Abstract
Aromatic π–π stacking interactions are ubiquitous in nature, medicinal chemistry and materials sciences. They play a crucial role in the stacking of nucleobases, thus stabilising the DNA double helix. The following paper describes a series of chimeric DNA–polycyclic aromatic hydrocarbon (PAH) hybrids. The PAH building blocks are electron-rich pyrene and electron-poor perylenediimide (PDI), and were incorporated into complementary DNA strands. The hybrids contain different numbers of pyrene–PDI interactions that were found to directly influence duplex stability. As the pyrene–PDI ratio approaches 1:1, the stability of the duplexes increases with an average value of 7.5 °C per pyrene–PDI supramolecular interaction indicating the importance of electrostatic complementarity for aromatic π–π stacking interactions.
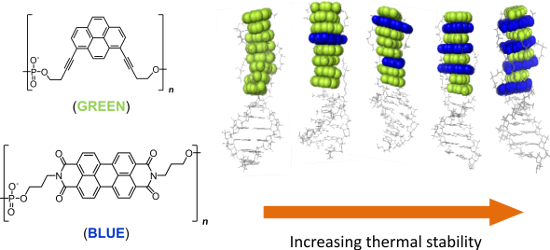
Graphical Abstract
Introduction
When two aromatic molecules are in close proximity they often have a tendency to interact non-covalently in a face-to-face stacking arrangement. Face-centered, parallel aromatic π–π stacking interactions have been studied and reviewed in great detail [1-5]. These interactions are especially important for polycyclic aromatic hydrocarbons (PAHs) [6,7]. The interaction is the result of solvophobicity, as well as van der Waals, electrostatic and charge transfer interactions that can lead to a thermodynamically favourable association [8]. It is an important interaction in biological systems, drug receptor interactions, materials sciences, and supramolecular chemistry [8-12]. Such interactions are strongly dependent on the electron density and distribution of the partners [2,9,13-16]. In particular, the interaction between electron-rich (donor) and electron-deficient (acceptor) aromatic rings results in stable aggregates [17-22].
In the DNA duplex, the interaction of the two complementary strands is governed mainly by aromatic π–π stacking interactions, hydrogen bonds, and electrostatic repulsion from the negatively-charged sugar phosphate backbone [10,23-28]. DNA can be regarded as an amphiphilic polymer in which aromatic residues are linked by negatively charged phosphodiester groups [29]. The importance of aromatic and hydrophobic factors for duplex stability was demonstrated by replacing the natural nucleobases by size expanded analogs [30-35].
A classic example of polymeric donor–acceptor complexes are the aedamers (aromatic electron donor acceptor oligomers) pioneered by Iverson and coworkers [18,36,37]. They consist of face-to-face stacked electron-rich naphthalene and electron-poor naphthalenediimide (NDI) chromophores and belong to the broader area of foldamers [38].
DNA has been described as a molecular scaffold for arranging various types of chromophores [39-44]. Recently, we reported that oligoarenotides (oligomers with an alternating phosphodiester-aromatic hydrocarbon motif) exhibit similar structural properties as nucleic acids, and although the aromatic hydrocarbons cannot engage in any sort of Watson–Crick related hydrogen bonding, the individual strands interact via an interstrand stacking motif [45-48]. Herein we describe a series of DNA-based hybrids (Figure 1 and Table 1) containing electron-rich 1,8-dialkynylpyrenes (Y) and electron-poor perylenediimides (PDI, E).
![[1860-5397-10-164-1]](/bjoc/content/figures/1860-5397-10-164-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (A) Structures of 1,8-dialkynylpyrene (Y) and PDI (E); (B) illustration of the electrostatic potential surface of 1,8-diprop-1-ynylpyrene (left) and N,N’-dimethyl-PDI (right); (C) illustration of duplex formation with chimeric oligomers; (D) hybrids 1*2 to 1*6. The number of pyrene–PDI interactions increases from left to right.
Figure 1: (A) Structures of 1,8-dialkynylpyrene (Y) and PDI (E); (B) illustration of the electrostatic potent...
Table 1: Tm values of the hybrids determined by thermal denaturation experiments.a
| Sequence | Tm (°C) | Number of pyrene–PDI interactions | |
|---|---|---|---|
| Ref |
5‘ GCGTTA
3‘ CGCAAT |
13.0 | |
|
1
2 |
5‘ GCGTTA YYYY
3‘ CGCAAT YYYY |
50.5 | 0 |
|
1
3 |
5‘ GCGTTA YYYY
3‘ CGCAAT YYEY |
54.5 | 2 |
|
1
4 |
5‘ GCGTTA YYYY
3‘ CGCAAT YEYE |
58.5 | 4 |
|
1
5 |
5‘ GCGTTA YYYY
3‘ CGCAAT YEEE |
61.0 | 6 |
|
1
6 |
5‘ GCGTTA YYYY
3‘ CGCAAT EEEE |
64.5 | 7 |
|
7
2 |
5‘ GCGTTA EEEE
3‘ CGCAAT YYYY |
66.5 | 7 |
|
7
6 |
5‘ GCGTTA EEEE
3‘ CGCAAT EEEE |
52.0 | 0 |
|
1
7 |
5‘ GCGTTA YYYY
5‘ GCGTTA EEEE |
–b | n/a |
|
2
6 |
3‘ CGCAAT YYYY
3‘ CGCAAT EEEE |
–b | n/a |
aConditions: oligomer conc. 2.5 μM single strand, 10 mM sodium phosphate buffer, pH 7.2, 0.1 M NaCl, absorption monitored at 260 nm; error ±0.5 °C; bno transition observed (see Supporting Information File 1).
