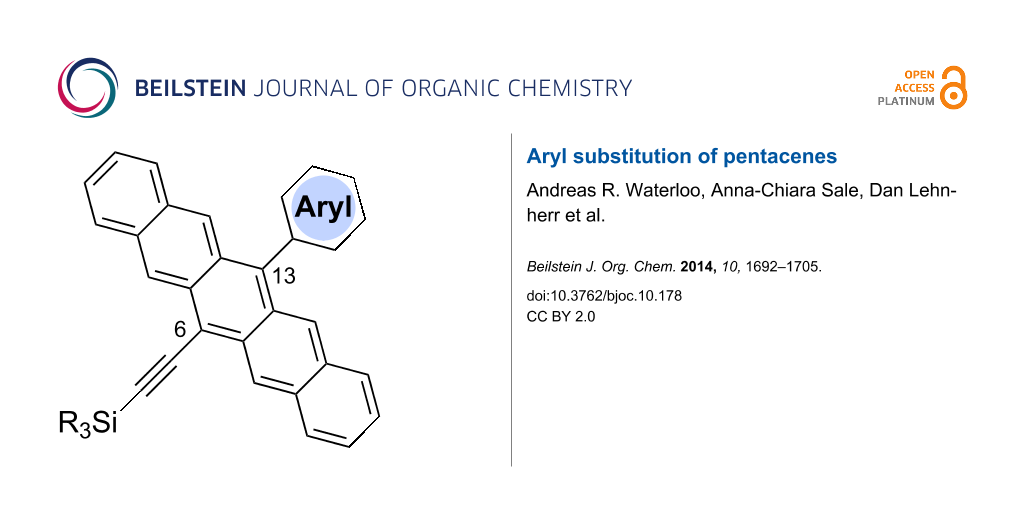
Abstract
A series of 11 new pentacene derivatives has been synthesized, with unsymmetrical substitution based on a trialkylsilylethynyl group at the 6-position and various aryl groups appended to the 13-position. The electronic and physical properties of the new pentacene chromophores have been analyzed by UV–vis spectroscopy (solution and thin films), thermoanalytical methods (DSC and TGA), cyclic voltammetry, as well as X-ray crystallography (for 8 derivatives). X-ray crystallography has been specifically used to study the influence of unsymmetrical substitution on the solid-state packing of the pentacene derivatives. The obtained results add to our ability to better predict substitution patterns that might be helpful for designing new semiconductors for use in solid-state devices.

Graphical Abstract
Introduction
Conjugated organic molecules are promising candidates for use in optoelectronic applications including OLEDs [1], photovoltaics [2], and OFETs [3]. Even though there is literally an infinite number of possibilities to chemically assemble small organic molecules for such applications, clever design and synthesis have rapidly offered new materials for the nascent era of molecular electronic [4-11]. Prominent p-type organic semiconductors include substituted acenes in general [12-15], and specifically 6,13-(triisopropylsilylethynyl)pentacene (TIPSPc) [13,14]. The latter was synthesized by Anthony over a decade ago [16], but it is still a leading focus of the scientific community. Substituted pentacenes can offer several advantages to other small molecule semiconductors, including stability, processability, and semiconductor device performance. Intermolecular π–π-interactions between chromophores can be dramatically improved upon functionalization at the 6- and 13-positions of pentacene, as demonstrated by the two-dimensional (2D) bricklayer-packing motif for TIPSPc [13,14]. A number of well-designed substitution patterns for the pentacene skeleton have been realized to date [17-25], and pentacene derivatives that provide good semiconductor device performance often show similar 6,13-substitution patterns (Figure 1a) [12,26]. The most reactive positions of the acene core are the 6- and 13-positions [27-29], and these positions can be effectively blocked by different trialkylsilylethynyl units [30]. Inspired by previous studies in which we [18,31], and others [32-34], observed promising solid-state arrangement and properties of aryl-substituted pentacenes, we were interested in the influence of different aryl moieties directly linked to the pentacene core. In this work we present a simple synthetic approach to unsymmetrically substituted pentacenes via stepwise functionalization of pentacenequinone, using a nucleophilic aryl group (Figure 1b). To determine the influence of aryl substitution, the obtained pentacenes have been studied by a variety of spectroscopic characterization methods as well as X-ray crystallography of eight derivatives. The studies reported herein offer an opportunity to interpret various properties of acenes based on their substitution and should contribute toward the use of such derivatives in materials science.
