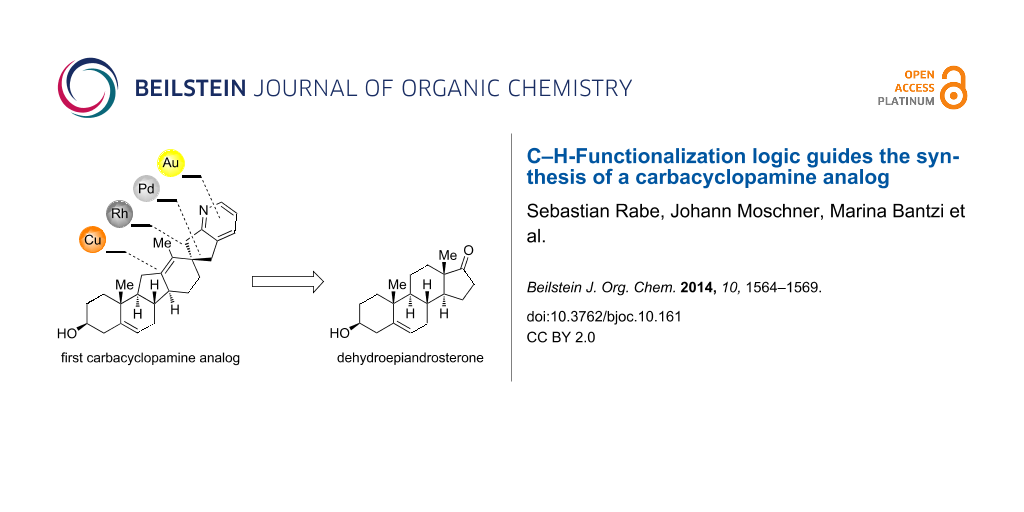
Abstract
The chemical synthesis of carbacyclopamine analog 2, a cyclopamine analog with an all-carbon E-ring, is reported. The use of C–H-functionalization logic and further metal-catalyzed transformations allows for a concise entry to this new class of acid-stable cyclopamine analogs.

Graphical Abstract
Introduction
The isolation of cyclopamine (1, Figure 1) nearly 50 years ago followed by the determination of its biological target and pharmacological profile has drawn considerable interest from research groups in biology, chemistry and medicine [1-4]. As the first identified hedgehog signaling inhibitor, cyclopamine exerts its anticancer activity through a novel mechanism of action, which manifests itself in its remarkable activity against several types of human cancer, including medulloblastoma, basal cell carcinoma, and rhabdomyosarcoma [5-10]. The impressive biological activity combined with a unique molecular architecture and the synthetic challenge it poses already let to the first synthesis of cyclopamine by this laboratory employing C–H-functionalization logic [11]. In addition, the rational design and chemical synthesis of several analogs and derivatives by this [12,13] and other groups [14-17] have been disclosed. Here, we report the synthesis of a carbacyclopamine analog (2, Figure 1), an analog of the natural product with an all-carbon E-ring and a pyridine F-ring.
![[1860-5397-10-161-1]](/bjoc/content/figures/1860-5397-10-161-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of cyclopamine (1) and carbacyclopamine analog 2.
Figure 1: Structures of cyclopamine (1) and carbacyclopamine analog 2.
Cyclopamine inhibits the 7-transmembrane protein smoothened from converting into its active form [18,19]. Without active smoothened, protein kinases like PKA, GSK3β, and CK1ε phosphorylate the transcription factors Gli2 and Gli3, thereby creating binding sites for the adapter protein β-TrCP [20]. The Gli family of transcription factors acts as an effector of the hedgehog signaling pathway and thus, is associated with a wide array of physiological effects, including cell fate determination, proliferation and patterning. The so-formed Gli/β-TrCP complex becomes subject to ubiquitinylation, mediated by the Cul1-based E3 ligase. Eventually, this results in partial proteosomal degradation to form Gli3-R [21], or in the case of Gli2 [22,23], in complete degradation. In addition, the Gli3-R factor acts as a transcription inhibitor of hedgehog-response genes [10].
As part of our continuing work on acid-stable cyclopamine analogs [12,13] we have now focused on the role of the allylic ether oxygen in the acid-mediated E-ring cleavage and decomposition of cyclopamine [24]. We envisioned that its replacement by a methylene group would create an acid-stable cyclopamine analog that still exhibits similar inhibitory activity on hedgehog-signaling. For the sake of brevity of the overall synthetic sequence we defined carbacyclopamine analog 2 (see Figure 1) as our primary target.