PDIs (Figure 1A) are some of the most widely studied organic chromophores [49-52]. Moreover, we have reported on the aggregation and stacking properties of 1,8- and 1,6-dialkynylpyrene [53,54]. Figure 1B shows the electrostatic potential surface of 1,8-diprop-1-ynylpyrene and N,N’-dimethyl-PDI. The former is considerably more electron-rich/higher electron density (red) than the latter, which is expected to favour an alternating aromatic π–π stacking arrangement of E and Y due to electrostatic complementarity.
We show herein that duplex formation by our chimeric DNA-oligoarenotide strands proceeds in a selective manner, the chromophores on opposite strands interdigitate and stack face-to-face in an organised controlled assembly.
Results and Discussion
The principle of the system is illustrated in Figure 1. All oligomers are composed of a DNA part and a modified section containing a total of four PDIs (blue) and/or pyrenes (green). Oligomers 1–7 consisting of varying numbers of pyrene or PDI moieties covalently linked to complementary DNA strands were prepared by automated oligonucleotide synthesis using the previously described phosphoramidite pyrene [53] and PDI [55] building blocks.
The DNA stem acts as a supramolecular scaffold, and together with the flexible, negatively-charged phosphate linker allows the chromphores to adopt optimal conformations in aqueous solution and increases the solubility. The strands were hybridised in various combinations (Table 1), and their stability and photophysical properties were investigated. Since the DNA duplex is identical in all hybrids, differences in stability must originate from the modified section. The sequence of the modified part is changed in such a way that annealing of different strands leads to a varying number of pyrene–PDI stacking interactions. Strand 1 is common to all hybrids. The complementary strands 2–6 possess an increasing number of PDIs. Thus, in the resultant hybrids, the number of pyrene–PDI face-to-face stacking interactions also increases steadily from left to right, e.g., duplex 1*2 contains only pyrene–pyrene interactions, whereas duplex 1*6 has the maximum number pyrene–PDI interactions.
Thermal denaturation experiments
Thermal denaturation experiments revealed a clear trend in duplex stability (Figure 2). The thermal stability correlates with the number of pyrene–PDI interactions [56] and increases linearly in the series. The melting temperature (Tm) values are summarized in Table 1.
![[1860-5397-10-164-2]](/bjoc/content/figures/1860-5397-10-164-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A plot of melting temperature (Tm) versus the number of pyrene–PDI interactions for duplexes 1*2 to 1*6 (from left to right) presented in Table 1. The Tm was recorded at 260 nm; R2 = 0.987. The red triangle represents the Tm of the control hybrid 7*6.
Figure 2: A plot of melting temperature (Tm) versus the number of pyrene–PDI interactions for duplexes 1*2 to ...
Hybrid 1*2 has a Tm of 50.5 °C which is 37.5 °C higher than the reference DNA duplex (Tm = 13 °C). Since hybrid 1*2 has seven pyrene–pyrene interactions, one of these interactions (ΔTm/(Y−Y)) contributes ≈ 5.4 °C to hybrid stability. Likewise, a value for ΔTm/(E−E) = 5.6 °C is calculated for hybrid 7*6. The average contribution of a pyrene–PDI interaction can be calculated from the Tm difference (Tm = 51.5 °C) between 1*6 and the reference duplex. A value of ΔTm/(Y−E) = 7.4 °C is obtained in this way. Hybrid 7*2 serves as a further control. In this duplex, the DNA and the modified parts of the two strands have been interchanged relative to 1*6. The Tm value of 7*2 is in the same range as 1*6 (66.5 and 64.5 °C, respectively), which translates into ΔTm/(Y−E) = 7.7 °C. Thus, the average contributions to the hybrid stabilities are as follows: ΔTm/(Y−E) ≈ 7.5 °C, whereas ΔTm/(Y−Y) and ΔTm/(E−E) ≈ 5.5 °C.
The results of electrostatic complementarity between an electron-rich pyrene and an electron-poor PDI is highlighted by the fact that duplexes with only pyrene or PDI are considerably less stable (Table 1, hybrids 1*2 and 7*6) than hybrids containing both types of aromatic compounds. Although the actual stability of such duplexes strongly depends on several parameters like, e.g., the geometry of the building blocks and the flexibility of the linkers, a general trend can be deduced from the thermal denaturation results that accounts for the above mentioned design of building blocks and sequences.
UV–vis absorption spectroscopy
The stacking interactions of Y and E in the hybrids could be followed by UV–vis absorption spectroscopy. A significant change in the vibronic band ratio supports the model of an alternating interstrand interdigitation interaction between pyrene and PDI chromophores as proposed in Figure 1. In general, the vibronic band ratio of PAHs gives valuable information on the aggregation state of the molecules [57]. In a stack of only pyrenes (1*2) the vibronic band at 370 nm is higher than that at 390 nm (Figure 3), indicating that the pyrenes are stacked parallel and face-to-face. In contrast, in duplex 1*6 the intensity of the vibronic band 390 nm is higher indicating that the pyrenes are separated from each other by intercalating PDIs [58]. The same absorption behaviour is seen for the PDI vibronic band intensities.