![[1860-5397-10-178-1]](/bjoc/content/figures/1860-5397-10-178-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Examples of common pentacene functionalization patterns and b) unsymmetrically aryl-substituted pentacenes reported in this work.
Figure 1: a) Examples of common pentacene functionalization patterns and b) unsymmetrically aryl-substituted ...
Results and Discussion
Synthesis and Characterization
Synthesis
The synthesis of arylpentacenes was developed based on the known ketone derivatives 4a and 4b, formed through the addition of an acetylide to pentacenequinone (Scheme 1) [21,35-38]. With these two ketones in hand, a second nucleophilic addition was initiated. Thus, commercially available aryl halides dissolved in dry THF were subjected to lithium halogen exchange at −78 °C using n-butyllithium. A substoichiometric amount of n-BuLi was used in each case to ensure complete consumption of the n-BuLi through Li–halogen exchange and thus avoid the possibility of competitive addition of the nucleophilic n-Bu anion to the ketone group of either 4a or 4b. After reaction with the appropriate aryl lithium species, the reaction was quenched with a proton source, and the resulting diol intermediates 5a–h were carried on directly to reductive aromatization with SnCl2/H2SO4 without further purification, ultimately yielding pentacene products 3a–h. While the isolation and characterization of diol products resulting from nucleophilic additions to pentacenequinone has been possible [17,18,34], previous work has shown that aromatized products were more easily purified by column chromatography and recrystallization following the last step [30,31,36]. Thus, it was deemed procedurally more efficient to eliminate the purification step of the intermediate diols. Once formed, pentacenes 3a–h were obtained in moderate yield over two steps, as deep-blue solids.
![[1860-5397-10-178-i1]](/bjoc/content/inline/1860-5397-10-178-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of unsymmetrically substituted pentacenes by nucleophilic addition (yields given are for the pentacene product 3, over the two steps from either 4a or 4b).
Scheme 1: Synthesis of unsymmetrically substituted pentacenes by nucleophilic addition (yields given are for ...
To expand the π-system in a linear fashion along the short molecular axis of the pentacene core, the general procedure described above was changed slightly, and ketone 4a was treated with a solution of biphenylmagnesium bromide in THF. After work-up and isolation of the intermediate diol 5i, reductive aromatization gave pentacene 3i in moderate yield over the two steps. Elaborating on the general idea of lateral functionalization, iodoaryl pentacene 3g offered an opportunity to vary the pendent substituent through a Pd-catalyzed cross-coupling protocol (Scheme 2). Thus, pentacene 3g was treated under Suzuki–Miyaura coupling conditions with arylboronic acids, and the desired pentacenes 3j,k were obtained in yields of 92 and 68%, respectively. Notably, anthracenyl-substituted pentacene 3k was the least stable of all derivatives synthesized in this study. It slowly decomposed in solution when exposed to ambient laboratory conditions and was unstable toward silica gel chromatography. Compound 3k could, however, be purified by recrystallization from a mixture of MeOH and acetone.
![[1860-5397-10-178-i2]](/bjoc/content/inline/1860-5397-10-178-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Functionalization of iodoaryl pentacene 3g using the Suzuki–Miyaura cross-coupling reaction.
Scheme 2: Functionalization of iodoaryl pentacene 3g using the Suzuki–Miyaura cross-coupling reaction.
UV–vis spectroscopy
Aryl pentacenes 3a–k have been studied by UV–vis absorption spectroscopy in CH2Cl2 solutions and as thin films cast from CH2Cl2 onto quartz. Solution-state UV–vis spectra show characteristic absorptions in the high-energy region with a maximum intensity absorption centered at ~310 nm, as well as low-energy absorptions at ~578 nm and ~622 nm. In comparison, unsubstituted pentacene (PEN) shows a λmax = 576 nm (measured in benzene) [39], while the symmetrically substituted analogue TIPSPc shows a low-energy absorption at 643 nm (measured in CH2Cl2) due to extended conjugation through the two alkyne substituents [11].
As can be seen in the representative spectra in Figure 2, the nature and size of aryl substituents at the 13-position does not alter the basic absorption wavelengths as one progresses, for example, through the series of phenyl (3a), naphthyl (3b) and anthracenyl (3c), although some differences in molar absorptivity are observed. Similar trends are observed within the series of pentacenes 3i, 3j, and 3k. Thus, these UV–vis spectra clearly document the lack of communication between the aryl substituent and the pentacene unit, as a result of hindered rotation about the aryl–pentacene C–C bond and a preferred conformation in which the π-system of the aryl group is orthogonal to that of the pentacene. The orthogonal orientation of the aryl groups is also well-documented in the solid state by X-ray crystallographic analysis (vide infra). The fluorescence characteristics of 3a–k are unremarkable, showing only minor variances in emission wavelength ranging from 652–671 nm, as measured in CH2Cl2 (see Supporting Information File 1 for details and spectra).