Results and Discussion
A retrosynthetic analysis identified diazo compound 3 as a key intermediate in the synthesis of 2 (see Scheme 1). We envisioned a rhodium-catalyzed C–H-insertion into the C17–H bond to occur with a high degree of selectivity (both regio- and stereoselectivity) to form the all-carbon E-ring (for its structure see 11, Scheme 2). Furthermore, a Wagner–Meerwein rearrangement was thought to establish the C-nor-D-homo steroid system, and a gold-catalyzed amination/annulation/aromatization sequence was planned to install the pyridine F-ring. Diazo compound 3 originates from diene 4 through standard transformations, the latter being accessible from commercially available and inexpensive dehydroepiandrosterone (5) by the means of a copper-mediated C–H hydroxylation in the 12-position and a palladium-catalyzed coupling of methyl acrylate to an activated enol ether in the 17-position.
![[1860-5397-10-161-i1]](/bjoc/content/inline/1860-5397-10-161-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of carbacyclopamine analog 2.
Scheme 1: Retrosynthetic analysis of carbacyclopamine analog 2.
![[1860-5397-10-161-i2]](/bjoc/content/inline/1860-5397-10-161-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of carbacyclopamine analog 2.
Scheme 2: Synthesis of carbacyclopamine analog 2.
In synthetic direction (Scheme 2), dehydroepiandrosterone (5) was protected as its tetrahydropyranyl ether (3,4-dihydro-2H-pyran, cat. pyridinium para-toluenesulfonate, CH2Cl2, 25 °C, quantiative, inconsequential mixture of diastereoisomers), and the C17 carbonyl group was transformed into its 2-picolylimine (2-picolylamine, cat. para-toluenesulfonic acid, toluene, 111 °C, 92% yield). Subjecting the latter to modified Schönecker conditions ([Cu(MeCN)4]PF6, O2, acetone, 25 °C) [25-27], gave, after acidic work-up (AcOH/MeOH, 1:1, 90 °C), diol 6 in 46% yield as the only isolable product. Protection of the newly installed 12β-hydroxy group as a triethylsilyl ether via the bis-protected intermediate (not shown, triethylsilyl trifluoromethanesulfonate, 2,6-lutidine, CH2Cl2, 0 °C; then HF·pyridine, THF, 0 °C, 67% yield for the two steps) [28] gave hydroxy ketone 7.
The remaining homoallylic alcohol was then masked as an i-steroid [29,30] (para-toluenesulfonyl chloride, cat. 4-dimethylaminopyridine, pyridine, 25 °C; then KOAc, MeOH, 64 °C, 85% yield for the two steps) to give 8. Generation of an enol triflate from the carbonyl group in 8 (potassium hexamethyldisilazide, phenyl bis-triflimide, THF, −20 → −10 °C, 85% yield) set stage for a Heck-reaction with methyl acrylate (cat. Pd(OAc)2, cat. PPh3, Et3N, DMF, 70 °C, 64% yield). The hydrogenation of the so-obtained diene 4 (for its structure see Scheme 1) proceeded smoothly (H2, cat. Pd/C, MeOH, 25 °C, 82% yield) under concomitant removal of the triethylsilyl ether [31] in 12-position to give methyl ester 9 as a single diastereoisomer. At this point, extensive experimentation suggested a change of protecting groups to enable the pending C–H insertion reaction at C17.
Therefore, the i-steroid was reverted to the homoallylic alcohol (cat. para-toluenesulfonic acid, 1,4-dioxane/H2O, 10:1, 65 °C), the methyl ester was hydrolyzed under basic conditions (LiOH, THF/H2O, 1:1, 68% yield for the two steps), and the alcohol moieties were protected as formyl esters (formic acid, 50 °C, 85% yield) to give key intermediate 10.