![[1860-5397-10-164-3]](/bjoc/content/figures/1860-5397-10-164-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis absorption spectra (scaled) of duplexes 1*2 (blue) and 1*6 (red) at 20 °C. Conditions: see Table 1.
Figure 3: UV–vis absorption spectra (scaled) of duplexes 1*2 (blue) and 1*6 (red) at 20 °C. Conditions: see Table 1.
Figure 4 focuses on the vibronic bands of pyrene’s S0→S1 absorption band in duplexes 1*2 to 1*6. An increasing number of PDIs in a stack leads to a stronger vibronic band at 390 nm.
![[1860-5397-10-164-4]](/bjoc/content/figures/1860-5397-10-164-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis absorption spectra (scaled) of duplexes 1*2 to 1*6 at 20 °C. Conditions: see Table 1.
Figure 4: UV–vis absorption spectra (scaled) of duplexes 1*2 to 1*6 at 20 °C. Conditions: see Table 1.
This is in stark contrast to the effect of thermally denaturing duplex 1*2 into two single strands (Figure 5). There, the vibronic band at 370 nm is always the highest indicating that the pyrenes are stacked even at 90 °C in the single strands. Such behaviour was also observed in chrysene-modified DNA [59].
![[1860-5397-10-164-5]](/bjoc/content/figures/1860-5397-10-164-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Temperature-dependent UV–vis absorption spectrum of 1*2. Conditions: see Table 1.
Figure 5: Temperature-dependent UV–vis absorption spectrum of 1*2. Conditions: see Table 1.
Fluorescence spectroscopy
The interaction of two or more dialkynylpyrenes (Y) results in a pronounced excimer fluorescence [53]. Hybridization of single strands 1 and 2 increases the intensity of the excimer (Figure 6), whereas hybridization of single strands 1 and 6 results in an extinction of excimer fluorescence. Such behaviour was also observed in previous work and was explained by an alternating interdigitation interaction of the pyrene with the PDI building blocks [60].
![[1860-5397-10-164-6]](/bjoc/content/figures/1860-5397-10-164-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fluorescence spectra of oligomer 1 (black), duplex 1*2 (blue) and duplex 1*6 (red) at 20 °C. Excitation: 370 nm. Conditions: see Table 1.
Figure 6: Fluorescence spectra of oligomer 1 (black), duplex 1*2 (blue) and duplex 1*6 (red) at 20 °C. Excita...
Gel migration experiments
The electrophoretic mobility of relatively small (<1000 kbp), linear DNA strands is inversely proportional to their molecular weight [61]. It serves as a reliable method to demonstrate the formation of double versus single stranded DNA structure. Therefore, the formation of defined short duplexes has been further investigated using polyacrylamide gel electrophoresis (PAGE) experiments. Oligomer single strands 1, 6 and 7 migrate with the same velocity as the 13 bp reference (Figure 7). Strands 1 and 6, however, form a duplex and thus have lower electrophoretic mobility, similar to an 18–20 bp reference. Oligomer 7 has the same DNA sequence as 6, but with 4 PDI molecules instead of 4 pyrenes (see Table 1). Thus when combined, single strands 1 and 7 do not form a duplex due to having non-complementary DNA parts, and migrate on the gel like single strands 1, 6, and 7. These results underline the importance of the complementary DNA segments in aligning the pyrene and PDI chromophores for optimal interaction.
![[1860-5397-10-164-7]](/bjoc/content/figures/1860-5397-10-164-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: PAGE experiment. All oligomers were used in a total amount of 150 pmol in 10 mM sodium phosphate buffer, 100 mM NaCl and 10% loading buffer, 20% polyacrylamide gel with a 10% loading gel, 1 h 40 min, 4 °C, 170 V, 6 mA, 2 W. Left lane: DNA ladder.
Figure 7: PAGE experiment. All oligomers were used in a total amount of 150 pmol in 10 mM sodium phosphate bu...