![[1860-5397-10-178-2]](/bjoc/content/figures/1860-5397-10-178-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis spectra of pentacenes a) 3a–c and b) 3i–k (measured in CH2Cl2).
Figure 2: UV–vis spectra of pentacenes a) 3a–c and b) 3i–k (measured in CH2Cl2).
The major electronic absorptions found in solution are also reproduced to a large extent in spectra obtained from solid-state films. Pentacene samples 3a–k were drop-cast from a concentrated CH2Cl2 solution onto a quartz surface, and after air-drying, the absorption spectra were measured by UV–vis spectroscopy (Figure 3 and Table 1). While this method sometimes results in rather significant scattering versus the formation of films by spin-casting, only milligram quantities of material are required and the results are qualitatively informative (i.e., absorption wavelengths can be readily discerned, while determination of molar absorptivity is not possible). As can be seen in Figure 3, spectra from thin films show absorption profiles similar to those from solution-state measurements, although signals are broadened and absorptivities vary dramatically due to scattering. In the solid state, there are no observed absorptions at wavelengths beyond ca. 660 nm, and absorptions in the lower energy region show a red shift (7 to 33 nm) in comparison to analogous absorptions in solution. A red shift in the absorption features of samples in the solid state relative to those in solution is typically ascribed to a local electronic interaction between the respective pentacene molecules in the solid state. Of the aryl pentacenes studied here, veratrole derivative 3f shows the largest red shift (33 nm) as a film compared to its solution-state UV–vis spectrum, although the origin of change in the solid state is not understood. It is worth noting, however, that more significant red-shifted λmax-values are often observed for samples which give solution cast films that result in significant π-stacking between molecules, such as TIPSPc, in which λmax shifts from 643 nm in solution to ca. 705 nm in the solid film [11,35,40]. This same logic also suggests that the minimal difference between the absorption characteristics of 3c and 3h results from both a lack of influence from the different silyl groups, as well as the absence of strong π-stacking for both derivatives in the films, even though X-ray crystallographic analysis suggests that strong interactions might be possible for some derivatives in the solid state (vide infra).
![[1860-5397-10-178-3]](/bjoc/content/figures/1860-5397-10-178-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: UV–vis spectra of thin films (drop cast on quartz from a CH2Cl2 solution) for pentacenes a) 3a–c and b) 3i–k.
Figure 3: UV–vis spectra of thin films (drop cast on quartz from a CH2Cl2 solution) for pentacenes a) 3a–c an...
Table 1: Optical properties of pentacenes 3a–k, unsubstituted pentacene (PEN), and TIPSPc.
| Compound | λmax (in CH2Cl2)a [nm] | λmax (film)b [nm] | red shift [nm] (meV) | Egap,opt [eV]c |
|---|---|---|---|---|
| 3a | 621 | 637 | 16 (50) | 1.89 |
| 3b | 621 | 635 | 14 (44) | 1.91 |
| 3c | 623 | 637 | 14 (44) | 1.92 |
| 3d | 623 | 637 | 14 (44) | 1.91 |
| 3e | 621 | 634 | 13 (41) | 1.89 |
| 3f | 622 | 655 | 33 (100) | 1.87 |
| 3g | 621 | 628 | 7 (22) | 1.90 |
| 3h | 622 | 638 | 16 (50) | 1.91 |
| 3i | 623 | 641 | 18 (56) | 1.88 |
| 3j | 623 | 633 | 10 (31) | 1.89 |
| 3k | 623 | 635 | 12 (38) | 1.89 |
| PEN | 576d | 673e | 97 (310) | 2.15 |
| TIPSPc | 643f | 705g | 62 (170) | 1.84f |
aLowest energy absorption maxima. bCast from CH2Cl2 onto quartz. cDetermined from solution-state spectra, based on a tangent line applied to the lower edge of the longest wavelength absorption peak and the intercept with the x-axis. dMeasured in benzene and data taken from [39]. eData taken from [41,42]. fData taken from [11]. gData taken from [35].