Employing the formyl protecting groups [32], diazoketone 3 (for its structure see Scheme 1) was readily obtained from acid 10 via the corresponding acid chloride (oxalyl chloride, CH2Cl2, 25 °C; then diazomethane, THF, 25 °C, 85% yield for the two steps) [33]. The anticipated C–H insertion then proceeded uneventfully under oxygen-free conditions (cat. Rh2(OAc)4, CH2Cl2, 41 °C) [34,35] to give cyclopentanone 11 in 52% yield as a single isomer. Removal of the formyl ester protecting groups (LiOH, THF/H2O, 1:1, 30 °C, 94% yield) and selective protection of the 3-hydroxy group as a tert-butyldimethylsilyl ether (tert-butyldimethylsilyl chloride, imidazole, DMF, 25 °C, 95% yield) furnished alcohol 12.
Treatment of the latter under our previously reported conditions [36] (Comins’ reagent, 4-dimethylaminopyridine, toluene, 111 °C) gave an inseparable mixture of the rearranged olefin products, with the desired endo-product 13 as the major constituent (89% combined yield, endo-product 13:exo-product 14, 4:1). Isolation of desired product 13 by chromatography was then achieved after selective hydrogenation of only the exocyclic olefin in 14 (H2, cat. Rh/C, EtOAc, 25 °C, structure of hydrogenation product not shown) employing the mixture of the isomers 13 and 14 from the previous step.
Finally, a gold-catalyzed amination/annulation/aromatization sequence (propargylamine, cat. Na[AuCl4]·2H2O, EtOH, 100 °C) [37,38] furnished, after removal of the tert-butyldimethylsilyl ether (tetrabutylammonium fluoride, THF, 25 °C), carbacyclopamine analog 2 in 36% overall yield for the two steps [39].
Carbacyclopamine analog 2 was then tested for its stability towards acid. Therefore, 2 was treated with a mixture of aqueous HCl/THF (pH of approx. 0.3) for 1 h and 1H NMR spectra were recorded before and afterwards. While at this pH natural cyclopamine (1) was shown to decompose rapidly [12], carbacyclopamine analog 2 showed no signs of decomposition. This result was in full agreement with our initial considerations.
To determine the ability of carbacyclopamine analog 2 to inhibit Gli1-dependent luciferase expression, we employed Shh-LIGHTII cells, a clonal mouse fibroblast cell line which stably incorporates a Gli-dependent firefly luciferase reporter and a constitutive Renilla luciferase reporter [18]. The analog was tested in a concentration range from 0.01 μM to 100 μM but showed no activity (data not shown).
Conclusion
We have reported the synthesis of a new, acid stable cyclopamine analog. The implementation of C–H-functionalization logic in the synthetic planning and further metal-catalyzed transformations allows for a fast access to carbocyclopamine analog 2. At the same time, these investigations strongly suggest the role the ether oxygen plays in the acid-catalyzed decomposition of cyclopamine. This study may find further application in the rational design of new hedgehog inhibitors based on lead structure 2. Future work will focus on the synthesis of carbacyclopamine analogs with a piperidine F-ring and their biological investigation.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for new compounds. | ||
| Format: PDF | Size: 7.2 MB | Download |
References
-
Keeler, R. F. Phytochemistry 1968, 7, 303–306. doi:10.1016/S0031-9422(00)86328-1
Return to citation in text: [1] -
Keeler, R. F. Teratology 1970, 3, 169–174. doi:10.1002/tera.1420030209
Return to citation in text: [1] -
Beachy, P. A.; Karhadkar, S. S.; Berman, D. M. Nature 2004, 432, 324–331. doi:10.1038/nature03100
Return to citation in text: [1] -
Taipale, J.; Beachy, P. A. Nature 2001, 411, 349–354. doi:10.1038/35077219
Return to citation in text: [1] -
Hidalgo, M.; Maitra, A. N. Engl. J. Med. 2009, 361, 2094–2096. doi:10.1056/NEJMcibr0905857
Return to citation in text: [1] -
Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944–951. doi:10.1038/nm1614
Return to citation in text: [1] -
Hedge, G. V.; Munger, C. M.; Emanuel, K.; Joshi, A. D.; Greiner, T. C.; Weisenburger, D. D.; Vose, J. M.; Joshi, S. S. Mol. Cancer Ther. 2008, 7, 1450–1460. doi:10.1158/1535-7163.MCT-07-2118
Return to citation in text: [1] -
Kawahara, T.; Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Anticancer Res. 2009, 29, 4629–4632.
Return to citation in text: [1] -
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Angew. Chem., Int. Ed. 2010, 49, 3418–3427. doi:10.1002/anie.200906967
Return to citation in text: [1] -
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Bioorg. Med. Chem. 2010, 18, 6613–6624. doi:10.1016/j.bmc.2010.07.038
And references cited therein.