Conclusion
A series of DNA oligonucleotides functionalised with electron-poor perylenediimide (PDI, E) and electron-rich 1,8-dialkynylpyrene (Y) chromophores has been synthesized and their photophysical and thermal melting properties were investigated. UV–vis absorption and fluorescence spectra indicate an alternate, face-to-face, stacking of PDI and pyrene moieties. The DNA portion serves as an ideal scaffold to align the pyrene and PDI chromophores and to study their interaction in solution. The stability of the duplexes shows a clear dependence on the number of pyrene–PDI interactions within the duplex. As the pyrene–PDI ratio approaches 1:1, the stability of the duplexes increases with up to 7.5 °C per pyrene–PDI supramolecular interaction underlining the importance of electrostatic complementarity for aromatic π–π stacking interactions.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and supplementary spectroscopic data. | ||
| Format: PDF | Size: 961.2 KB | Download |
References
-
Hunter, C. A.; Sanders, J. K. M. J. Am. Chem. Soc. 1990, 112, 5525–5534. doi:10.1021/ja00170a016
Return to citation in text: [1] -
Hunter, C. A.; Lawson, K. R.; Perkins, J.; Urch, C. J. J. Chem. Soc., Perkin Trans. 2 2001, 651–669. doi:10.1039/b008495f
Return to citation in text: [1] [2] -
Salonen, L. M.; Ellermann, M.; Diederich, F. Angew. Chem., Int. Ed. 2011, 50, 4808–4842. doi:10.1002/anie.201007560
Return to citation in text: [1] -
Grimme, S. Angew. Chem., Int. Ed. 2008, 47, 3430–3434. doi:10.1002/anie.200705157
Return to citation in text: [1] -
Martinez, C. R.; Iverson, B. L. Chem. Sci. 2012, 3, 2191–2201. doi:10.1039/c2sc20045g
Return to citation in text: [1] -
Watson, M. D.; Fechtenkötter, A.; Müllen, K. Chem. Rev. 2001, 101, 1267–1300. doi:10.1021/cr990322p
Return to citation in text: [1] -
Feng, C.; Lin, C. S.; Fan, W.; Zhang, R. Q.; Van Hove, M. A. J. Chem. Phys. 2009, 131, 194702. doi:10.1063/1.3251785
Return to citation in text: [1] -
Hunter, C. A. Chem. Soc. Rev. 1994, 23, 101–109. doi:10.1039/cs9942300101
Return to citation in text: [1] [2] -
Meyer, E. A.; Castellano, R. K.; Diederich, F. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. doi:10.1002/anie.200390319
Return to citation in text: [1] [2] -
Hunter, C. A. J. Mol. Biol. 1993, 230, 1025–1054. doi:10.1006/jmbi.1993.1217
Return to citation in text: [1] [2] -
Philp, D.; Stoddart, J. F. Angew. Chem., Int. Ed. Engl. 1996, 35, 1154–1196. doi:10.1002/anie.199611541
Return to citation in text: [1] -
Sakai, N.; Matile, S. Beilstein J. Org. Chem. 2012, 8, 897–904. doi:10.3762/bjoc.8.102
Return to citation in text: [1] -
Cockroft, S. L.; Perkins, J.; Zonta, C.; Adams, H.; Spey, S. E.; Low, C. M. R.; Vinter, J. G.; Lawson, K. R.; Urch, C. J.; Hunter, C. A. Org. Biomol. Chem. 2007, 5, 1062–1080. doi:10.1039/b617576g
Return to citation in text: [1] -
Wheeler, S. E. Acc. Chem. Res. 2013, 46, 1029–1038. doi:10.1021/ar300109n
Return to citation in text: [1] -
Collings, J. C.; Roscoe, K. P.; Robins, E. G.; Batsanov, A. S.; Stimson, L. M.; Howard, J. A. K.; Clark, S. J.; Marder, T. B. New J. Chem. 2002, 26, 1740–1746. doi:10.1039/b207102a
Return to citation in text: [1] -
Ponzini, F.; Zagha, R.; Hardcastle, K.; Siegel, J. S. Angew. Chem., Int. Ed. 2000, 39, 2323–2325. doi:10.1002/1521-3773(20000703)39:13<2323::AID-ANIE2323>3.0.CO;2-X
Return to citation in text: [1] -
Lokey, R. S.; Iverson, B. L. Nature 1995, 375, 303–305. doi:10.1038/375303a0
Return to citation in text: [1] -
Gabriel, G. J.; Sorey, S.; Iverson, B. L. J. Am. Chem. Soc. 2005, 127, 2637–2640. doi:10.1021/ja046722y
Return to citation in text: [1] [2] -
Mathis, G.; Hunziker, J. Angew. Chem., Int. Ed. 2002, 41, 3203–3205. doi:10.1002/1521-3773(20020902)41:17<3203::AID-ANIE3203>3.0.CO;2-K
Return to citation in text: [1] -
Tanaka, H.; Bollot, G.; Mareda, J.; Litvinchuk, S.; Tran, D.-H.; Sakai, N.; Matile, S. Org. Biomol. Chem. 2007, 5, 1369–1380. doi:10.1039/b702255g
Return to citation in text: [1] -
Das, A.; Molla, M. R.; Maity, B.; Koley, D.; Ghosh, S. Chem.–Eur. J. 2012, 18, 9849–9859. doi:10.1002/chem.201201140
Return to citation in text: [1] -
Ghosh, S.; Ramakrishnan, S. Macromolecules 2005, 38, 676–686. doi:10.1021/ma0478759
Return to citation in text: [1] -
Hunter, C. A. BioEssays 1996, 18, 157–162. doi:10.1002/bies.950180212
Return to citation in text: [1] -
Eschenmoser, A. Science 1999, 284, 2118–2124. doi:10.1126/science.284.5423.2118
Return to citation in text: [1] -
Kool, E. T. Chem. Rev. 1997, 97, 1473–1488. doi:10.1021/cr9603791
Return to citation in text: [1] -
Herdewijn, P. Biochim. Biophys. Acta, Gene Struct. Expression 1999, 1489, 167–179. doi:10.1016/S0167-4781(99)00152-9