Thermal analysis
The thermal stability of selected aryl pentacenes has been explored by traditional melting point analysis (MPA) in open capillary tubes, thermal gravimetric analysis (TGA), and differential scanning calorimetry (DSC) measurements; the results are summarized in Table 2. TGA shows clearly that significant mass loss occurs in the range of 400 °C, which is also common for ethynylated pentacenes such as 1 and 2 [17,18]. There appears to be little evidence of a trend based on the size of the aryl group versus the temperature of observed mass loss in the TGA. By comparing the TGA data with that of MPA made in open capillary tubes, however, it is clear that all pentacene derivatives undergo either a phase change or decomposition prior to the mass loss observed in the TGA. This premise is also confirmed by DSC analyses, which show a melting point in all cases except for thienyl derivative 3d (which decomposed directly in the solid state). In the case of 3a and 3h, melting is followed immediately by decomposition (DSC and TGA scans are provided in Supporting Information File 1).
Table 2: Thermal properties of a representative selection of pentacenes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-178-i3.svg?max-width=637&scale=1.0)
|
|||||
| Compound | R | Aryl |
TGA Td
/°Ca |
MPA
mp /°Cb |
DSC mp
(DSC dp) /°Cc |
|---|---|---|---|---|---|
| 3a | iPr |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-178-i4.svg?max-width=637&scale=1.0)
|
370 | 162–165 |
177
(178/179) |
| 3b | iPr |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-178-i5.svg?max-width=637&scale=1.0)
|
370 | 244–246 | 248 |
| 3c | iPr |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-178-i6.svg?max-width=637&scale=1.0)
|
375 | 306–308d | 197e |
| 3d | iPr |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-178-i7.svg?max-width=637&scale=1.0)
|
372 | 316–318d |
–
(206/247) |
| 3h | Et |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-178-i8.svg?max-width=637&scale=1.0)
|
410 | 291–293 |
287
(288/290) |
| 3i | iPr |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-178-i9.svg?max-width=637&scale=1.0)
|
380 | 211–214 | 225 |
| 3j | iPr |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-178-i10.svg?max-width=637&scale=1.0)
|
380 | 233–235 | 220f |
aMeasured under a nitrogen atmosphere. Td = decomposition temperature, see Supporting Information File 1 for details. bTraditional open capillary melting point analysis (MPA), measured under ambient conditions; uncorrected. cMeasured under a nitrogen atmosphere; dp = decomposition point, shown as onset/peak temperatures. dDecomposition observed in that temperature range, with no indication of melting or decomposition at lower temperature. eEndotherm, although apparently not a true mp based on traditional mp analysis. Exotherm at 286 °C (peak) likely corresponds to dp in DSC. fThe strongest of several endotherms.
While no correlation between pendent substituent and stability emerges from this analysis, an important point is nevertheless noted, as exemplified by the examination of 3c and 3d. Traditional MPA is often insufficient for characterization of pentacene derivatives, in which subtle changes in the samples can be difficult to discern because of the deep, dark color of the sample, and conflicting results are often observed between MPA and DSC.
Cyclic voltammetry
Electrochemical analysis by cyclic voltammetry (CV) was used to investigate the electronic properties of pentacene derivatives 3a–k in CH2Cl2 (ca. 1.5 mM) using tetrabutylammonium hexafluorophosphate as supporting electrolyte and ferrocene as internal standard (all potentials reported are thus given versus Fc/Fc+). Aryl-substituted pentacenes 3a–k each show a one-electron reversible oxidation event in the range between 0.30–0.37 V (Table 3), and a second oxidation process (quasi-reversible) in the range of 0.80–0.99 V. There is, unfortunately, no clear trend observed for the oxidation potentials based on the substitution pattern of the aryl moieties, although both oxidations appear somewhat easier for pentacene 3f as a result of the two electron-donating methoxy groups attached to the pendent phenyl ring.