Return to citation in text: [1] [2] -
Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem., Int. Ed. 2009, 48, 7911–7914. doi:10.1002/anie.200902520
Return to citation in text: [1] -
Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362–7364. doi:10.1039/C1CC11782C
Return to citation in text: [1] [2] [3] -
Moschner, J.; Chentsova, A.; Eilert, N.; Rovardi, I.; Heretsch, P.; Giannis, A. Beilstein J. Org. Chem. 2013, 9, 2328–2335. doi:10.3762/bjoc.9.267
Return to citation in text: [1] [2] -
Tremblay, M. R.; Nevalainen, M.; Nair, S. J.; Porter, J. R.; Castro, A. C.; Behnke, M. L.; Yu, L.-C.; Hagel, M.; White, K.; Faia, K.; Grenier, L.; Campbell, M. J.; Cushing, J.; Woodward, C. N.; Hoyt, J.; Foley, M. A.; Read, M. A.; Sydor, J. R.; Tong, J. K.; Palombella, V. J.; McGovern, K.; Adams, J. J. Med. Chem. 2008, 51, 6646–6649. doi:10.1021/jm8008508
Return to citation in text: [1] -
Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.; Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.; Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.; Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400–4418. doi:10.1021/jm900305z
Return to citation in text: [1] -
Zhang, Z.; Baubet, V.; Ventocilla, C.; Xiang, C.; Dahmane, N.; Winkler, J. D. Org. Lett. 2011, 13, 4786–4789. doi:10.1021/ol2017966
Return to citation in text: [1] -
Winkler, J. D.; Isaacs, A.; Holderbaum, L.; Tatard, V.; Dahmane, N. Org. Lett. 2009, 11, 2824–2827. doi:10.1021/ol900974u
Return to citation in text: [1] -
Taipale, J.; Chen, J. K.; Cooper, M. K.; Wang, B.; Mann, R. K.; Milenkovic, L.; Scott, M. P.; Beachy, P. A. Nature 2000, 406, 1005–1009. doi:10.1038/35023008
Return to citation in text: [1] [2] -
Wang, Y.; Zhou, Z.; Walsh, C. T.; McMahon, A. P. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2623–2628. doi:10.1073/pnas.0812110106
Return to citation in text: [1] -
Pearse, R. V., II; Collier, L.S.; Scott, M. P.; Tabin, C. J. Dev. Biol. 1999, 212, 323–336. doi:10.1006/dbio.1999.9335
Return to citation in text: [1] -
Wen, X.; Lai, C. K.; Evangelista, M.; Hongo, J.-A.; de Sauvage, F. J.; Scales, S. J. Mol. Cell. Biol. 2010, 30, 1910–1922. doi:10.1128/MCB.01089-09
Return to citation in text: [1] -
Pan, Y.; Bai, C. B.; Joyner, A. L.; Wang, B. Mol. Cell. Biol. 2006, 26, 3365–3377. doi:10.1128/MCB.26.9.3365-3377.2006
Return to citation in text: [1] -
Wang, B.; Fallon, J. F.; Beachy, P. A. Cell 2000, 100, 423–434. doi:10.1016/S0092-8674(00)80678-9
Return to citation in text: [1] -
Wilson, S. R.; Strand, M. F.; Krapp, A.; Rise, F.; Petersen, D.; Krauss, S. J. Pharm. Biomed. Anal. 2010, 52, 707–713. doi:10.1016/j.jpba.2010.02.017
See for a proposed mechanism of the decomposition of cyclopamine. For a discussion of the molecular orbitals involved in the process, see ref. [12.]
