. E. Drug Dev. Res. 2000, 51, 273–286. doi:10.1002/ddr.9 |
| 45. | Grundmann, C. Angew. Chem., Int. Ed. Engl. 1963, 2, 260–261. doi:10.1002/anie.196302601 |
| 38. | Jäger, V.; Grund, H.; Buß, V.; Schwab, W.; Müller, I.; Schohe, R.; Franz, R.; Ehrler, R. Bull. Soc. Chim. Belg. 1983, 92, 1039–1054. doi:10.1002/bscb.19830921113 |
| 39. | Evans, D. A.; Ripin, D. H. B.; Halstead, D. P.; Campos, K. R. J. Am. Chem. Soc. 1999, 121, 6816–6826. doi:10.1021/ja990789h |
| 26. | Gałȩzowski, M.; Gryko, D. T. J. Org. Chem. 2006, 71, 5942–5950. doi:10.1021/jo060545x |
| 27. | Liu, X.; Feng, Y.; Hu, X.; Li, X. Synthesis 2005, 3632–3638. doi:10.1055/s-2005-918415 |
| 28. | Liu, X.; Feng, Y.; Chen, X.; Li, F.; Li, X. Synlett 2005, 1030–1032. doi:10.1055/s-2005-864811 |
| 29. | Li, X.; Zhuang, J.; Li, Y.; Liu, H.; Wang, S.; Zhu, D. Tetrahedron Lett. 2005, 46, 1555–1559. doi:10.1016/j.tetlet.2004.12.138 |
| 30. | Almeida, J.; Aguiar, A.; Leite, A.; Silva, A. M. N.; Cunha-Silva, L.; de Castro, B.; Rangel, M.; Barone, G.; Tomé, A. C.; Silva, A. M. G. Org. Chem. Front. 2017, 4, 534–544. doi:10.1039/c6qo00771f |
| 31. | Wyrębek, P.; Mikus, A.; Ostrowski, S. Heterocycles 2012, 85, 57–64. doi:10.3987/com-11-12347 |
| 32. | Tomé, A. C.; Neves, M. G. P. M. S.; Cavaleiro, J. A. S. J. Porphyrins Phthalocyanines 2009, 13, 408–414. doi:10.1142/s1088424609000619 |
| 33. | Ostrowski, S.; Wyrebek, P.; Mikus, A. Heterocycles 2006, 68, 885–888. doi:10.3987/com-06-10691 |
| 34. | Silva, A. M. G.; Tomé, A. C.; Neves, M. G. P. M. S.; Silva, A. M. S.; Cavaleiro, J. A. S.; Perrone, D.; Dondoni, A. Tetrahedron Lett. 2002, 43, 603–605. doi:10.1016/s0040-4039(01)02243-2 |
| 35. | Silva, A. M. G.; Tomé,, A. C.; Neves, M. G. P. M. S.; Silva, A. M. S.; Cavaleiro, J. A. S. J. Org. Chem. 2005, 70, 2306–2314. doi:10.1021/jo048349i |
| 36. | Séverac, M.; Pleux, L. L.; Scarpaci, A.; Blart, E.; Odobel, F. Tetrahedron Lett. 2007, 48, 6518–6522. doi:10.1016/j.tetlet.2007.07.049 |
| 37. | Flemming, J.; Dolphin, D. Tetrahedron Lett. 2002, 43, 7281–7283. doi:10.1016/s0040-4039(02)01619-2 |
| 43. | Wang, J.-J.; Li, J.-Z.; Li, Y.-W.; Jakus, J.; Shim, Y. K. J. Porphyrins Phthalocyanines 2010, 14, 859–865. doi:10.1142/s1088424610002690 |
| 44. | Bonfantini, E. E.; Officer, D. L. Tetrahedron Lett. 1993, 34, 8531–8534. doi:10.1016/s0040-4039(00)61377-1 |
| 49. | Banala, S.; Wurst, K.; Kräutler, B. J. Porphyrins Phthalocyanines 2014, 18, 115–122. doi:10.1142/s1088424613501204 |
| 50. | Zhang, W.; Li, G.; Xu, L.; Zhuo, Y.; Wan, W.; Yan, N.; He, G. Chem. Sci. 2018, 9, 4444–4450. doi:10.1039/c8sc00688a |
| 51. | Jiang, T.; Wang, X.; Wang, J.; Hu, G.; Ma, X. ACS Appl. Mater. Interfaces 2019, 11, 14399–14407. doi:10.1021/acsami.9b03112 |
| 52. | Xu, C.; Xu, L.; Ma, X. Chin. Chem. Lett. 2018, 29, 970–972. doi:10.1016/j.cclet.2017.11.045 |
| 53. | Li, D.; Wang, J.; Ma, X. Adv. Opt. Mater. 2018, 6, 1800273. doi:10.1002/adom.201800273 |
| 45. | Grundmann, C. Angew. Chem., Int. Ed. Engl. 1963, 2, 260–261. doi:10.1002/anie.196302601 |
| 48. | Vail, S. A.; Schuster, D. I.; Guldi, D. M.; Isosomppi, M.; Tkachenko, N.; Lemmetyinen, H.; Palkar, A.; Echegoyen, L.; Chen, X.; Zhang, J. Z. H. J. Phys. Chem. B 2006, 110, 14155–14166. doi:10.1021/jp061844t |
| 55. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 56. | Bourhis, L. J.; Dolomanov, O. V.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 59–75. doi:10.1107/s2053273314022207 |
| 45. | Grundmann, C. Angew. Chem., Int. Ed. Engl. 1963, 2, 260–261. doi:10.1002/anie.196302601 |
| 54. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
© 2019 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)