Table 3: Electrochemical properties of 3a–k compared to TIPSPc and 2a–c.a
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-178-i11.svg?max-width=637&scale=1.0)
|
||||||
| Compound | R | Aryl | Eox1 [V] | Eox2 [V] | Ered1 [V] | Egap,el [eV]b |
|---|---|---|---|---|---|---|
| 3a | iPr |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-178-i12.svg?max-width=637&scale=1.0)
|
0.34 | 0.87 | −1.63 | 1.97 |
| 3b | iPr |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-178-i13.svg?max-width=637&scale=1.0)
|
0.37 | 0.99 | −1.61 | 1.98 |
| 3c | iPr |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-178-i14.svg?max-width=637&scale=1.0)
|
0.36 | 0.93 | −1.65 | 2.01 |
| 3h | Et |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-178-i15.svg?max-width=637&scale=1.0)
|
0.32 | 0.91 | −1.68 | 2.00 |
| 3d | iPr |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-178-i16.svg?max-width=637&scale=1.0)
|
0.35 | 0.87 | −1.59 | 1.94 |
| 3e | iPr |
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-178-i17.svg?max-width=637&scale=1.0)
|
0.32 | 0.87 | −1.68 | 2.00 |
| 3f | iPr |
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-178-i18.svg?max-width=637&scale=1.0)
|
0.30 | 0.80 | −1.67 | 1.97 |
| 3g | iPr |
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-178-i19.svg?max-width=637&scale=1.0)
|
0.34 | 0.87 | −1.65 | 1.99 |
| 3i | iPr |
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-178-i20.svg?max-width=637&scale=1.0)
|
0.32 | 0.87 | −1.66 | 1.98 |
| 3j | iPr |
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-178-i21.svg?max-width=637&scale=1.0)
|
0.32 | 0.93 | −1.67 | 1.99 |
| 3k | iPr |
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-178-i22.svg?max-width=637&scale=1.0)
|
0.35 | 0.88 | −1.67 | 2.02 |
| 2ac | iPr |
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-178-i23.svg?max-width=637&scale=1.0)
|
0.39 | – | −1.44 | 1.83 |
| 2bc | iPr |
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-178-i24.svg?max-width=637&scale=1.0)
|
0.39 | – | −1.42 | 1.81 |
| 2cc | iPr |
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-178-i25.svg?max-width=637&scale=1.0)
|
0.33 | – | −1.38 | 1.71 |
| TIPSPc | iPr |
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-178-i26.svg?max-width=637&scale=1.0)
|
0.39 | 0.99 | −1.52 | 1.91 |
aCyclic voltammetry was performed in CH2Cl2 solutions (1.5 mM) containing 0.1 M n-Bu4NPF6 as supporting electrolyte at a scan rate of 150 mV/s. Platinum wire was used as counter electrode, Ag/AgNO3 as reference electrode, and Pt working electrode. The potential values (E) were calculated using the following equation E = (Epc + Epa)/2, where Epc and Epa correspond to the cathodic and anodic peak potentials, respectively. Potentials are referenced to the ferrocene/ferrocenium (Fc/Fc+) couple used as an internal standard. All potentials represent a reversible one-electron reduction or oxidation event. bElectrochemical HOMO–LUMO gaps determined by Egap,el = Eox1 − Ered1. cData taken from [18].
Aryl-substituted pentacenes 3a–k each show one reversible reduction event in a rather narrow range of −1.59 to −1.68 V. Similar to that observed for the oxidation potentials, there is no obvious trend that can be identified in the reduction potentials based on substitution pattern, aside from the observation that the silyl substituent might have a slight impact on reduction (3h is slightly harder to reduce than 3c), and the reduction of thienyl derivative 3d (−1.59 V) stands out as lower than the others.
Substituted pentacenes 3a–k are slightly easier to oxidize than TIPSPc (Eox1 = 0.39 V), and the Eox1 values of 3a–k fall into a similar range as found for pentacene-based PAH dyads 2a–c in which the pendant aryl groups are linked by an ethynyl spacer that allows electronic communication between the two arenes [18]. The range of oxidation potentials between TIPSPc, 2a–c, and 3a–k is, however, quite narrow, suggesting that the pendent substituent offers little influence on the HOMO level. On the other hand, there is a marked difference in the observed reduction potentials. Compounds 2a–c are most easily reduced while 3a–k are the most difficult, and the reduction of TIPSPc falls at approximately a midpoint between the two other classes. Thus, the biggest influence of the pendent substituent appears to be related to the energy of the LUMO.
As suggested by the UV–vis analyses (vide infra), the HOMO–LUMO gap estimated for pentacenes 3a–k by CV (1.94–2.02 eV) is larger than the HOMO–LUMO gap of TIPSPc (1.91 eV), while incorporation of the ethynyl spacer in 2a–c provides for the lowest HOMO–LUMO gap of the molecules discussed here.