Return to citation in text: [1] -
Schönecker, B.; Zheldakova, T.; Lange, C.; Günther, W.; Görls, H.; Bohl, M. Chem.–Eur. J. 2004, 10, 6029–6042. doi:10.1002/chem.200306054
Return to citation in text: [1] -
Schönecker, B.; Zheldakova, T.; Liu, Y.; Kötteritzsch, M.; Günther, W.; Görls, H. Angew. Chem., Int. Ed. 2003, 42, 3240–3244. doi:10.1002/anie.200250815
Return to citation in text: [1] -
Schönecker, B.; Lange, C.; Zheldakova, T.; Günther, W.; Görls, H.; Vaughan, G. Tetrahedron 2005, 61, 103–114. doi:10.1016/j.tet.2004.10.055
Return to citation in text: [1] -
Ojima, I.; Chen, J.; Sun, L.; Borella, C. P.; Wang, T.; Miller, M. L.; Lin, S.; Geng, X.; Kuznetsova, L.; Qu, C.; Gallager, D.; Zhao, X.; Zanardi, I.; Xia, S.; Horwitz, S. B.; Mallen-St. Clair, J.; Guerriero, J. L.; Bar-Sagi, D.; Veith, J. M.; Pera, P.; Bernacki, R. J. J. Med. Chem. 2008, 51, 3203–3221. doi:10.1021/jm800086e
Return to citation in text: [1] -
Fernholz, E.; Ruigh, W. L. J. Am. Chem. Soc. 1940, 62, 3346–3348. doi:10.1021/ja01869a022
Return to citation in text: [1] -
Wallis, E. S.; Fernholz, E.; Gephart, F. T. J. Am. Chem. Soc. 1937, 59, 137–140. doi:10.1021/ja01280a034
Return to citation in text: [1] -
Rotulo-Sims, D.; Prunet, J. Org. Lett. 2002, 4, 4701–4704. doi:10.1021/ol0271382
Return to citation in text: [1] -
The formyl groups were chosen both for the ease of their installation, and a considerably lower sterical demand in the pending cyclization event when compared to other protective groups. Using triethylsilyl ethers or allyl ethers instead did not allow for the generation of the desired diazo-compound. The use of acetyl protecting groups led to the formation of seven-membered lactone 25 (for experimental details see Supporting Information File 1).
Return to citation in text: [1] -
A similar reaction employing diazoethane instead of diazomethane gave the methyl homolog of compound 10. All attempts to effect the C–H insertion were met with failure, though (see Supporting Information File 1 for experimental details).
Return to citation in text: [1] -
Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o
Return to citation in text: [1] -
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n
Return to citation in text: [1] -
Heretsch, P.; Rabe, S.; Giannis, A. J. Am. Chem. Soc. 2010, 132, 9968–9969. doi:10.1021/ja103152k
Return to citation in text: [1] -
Abbiati, G.; Arcadi, A.; Bianchi, G.; Di Giuseppe, S.; Marinelli, F.; Rossi, E. J. Org. Chem. 2003, 68, 6959–6966. doi:10.1021/jo0347260
Return to citation in text: [1] -
The regioisomeric cyclization product (theoretically obtainable by fusion with the C20 methylene) was not detected in the crude reaction mixture.
Return to citation in text: [1] -
After chromatographic purification the final product was obtained in ca. 90% purity (judged by 1H NMR, see Supporting Information File 1). We were not able to determine the structures of the impurities or completely remove them. The yield reported takes this into account; the biological assay and the stability assay were run with this material.
Return to citation in text: [1]
| 34. | Nakamura, E.; Yoshikai, N.; Yamanaka, M. J. Am. Chem. Soc. 2002, 124, 7181–7192. doi:10.1021/ja017823o |
| 35. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 32. | The formyl groups were chosen both for the ease of their installation, and a considerably lower sterical demand in the pending cyclization event when compared to other protective groups. Using triethylsilyl ethers or allyl ethers instead did not allow for the generation of the desired diazo-compound. The use of acetyl protecting groups led to the formation of seven-membered lactone 25 (for experimental details see Supporting Information File 1). |