Return to citation in text: [1] -
Benner, S. A. Acc. Chem. Res. 2004, 37, 784–797. doi:10.1021/ar040004z
Return to citation in text: [1] -
Wengel, J. Acc. Chem. Res. 1999, 32, 301–310. doi:10.1021/ar980051p
Return to citation in text: [1] -
Benner, S. A.; Hutter, D. Bioorg. Chem. 2002, 30, 62–80. doi:10.1006/bioo.2001.1232
Return to citation in text: [1] -
Yang, Z. Y.; Hutter, D.; Sheng, P.; Sismour, A. M.; Benner, S. A. Nucleic Acids Res. 2006, 34, 6095–6101. doi:10.1093/nar/gkl633
Return to citation in text: [1] -
Hirao, I.; Kimoto, M.; Yamashige, R. Acc. Chem. Res. 2012, 45, 2055–2065. doi:10.1021/ar200257x
Return to citation in text: [1] -
Wu, Y. Q.; Ogawa, A. K.; Berger, M.; McMinn, D. L.; Schultz, P. G.; Romesberg, F. E. J. Am. Chem. Soc. 2000, 122, 7621–7632. doi:10.1021/ja0009931
Return to citation in text: [1] -
Berger, M.; Ogawa, A. K.; McMinn, D. L.; Wu, Y.; Schultz, P. G.; Romesberg, F. E. Angew. Chem., Int. Ed. 2000, 39, 2940–2942. doi:10.1002/1521-3773(20000818)39:16<2940::AID-ANIE2940>3.0.CO;2-#
Return to citation in text: [1] -
Liu, H.; Gao, J.; Maynard, L.; Saito, Y. D.; Kool, E. T. J. Am. Chem. Soc. 2004, 126, 1102–1109. doi:10.1021/ja038384r
Return to citation in text: [1] -
Brotschi, C.; Häberli, A.; Leumann, C. J. Angew. Chem., Int. Ed. 2001, 40, 3012–3014. doi:10.1002/1521-3773(20010817)40:16<3012::AID-ANIE3012>3.0.CO;2-Y
Return to citation in text: [1] -
Lokey, R. S. Curr. Opin. Chem. Biol. 2003, 7, 91–96. doi:10.1016/S1367-5931(02)00002-9
Return to citation in text: [1] -
Ikkanda, B. A.; Samuel, S. A.; Iverson, B. L. J. Org. Chem. 2014, 79, 2029–2037. doi:10.1021/jo402704z
Return to citation in text: [1] -
Hecht, S.; Huc, I., Eds. Foldamers - Structure, Properties, and Applications; Wiley-VCH: Weinheim, 2007.
Return to citation in text: [1] -
Filichev, V. V.; Pedersen, E. B. DNA-Conjugated Organic Chromophores in DNA Stacking Interactions. In Wiley Encyclopedia of Chemical Biology; Begley, T. P., Ed.; Wiley: Hoboken, 2009; Vol. 1.
Return to citation in text: [1] -
Varghese, R.; Wagenknecht, H.-A. Chem. Commun. 2009, 2615–2624. doi:10.1039/b821728a
Return to citation in text: [1] -
Malinovskii, V. L.; Wenger, D.; Häner, R. Chem. Soc. Rev. 2010, 39, 410–422. doi:10.1039/b910030j
Return to citation in text: [1] -
Østergaard, M. E.; Hrdlicka, P. J. Chem. Soc. Rev. 2011, 40, 5771–5788. doi:10.1039/c1cs15014f
Return to citation in text: [1] -
Stulz, E. Chem.–Eur. J. 2012, 18, 4456–4469. doi:10.1002/chem.201102908
Return to citation in text: [1] -
Asanuma, H.; Fujii, T.; Kato, T.; Kashida, H. J. Photochem. Photobiol., C 2012, 13, 124–135. doi:10.1016/j.jphotochemrev.2012.04.002
Return to citation in text: [1] -
Häner, R.; Garo, F.; Wenger, D.; Malinovskii, V. L. J. Am. Chem. Soc. 2010, 132, 7466–7471. doi:10.1021/ja102042p
Return to citation in text: [1] -
Rudnev, A. V.; Malinovskii, V. L.; Nussbaumer, A. L.; Mishchenko, A.; Häner, R.; Wandlowski, T. Macromolecules 2012, 45, 5986–5992. doi:10.1021/ma3007619
Return to citation in text: [1] -
Simona, F.; Nussbaumer, A. L.; Häner, R.; Cascella, M. J. Phys. Chem. B 2013, 117, 2576–2585. doi:10.1021/jp310320f
Return to citation in text: [1] -
Häner, R.; Samain, F.; Malinovskii, V. L. Chem.–Eur. J. 2009, 15, 5701–5708. doi:10.1002/chem.200900369
Return to citation in text: [1] -
Langhals, H. Helv. Chim. Acta 2005, 88, 1309–1343. doi:10.1002/hlca.200590107
Return to citation in text: [1] -
Weil, T.; Vosch, T.; Hofkens, J.; Peneva, K.; Müllen, K. Angew. Chem., Int. Ed. 2010, 49, 9068–9093. doi:10.1002/anie.200902532
Return to citation in text: [1] -
Würthner, F. Chem. Commun. 2004, 1564–1579. doi:10.1039/b401630k
Return to citation in text: [1] -
Neelakandan, P. P.; Zeidan, T. A.; McCullagh, M.; Schatz, G. C.; Vura-Weis, J.; Kim, C. H.; Wasielewski, M. R.; Lewis, F. D. Chem. Sci. 2014, 5, 973–981. doi:10.1039/c3sc52908h
Return to citation in text: [1] -
Bittermann, H.; Siegemund, D.; Malinovskii, V. L.; Häner, R. J. Am. Chem. Soc. 2008, 130, 15285–15287. doi:10.1021/ja806747h
Return to citation in text: [1] [2] [3] -
Vybornyi, M.; Rudnev, A. V.; Langenegger, S. M.; Wandlowski, T.; Calzaferri, G.; Häner, R. Angew. Chem., Int. Ed. 2013, 52, 11488–11493. doi:10.1002/anie.201307029
Return to citation in text: [1] -
Rahe, N.; Rinn, C.; Carell, T. Chem. Commun. 2003, 2120–2121. doi:10.1039/B307395E
Return to citation in text: [1] -
It must be noted at this point, that the exact sequence of pyrene/PDI alternation can only be guessed, since the first residue stacking on the DNA stem could belong to either of the two strands. To simplify all subsequent description and discussion we have chosen that the first modified residue is the one attached at the nucleotide 5'-position.