X-ray crystallographic analysis
Typically, three predominant solid-state packing patterns are found by X-ray crystallographic analysis of pentacene and its derivatives [13]: a) a herringbone packing, b) a one-dimensional (1D) slipped-stack, and c) a 2D “bricklayer” packing, as schematically summarized in Figure 4. While several polymorphs have been reported for unsubstituted pentacene [43-46], the arrangement in the solid state is commonly the edge-to-face, herringbone motif [47]. This packing arrangement provides strong electronic coupling in the solid state, and therefore makes this material interesting as the active component for semiconducting devices [48]. It has been shown that functionalization of the pentacene framework, particular by trialkylsilylethynyl groups, drastically alters the solid-state arrangements of the acenes [30]. In certain cases, this leads to a 2D face-to-face “bricklayer” arrangement, which can potentially facilitate charge transport in an electric device by several orders of magnitude [49].
![[1860-5397-10-178-4]](/bjoc/content/figures/1860-5397-10-178-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Schematic classification of three common solid-state arrangements of pentacene derivatives a) herringbone packing, b) 1D slipped-stack packing, and c) 2D “bricklayer” packing (as viewed side-on, approximately down the short-molecular axis).
Figure 4: Schematic classification of three common solid-state arrangements of pentacene derivatives a) herri...
With the understanding that the solid-state packing of acenes can provide vital information about intermolecular interactions, single crystals of pentacenes 3a–d and 3g–j were grown and their solid-state arrangements determined by single crystal X-ray diffraction analysis (crystallographic details are provided in Supporting Information File 1). Pentacene 3a crystallizes in the space group C2/c with eight molecules in the unit cell (Figure 5). Within the solid-state structure, the pendant phenyl ring and the pentacene core are slightly twisted, with a torsion angle of ~71°. Molecules of pentacene 3a arrange as dimeric pairs, which then pack into a so-called sandwich herringbone motif (Figure 5c) [50]. Each dimeric pair of pentacenes shows face-to-face π-stacking interactions with an interplanar distance of ~3.40 Å and a total overlap of nearly four of the aromatic pentacene rings.
![[1860-5397-10-178-5]](/bjoc/content/figures/1860-5397-10-178-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: X-ray crystallographic analysis of 3a showing a) molecular structure and b) packing motif (triisopropylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level. c) Schematic representation of “sandwich herringbone” packing arrangement.
Figure 5: X-ray crystallographic analysis of 3a showing a) molecular structure and b) packing motif (triisopr...
Pentacene 3b crystallizes in the space group P-1 with two molecules in the unit cell (Figure 6). Notably, the naphthyl unit is significantly disordered over two unique positions in the solid state but is nearly perpendicular to the pentacene core with a twist angle of ~81°. Pentacene 3b assembles in dimeric pairs, which then arrange in a 1D π-slipped stack motif, and the pentacene molecules are separated by interplanar distances of ~3.42 Å and ~3.52 Å.
![[1860-5397-10-178-6]](/bjoc/content/figures/1860-5397-10-178-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: X-ray crystallographic analysis of 3b showing a) molecular structure and b) packing motif (triisopropylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level.
Figure 6: X-ray crystallographic analysis of 3b showing a) molecular structure and b) packing motif (triisopr...
Pentacene 3c crystallizes in the space group P21/c with four molecules in the unit cell (Figure 7) [31]. The pentacene skeleton and the anthracenyl substituent are nearly perpendicular to each other with a twist angle of ~90°. This motif also places the anthracene moieties in a face-to-face packing 1D slipped stack arrangement, although the interplanar distance of ~3.61 Å is sizable. The aromatic pentacene cores pack in a face-to-face 2D bricklayer arrangement, with approximately two pentacene rings overlapping and interplanar distances of ~3.52 Å and 3.46 Å.
![[1860-5397-10-178-7]](/bjoc/content/figures/1860-5397-10-178-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: X-ray crystallographic analysis of 3c showing a) molecular structure and b) packing motif (triisopropylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level.
Figure 7: X-ray crystallographic analysis of 3c showing a) molecular structure and b) packing motif (triisopr...
Pentacene 3d crystallizes in the space group P21/n with four molecules in each unit cell (Figure 8), and the thienyl and trialkylsilyl groups show disorder in the structure. With a twist angle of ~90°, the thienyl unit is essentially perpendicular to the pentacene skeleton. Centrosymmetric dimeric pairs of pentacene 3d pack with an interplanar distance of 3.52 Å and these pairs then arrange into a sandwich herringbone stacking pattern, similar to phenyl derivative 3a.