| 33. | A similar reaction employing diazoethane instead of diazomethane gave the methyl homolog of compound 10. All attempts to effect the C–H insertion were met with failure, though (see Supporting Information File 1 for experimental details). |
| 1. | Keeler, R. F. Phytochemistry 1968, 7, 303–306. doi:10.1016/S0031-9422(00)86328-1 |
| 2. | Keeler, R. F. Teratology 1970, 3, 169–174. doi:10.1002/tera.1420030209 |
| 3. | Beachy, P. A.; Karhadkar, S. S.; Berman, D. M. Nature 2004, 432, 324–331. doi:10.1038/nature03100 |
| 4. | Taipale, J.; Beachy, P. A. Nature 2001, 411, 349–354. doi:10.1038/35077219 |
| 14. | Tremblay, M. R.; Nevalainen, M.; Nair, S. J.; Porter, J. R.; Castro, A. C.; Behnke, M. L.; Yu, L.-C.; Hagel, M.; White, K.; Faia, K.; Grenier, L.; Campbell, M. J.; Cushing, J.; Woodward, C. N.; Hoyt, J.; Foley, M. A.; Read, M. A.; Sydor, J. R.; Tong, J. K.; Palombella, V. J.; McGovern, K.; Adams, J. J. Med. Chem. 2008, 51, 6646–6649. doi:10.1021/jm8008508 |
| 15. | Tremblay, M. R.; Lescarbeau, A.; Grogan, M. J.; Tan, E.; Lin, G.; Austad, B. C.; Yu, L.-C.; Behnke, M. L.; Nair, S. J.; Hagel, M.; White, K.; Conley, J.; Manna, J. D.; Alvarez-Diez, T. M.; Hoyt, J.; Woodward, C. N.; Sydor, J. R.; Pink, M.; MacDougall, J.; Campbell, M. J.; Cushing, J.; Ferguson, J.; Curtis, M. S.; McGovern, K.; Read, M. A.; Palombella, V. J.; Adams, J.; Castro, A. C. J. Med. Chem. 2009, 52, 4400–4418. doi:10.1021/jm900305z |
| 16. | Zhang, Z.; Baubet, V.; Ventocilla, C.; Xiang, C.; Dahmane, N.; Winkler, J. D. Org. Lett. 2011, 13, 4786–4789. doi:10.1021/ol2017966 |
| 17. | Winkler, J. D.; Isaacs, A.; Holderbaum, L.; Tatard, V.; Dahmane, N. Org. Lett. 2009, 11, 2824–2827. doi:10.1021/ol900974u |
| 29. | Fernholz, E.; Ruigh, W. L. J. Am. Chem. Soc. 1940, 62, 3346–3348. doi:10.1021/ja01869a022 |
| 30. | Wallis, E. S.; Fernholz, E.; Gephart, F. T. J. Am. Chem. Soc. 1937, 59, 137–140. doi:10.1021/ja01280a034 |
| 12. | Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362–7364. doi:10.1039/C1CC11782C |
| 13. | Moschner, J.; Chentsova, A.; Eilert, N.; Rovardi, I.; Heretsch, P.; Giannis, A. Beilstein J. Org. Chem. 2013, 9, 2328–2335. doi:10.3762/bjoc.9.267 |
| 31. | Rotulo-Sims, D.; Prunet, J. Org. Lett. 2002, 4, 4701–4704. doi:10.1021/ol0271382 |
| 11. | Giannis, A.; Heretsch, P.; Sarli, V.; Stößel, A. Angew. Chem., Int. Ed. 2009, 48, 7911–7914. doi:10.1002/anie.200902520 |
| 25. | Schönecker, B.; Zheldakova, T.; Lange, C.; Günther, W.; Görls, H.; Bohl, M. Chem.–Eur. J. 2004, 10, 6029–6042. doi:10.1002/chem.200306054 |
| 26. | Schönecker, B.; Zheldakova, T.; Liu, Y.; Kötteritzsch, M.; Günther, W.; Görls, H. Angew. Chem., Int. Ed. 2003, 42, 3240–3244. doi:10.1002/anie.200250815 |
| 27. | Schönecker, B.; Lange, C.; Zheldakova, T.; Günther, W.; Görls, H.; Vaughan, G. Tetrahedron 2005, 61, 103–114. doi:10.1016/j.tet.2004.10.055 |
| 18. | Taipale, J.; Chen, J. K.; Cooper, M. K.; Wang, B.; Mann, R. K.; Milenkovic, L.; Scott, M. P.; Beachy, P. A. Nature 2000, 406, 1005–1009. doi:10.1038/35023008 |
| 5. | Hidalgo, M.; Maitra, A. N. Engl. J. Med. 2009, 361, 2094–2096. doi:10.1056/NEJMcibr0905857 |
| 6. | Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N. P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J. F.; Schultz, P.; Warmuth, M. Nat. Med. 2007, 13, 944–951. doi:10.1038/nm1614 |