Return to citation in text: [1] -
Clark, A. E.; Qin, C.; Li, A. D. Q. J. Am. Chem. Soc. 2007, 129, 7586–7595. doi:10.1021/ja0687724
Return to citation in text: [1] -
Biner, S. M.; Kummer, D.; Malinovskii, V. L.; Häner, R. Org. Biomol. Chem. 2011, 9, 2628–2633. doi:10.1039/c0ob01132k
Return to citation in text: [1] -
Khorev, O.; Bösch, C. D.; Probst, M.; Häner, R. Chem. Sci. 2014, 5, 1506–1512. doi:10.1039/c3sc53316f
Return to citation in text: [1] -
Bouquin, N.; Malinovskii, V. L.; Häner, R. Chem. Commun. 2008, 1974–1976. doi:10.1039/b802193g
Return to citation in text: [1] -
Bloomfield, V. A.; Crothers, D. M.; Tinoco, I., Jr. Nucleic Acids - Structures, Properties, and Functions; University Science Books: Sausalito, CA, 2000.
Return to citation in text: [1]
| 57. | Clark, A. E.; Qin, C.; Li, A. D. Q. J. Am. Chem. Soc. 2007, 129, 7586–7595. doi:10.1021/ja0687724 |
| 55. | Rahe, N.; Rinn, C.; Carell, T. Chem. Commun. 2003, 2120–2121. doi:10.1039/B307395E |
| 56. | It must be noted at this point, that the exact sequence of pyrene/PDI alternation can only be guessed, since the first residue stacking on the DNA stem could belong to either of the two strands. To simplify all subsequent description and discussion we have chosen that the first modified residue is the one attached at the nucleotide 5'-position. |
| 1. | Hunter, C. A.; Sanders, J. K. M. J. Am. Chem. Soc. 1990, 112, 5525–5534. doi:10.1021/ja00170a016 |
| 2. | Hunter, C. A.; Lawson, K. R.; Perkins, J.; Urch, C. J. J. Chem. Soc., Perkin Trans. 2 2001, 651–669. doi:10.1039/b008495f |
| 3. | Salonen, L. M.; Ellermann, M.; Diederich, F. Angew. Chem., Int. Ed. 2011, 50, 4808–4842. doi:10.1002/anie.201007560 |
| 4. | Grimme, S. Angew. Chem., Int. Ed. 2008, 47, 3430–3434. doi:10.1002/anie.200705157 |
| 5. | Martinez, C. R.; Iverson, B. L. Chem. Sci. 2012, 3, 2191–2201. doi:10.1039/c2sc20045g |
| 2. | Hunter, C. A.; Lawson, K. R.; Perkins, J.; Urch, C. J. J. Chem. Soc., Perkin Trans. 2 2001, 651–669. doi:10.1039/b008495f |
| 9. | Meyer, E. A.; Castellano, R. K.; Diederich, F. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. doi:10.1002/anie.200390319 |
| 13. | Cockroft, S. L.; Perkins, J.; Zonta, C.; Adams, H.; Spey, S. E.; Low, C. M. R.; Vinter, J. G.; Lawson, K. R.; Urch, C. J.; Hunter, C. A. Org. Biomol. Chem. 2007, 5, 1062–1080. doi:10.1039/b617576g |
| 14. | Wheeler, S. E. Acc. Chem. Res. 2013, 46, 1029–1038. doi:10.1021/ar300109n |
| 15. | Collings, J. C.; Roscoe, K. P.; Robins, E. G.; Batsanov, A. S.; Stimson, L. M.; Howard, J. A. K.; Clark, S. J.; Marder, T. B. New J. Chem. 2002, 26, 1740–1746. doi:10.1039/b207102a |
| 16. | Ponzini, F.; Zagha, R.; Hardcastle, K.; Siegel, J. S. Angew. Chem., Int. Ed. 2000, 39, 2323–2325. doi:10.1002/1521-3773(20000703)39:13<2323::AID-ANIE2323>3.0.CO;2-X |
| 53. | Bittermann, H.; Siegemund, D.; Malinovskii, V. L.; Häner, R. J. Am. Chem. Soc. 2008, 130, 15285–15287. doi:10.1021/ja806747h |
| 54. | Vybornyi, M.; Rudnev, A. V.; Langenegger, S. M.; Wandlowski, T.; Calzaferri, G.; Häner, R. Angew. Chem., Int. Ed. 2013, 52, 11488–11493. doi:10.1002/anie.201307029 |