![[1860-5397-10-178-8]](/bjoc/content/figures/1860-5397-10-178-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: X-ray crystallographic analysis of 3d showing a) molecular structure, and b) packing motif (triisopropylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level.
Figure 8: X-ray crystallographic analysis of 3d showing a) molecular structure, and b) packing motif (triisop...
Pentacene 3g crystallizes in the space group P21/n with four molecules in the unit cell (Figure 9). The phenyl substituent is twisted with an angle of ~70° relative to the pentacene skeleton. Two neighboring molecules of 3g arrange into a dimeric pair with an interplanar distance of 3.42 Å, and these pairs then pack in a sandwich herringbone arrangement. The overall solid-state arrangement is similar to that observed for 3a and 3d.
![[1860-5397-10-178-9]](/bjoc/content/figures/1860-5397-10-178-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: X-ray crystallographic analysis of 3g showing a) molecular structure and b) packing motif; ORTEP drawn at 50% probability level.
Figure 9: X-ray crystallographic analysis of 3g showing a) molecular structure and b) packing motif; ORTEP dr...
Pentacene 3h crystallizes in the space group P21/c with four molecules in each unit cell (Figure 10). The anthracenyl substituent is twisted relative to the pentacene skeleton with an angle of ~74°, less than that found for 3c (90°). Interestingly, pentacene 3h shows an unusual solid-state arrangement not typically observed for pentacene derivatives. Namely, the pentacene molecules form channels along the crystallographic a-axis, which are composed of only two tiers of a brick wall structure. The pentacene molecules within these channels are stacked with an interplanar distance of 3.57 Å. These channels are macroscopically arranged as staircases, dictated by the anthracenyl moieties that are oriented such that CH–π interactions of ~2.90 Å likely play a role in directing the packing (see Supporting Information File 1, Figure S12).
![[1860-5397-10-178-10]](/bjoc/content/figures/1860-5397-10-178-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: X-ray crystallographic analysis of 3h showing a) molecular structure and b) packing motif (triethylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level.
Figure 10: X-ray crystallographic analysis of 3h showing a) molecular structure and b) packing motif (triethyl...
Pentacene 3i crystallizes in the space group P-1 with two molecules in each unit cell (Figure 11). The benzene ring directly attached to the pentacene framework is nearly perpendicular to the pentacene core with an angle of ~81°, while the torsion angle between the two rings of the biphenyl unit is 32°. The biphenyl substituent is slightly bent from linearity with an angle of ~6° (as estimated from the three atoms designated with an asterisk *). Biphenyl-substituted pentacene 3i arranges in a 1D slipped stack motif along the crystallographic c-axis, with π-stacking distances of 3.28 Å and 3.35 Å.
![[1860-5397-10-178-11]](/bjoc/content/figures/1860-5397-10-178-11.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: X-ray crystallographic analysis of 3i showing a) molecular structure and b) packing motif (triisopropylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level.
Figure 11: X-ray crystallographic analysis of 3i showing a) molecular structure and b) packing motif (triisopr...
Pentacene derivative 3j crystallizes in the space group P-1 with two molecules in the unit cell (Figure 12). Interestingly, the pentacene core and the naphthyl group are nearly coplanar (4°), while the intervening benzene ring is nearly perpendicular to both the pentacene skeleton (~90°) and the naphthyl group (~86°). Compound 3j arranges in a 1D slipped stack arrangement along the crystallographic b-axis with two different interplanar distances of ~3.50 Å and 3.32 Å.
![[1860-5397-10-178-12]](/bjoc/content/figures/1860-5397-10-178-12.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: X-ray crystallographic analysis of 3j showing a) molecular structure and b) packing motif (triisopropylsilyl groups omitted for clarity); ORTEP drawn at 50% probability level.
Figure 12: X-ray crystallographic analysis of 3j showing a) molecular structure and b) packing motif (triisopr...