| 7. | Hedge, G. V.; Munger, C. M.; Emanuel, K.; Joshi, A. D.; Greiner, T. C.; Weisenburger, D. D.; Vose, J. M.; Joshi, S. S. Mol. Cancer Ther. 2008, 7, 1450–1460. doi:10.1158/1535-7163.MCT-07-2118 |
| 8. | Kawahara, T.; Kawaguchi-Ihara, N.; Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Anticancer Res. 2009, 29, 4629–4632. |
| 9. | Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Angew. Chem., Int. Ed. 2010, 49, 3418–3427. doi:10.1002/anie.200906967 |
| 10. |
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Bioorg. Med. Chem. 2010, 18, 6613–6624. doi:10.1016/j.bmc.2010.07.038
And references cited therein. |
| 28. | Ojima, I.; Chen, J.; Sun, L.; Borella, C. P.; Wang, T.; Miller, M. L.; Lin, S.; Geng, X.; Kuznetsova, L.; Qu, C.; Gallager, D.; Zhao, X.; Zanardi, I.; Xia, S.; Horwitz, S. B.; Mallen-St. Clair, J.; Guerriero, J. L.; Bar-Sagi, D.; Veith, J. M.; Pera, P.; Bernacki, R. J. J. Med. Chem. 2008, 51, 3203–3221. doi:10.1021/jm800086e |
| 22. | Pan, Y.; Bai, C. B.; Joyner, A. L.; Wang, B. Mol. Cell. Biol. 2006, 26, 3365–3377. doi:10.1128/MCB.26.9.3365-3377.2006 |
| 23. | Wang, B.; Fallon, J. F.; Beachy, P. A. Cell 2000, 100, 423–434. doi:10.1016/S0092-8674(00)80678-9 |
| 12. | Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362–7364. doi:10.1039/C1CC11782C |
| 13. | Moschner, J.; Chentsova, A.; Eilert, N.; Rovardi, I.; Heretsch, P.; Giannis, A. Beilstein J. Org. Chem. 2013, 9, 2328–2335. doi:10.3762/bjoc.9.267 |
| 39. | After chromatographic purification the final product was obtained in ca. 90% purity (judged by 1H NMR, see Supporting Information File 1). We were not able to determine the structures of the impurities or completely remove them. The yield reported takes this into account; the biological assay and the stability assay were run with this material. |
| 21. | Wen, X.; Lai, C. K.; Evangelista, M.; Hongo, J.-A.; de Sauvage, F. J.; Scales, S. J. Mol. Cell. Biol. 2010, 30, 1910–1922. doi:10.1128/MCB.01089-09 |
| 24. |
Wilson, S. R.; Strand, M. F.; Krapp, A.; Rise, F.; Petersen, D.; Krauss, S. J. Pharm. Biomed. Anal. 2010, 52, 707–713. doi:10.1016/j.jpba.2010.02.017
See for a proposed mechanism of the decomposition of cyclopamine. For a discussion of the molecular orbitals involved in the process, see ref. [12.] |
| 12. | Heretsch, P.; Büttner, A.; Tzagkaroulaki, L.; Zahn, S.; Kirchner, B.; Giannis, A. Chem. Commun. 2011, 47, 7362–7364. doi:10.1039/C1CC11782C |
| 20. | Pearse, R. V., II; Collier, L.S.; Scott, M. P.; Tabin, C. J. Dev. Biol. 1999, 212, 323–336. doi:10.1006/dbio.1999.9335 |
| 36. | Heretsch, P.; Rabe, S.; Giannis, A. J. Am. Chem. Soc. 2010, 132, 9968–9969. doi:10.1021/ja103152k |
| 18. | Taipale, J.; Chen, J. K.; Cooper, M. K.; Wang, B.; Mann, R. K.; Milenkovic, L.; Scott, M. P.; Beachy, P. A. Nature 2000, 406, 1005–1009. doi:10.1038/35023008 |
| 19. | Wang, Y.; Zhou, Z.; Walsh, C. T.; McMahon, A. P. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2623–2628. doi:10.1073/pnas.0812110106 |
| 10. |
Heretsch, P.; Tzagkaroulaki, L.; Giannis, A. Bioorg. Med. Chem. 2010, 18, 6613–6624. doi:10.1016/j.bmc.2010.07.038
And references cited therein. |
| 37. | Abbiati, G.; Arcadi, A.; Bianchi, G.; Di Giuseppe, S.; Marinelli, F.; Rossi, E. J. Org. Chem. 2003, 68, 6959–6966. doi:10.1021/jo0347260 |
| 38. | The regioisomeric cyclization product (theoretically obtainable by fusion with the C20 methylene) was not detected in the crude reaction mixture. |
© 2014 Rabe et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)