| 8. | Hunter, C. A. Chem. Soc. Rev. 1994, 23, 101–109. doi:10.1039/cs9942300101 |
| 9. | Meyer, E. A.; Castellano, R. K.; Diederich, F. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. doi:10.1002/anie.200390319 |
| 10. | Hunter, C. A. J. Mol. Biol. 1993, 230, 1025–1054. doi:10.1006/jmbi.1993.1217 |
| 11. | Philp, D.; Stoddart, J. F. Angew. Chem., Int. Ed. Engl. 1996, 35, 1154–1196. doi:10.1002/anie.199611541 |
| 12. | Sakai, N.; Matile, S. Beilstein J. Org. Chem. 2012, 8, 897–904. doi:10.3762/bjoc.8.102 |
| 53. | Bittermann, H.; Siegemund, D.; Malinovskii, V. L.; Häner, R. J. Am. Chem. Soc. 2008, 130, 15285–15287. doi:10.1021/ja806747h |
| 45. | Häner, R.; Garo, F.; Wenger, D.; Malinovskii, V. L. J. Am. Chem. Soc. 2010, 132, 7466–7471. doi:10.1021/ja102042p |
| 46. | Rudnev, A. V.; Malinovskii, V. L.; Nussbaumer, A. L.; Mishchenko, A.; Häner, R.; Wandlowski, T. Macromolecules 2012, 45, 5986–5992. doi:10.1021/ma3007619 |
| 47. | Simona, F.; Nussbaumer, A. L.; Häner, R.; Cascella, M. J. Phys. Chem. B 2013, 117, 2576–2585. doi:10.1021/jp310320f |
| 48. | Häner, R.; Samain, F.; Malinovskii, V. L. Chem.–Eur. J. 2009, 15, 5701–5708. doi:10.1002/chem.200900369 |
| 61. | Bloomfield, V. A.; Crothers, D. M.; Tinoco, I., Jr. Nucleic Acids - Structures, Properties, and Functions; University Science Books: Sausalito, CA, 2000. |
| 6. | Watson, M. D.; Fechtenkötter, A.; Müllen, K. Chem. Rev. 2001, 101, 1267–1300. doi:10.1021/cr990322p |
| 7. | Feng, C.; Lin, C. S.; Fan, W.; Zhang, R. Q.; Van Hove, M. A. J. Chem. Phys. 2009, 131, 194702. doi:10.1063/1.3251785 |
| 49. | Langhals, H. Helv. Chim. Acta 2005, 88, 1309–1343. doi:10.1002/hlca.200590107 |
| 50. | Weil, T.; Vosch, T.; Hofkens, J.; Peneva, K.; Müllen, K. Angew. Chem., Int. Ed. 2010, 49, 9068–9093. doi:10.1002/anie.200902532 |
| 51. | Würthner, F. Chem. Commun. 2004, 1564–1579. doi:10.1039/b401630k |
| 52. | Neelakandan, P. P.; Zeidan, T. A.; McCullagh, M.; Schatz, G. C.; Vura-Weis, J.; Kim, C. H.; Wasielewski, M. R.; Lewis, F. D. Chem. Sci. 2014, 5, 973–981. doi:10.1039/c3sc52908h |
| 30. | Yang, Z. Y.; Hutter, D.; Sheng, P.; Sismour, A. M.; Benner, S. A. Nucleic Acids Res. 2006, 34, 6095–6101. doi:10.1093/nar/gkl633 |
| 31. | Hirao, I.; Kimoto, M.; Yamashige, R. Acc. Chem. Res. 2012, 45, 2055–2065. doi:10.1021/ar200257x |
| 32. | Wu, Y. Q.; Ogawa, A. K.; Berger, M.; McMinn, D. L.; Schultz, P. G.; Romesberg, F. E. J. Am. Chem. Soc. 2000, 122, 7621–7632. doi:10.1021/ja0009931 |
| 33. | Berger, M.; Ogawa, A. K.; McMinn, D. L.; Wu, Y.; Schultz, P. G.; Romesberg, F. E. Angew. Chem., Int. Ed. 2000, 39, 2940–2942. doi:10.1002/1521-3773(20000818)39:16<2940::AID-ANIE2940>3.0.CO;2-# |
| 34. | Liu, H.; Gao, J.; Maynard, L.; Saito, Y. D.; Kool, E. T. J. Am. Chem. Soc. 2004, 126, 1102–1109. doi:10.1021/ja038384r |
| 35. | Brotschi, C.; Häberli, A.; Leumann, C. J. Angew. Chem., Int. Ed. 2001, 40, 3012–3014. doi:10.1002/1521-3773(20010817)40:16<3012::AID-ANIE3012>3.0.CO;2-Y |
| 38. | Hecht, S.; Huc, I., Eds. Foldamers - Structure, Properties, and Applications; Wiley-VCH: Weinheim, 2007. |
| 53. | Bittermann, H.; Siegemund, D.; Malinovskii, V. L.; Häner, R. J. Am. Chem. Soc. 2008, 130, 15285–15287. doi:10.1021/ja806747h |
| 29. | Benner, S. A.; Hutter, D. Bioorg. Chem. 2002, 30, 62–80. doi:10.1006/bioo.2001.1232 |
| 39. | Filichev, V. V.; Pedersen, E. B. DNA-Conjugated Organic Chromophores in DNA Stacking Interactions. In Wiley Encyclopedia of Chemical Biology; Begley, T. P., Ed.; Wiley: Hoboken, 2009; Vol. 1. |
| 40. | Varghese, R.; Wagenknecht, H.-A. Chem. Commun. 2009, 2615–2624. doi:10.1039/b821728a |
| 41. | Malinovskii, V. L.; Wenger, D.; Häner, R. Chem. Soc. Rev. 2010, 39, 410–422. doi:10.1039/b910030j |
| 42. | Østergaard, M. E.; Hrdlicka, P. J. Chem. Soc. Rev. 2011, 40, 5771–5788. doi:10.1039/c1cs15014f |
| 43. | Stulz, E. Chem.–Eur. J. 2012, 18, 4456–4469. doi:10.1002/chem.201102908 |
| 44. | Asanuma, H.; Fujii, T.; Kato, T.; Kashida, H. J. Photochem. Photobiol., C 2012, 13, 124–135. doi:10.1016/j.jphotochemrev.2012.04.002 |
| 60. | Bouquin, N.; Malinovskii, V. L.; Häner, R. Chem. Commun. 2008, 1974–1976. doi:10.1039/b802193g |
| 10. | Hunter, C. A. J. Mol. Biol. 1993, 230, 1025–1054. doi:10.1006/jmbi.1993.1217 |
| 23. | Hunter, C. A. BioEssays 1996, 18, 157–162. doi:10.1002/bies.950180212 |
| 24. | Eschenmoser, A. Science 1999, 284, 2118–2124. doi:10.1126/science.284.5423.2118 |
| 25. | Kool, E. T. Chem. Rev. 1997, 97, 1473–1488. doi:10.1021/cr9603791 |
| 26. | Herdewijn, P. Biochim. Biophys. Acta, Gene Struct. Expression 1999, 1489, 167–179. doi:10.1016/S0167-4781(99)00152-9 |
| 27. | Benner, S. A. Acc. Chem. Res. 2004, 37, 784–797. doi:10.1021/ar040004z |
| 28. | Wengel, J. Acc. Chem. Res. 1999, 32, 301–310. doi:10.1021/ar980051p |
| 58. | Biner, S. M.; Kummer, D.; Malinovskii, V. L.; Häner, R. Org. Biomol. Chem. 2011, 9, 2628–2633. doi:10.1039/c0ob01132k |
| 17. | Lokey, R. S.; Iverson, B. L. Nature 1995, 375, 303–305. doi:10.1038/375303a0 |
| 18. | Gabriel, G. J.; Sorey, S.; Iverson, B. L. J. Am. Chem. Soc. 2005, 127, 2637–2640. doi:10.1021/ja046722y |
| 19. | Mathis, G.; Hunziker, J. Angew. Chem., Int. Ed. 2002, 41, 3203–3205. doi:10.1002/1521-3773(20020902)41:17<3203::AID-ANIE3203>3.0.CO;2-K |
| 20. | Tanaka, H.; Bollot, G.; Mareda, J.; Litvinchuk, S.; Tran, D.-H.; Sakai, N.; Matile, S. Org. Biomol. Chem. 2007, 5, 1369–1380. doi:10.1039/b702255g |
| 21. | Das, A.; Molla, M. R.; Maity, B.; Koley, D.; Ghosh, S. Chem.–Eur. J. 2012, 18, 9849–9859. doi:10.1002/chem.201201140 |
| 22. | Ghosh, S.; Ramakrishnan, S. Macromolecules 2005, 38, 676–686. doi:10.1021/ma0478759 |
| 18. | Gabriel, G. J.; Sorey, S.; Iverson, B. L. J. Am. Chem. Soc. 2005, 127, 2637–2640. doi:10.1021/ja046722y |
| 36. | Lokey, R. S. Curr. Opin. Chem. Biol. 2003, 7, 91–96. doi:10.1016/S1367-5931(02)00002-9 |
| 37. | Ikkanda, B. A.; Samuel, S. A.; Iverson, B. L. J. Org. Chem. 2014, 79, 2029–2037. doi:10.1021/jo402704z |
| 59. | Khorev, O.; Bösch, C. D.; Probst, M.; Häner, R. Chem. Sci. 2014, 5, 1506–1512. doi:10.1039/c3sc53316f |
© 2014 Winiger et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)