Conclusion
In summary, a library of unsymmetrically substituted pentacenes has been synthesized by a straightforward procedure that requires only one purification step. Optical spectroscopy and cyclic voltammetry reveal that electronic communication between the pentacene core and the different substituents is limited, as a result of the orthogonal orientation of the pentacene backbone and the pendent aryl moieties. Thus, these results show that the nature of the substituent does not change the electronic properties of the pentacene skeleton itself. Aryl-substitution pattern does however, have a considerable effect on solid-state arrangement of the molecules, and X-ray crystallographic analysis afforded insight on the packing arrangements of the synthesized pentacenes. In spite of the large number of crystallographic analyses that have been examined here, general trends are difficult to establish based on, for example, either the number of π–π and CH–π interactions or the size of the aromatic group appended to the pentacene core. It does seem, however, that anthracenyl substitution (in 3c and 3h) affords the highest degree of π-stacking amongst the derivatives examined.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data for all new compounds. Copies of 1H and 13C NMR, UV–vis, and emission spectra; CV, DSC, and TGA scans. | ||
| Format: PDF | Size: 5.0 MB | Download |
Acknowledgements
The authors acknowledge funding for this work from the Energie Campus Nürnberg (EnCN), “Solar Technologies go Hybrid” (an initiative of the Bavarian State Ministry of Science, Research and Art), and the Deutsche Forschungsgemeinschaft (DFG) through the Cluster of Excellence “Engineering of Advanced Materials”. We thank Dr. Michael J. Ferguson for the X-ray crystallographic analysis of ketone 4b.
References
-
Figueira-Duarte, T. M.; Müllen, K. Chem. Rev. 2011, 111, 7260–7314. doi:10.1021/cr100428a
Return to citation in text: [1] -
Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689–6735. doi:10.1021/cr9002984
Return to citation in text: [1] -
Lucas, B.; Trigaud, T.; Videlot-Ackermann, C. Polym. Int. 2012, 61, 374–389. doi:10.1002/pi.3213
Return to citation in text: [1] -
Beaujuge, P. M.; Fréchet, J. M. J. J. Am. Chem. Soc. 2011, 133, 20009–20029. doi:10.1021/ja2073643
Return to citation in text: [1] -
Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Chem. Rev. 2009, 109, 5868–5923. doi:10.1021/cr900182s
Return to citation in text: [1] -
Mártin, N.; Sánchez, L.; Herranz, M. Á.; Illescas, B.; Guldi, D. M. Acc. Chem. Res. 2007, 40, 1015–1024. doi:10.1021/ar700026t
Return to citation in text: [1] -
Dang, M. T.; Hirsch, L.; Wantz, G.; Wuest, J. D. Chem. Rev. 2013, 113, 3734–3765. doi:10.1021/cr300005u
Return to citation in text: [1] -
Arias, A. C.; MacKenzie, J. D.; McCulloch, I.; Rivnay, J.; Salleo, A. Chem. Rev. 2010, 110, 3–24. doi:10.1021/cr900150b
Return to citation in text: [1] -
Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689–6735. doi:10.1021/cr9002984
Return to citation in text: [1] -
Mei, J.; Diao, Y.; Appleton, A. L.; Fang, L.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 6724–6746. doi:10.1021/ja400881n
Return to citation in text: [1] -
Lehnherr, D.; Tykwinski, R. R. Materials 2010, 3, 2772–2800. doi:10.3390/ma3042772
Return to citation in text: [1] [2] [3] [4] -
Lehnherr, D.; Tykwinski, R. R. Aust. J. Chem. 2011, 64, 919–929. doi:10.1071/CH11169
Return to citation in text: [1] [2] -
Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z
Return to citation in text: [1] [2] [3] [4] -
Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045
Return to citation in text: [1] [2] [3] -
Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4946. doi:10.1021/cr030666m
Return to citation in text: [1] -
Anthony, J. E.; Brooks, J. S.; Eaton, D. L.; Parkin, S. R. J. Am. Chem. Soc. 2001, 123, 9482–9483. doi:10.1021/ja0162459
Return to citation in text: [1] -
Lehnherr, D.; Murray, A. H.; McDonald, R.; Tykwinski, R. R. Angew. Chem., Int. Ed. 2010, 49, 6190–6194. doi:10.1002/anie.201000555
Return to citation in text: [1] [2] [3] -
Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lehnherr, D.; Gao, J.; Hegmann, F. A.; Tykwinski, R. R. J. Org. Chem. 2009, 74, 5017–5024. doi:10.1021/jo9007089
Return to citation in text: [1] -
Lehnherr, D.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Tetrahedron 2008, 64, 11449–11461. doi:10.1016/j.tet.2008.09.041
Return to citation in text: [1] -
Lehnherr, D.; Gao, J.; Hegmann, F. A.; Tykwinski, R. R. Org. Lett. 2008, 10, 4779–4782. doi:10.1021/ol801886h
Return to citation in text: [1] [2] -
Lehnherr, D.; Tykwinski, R. R. Org. Lett. 2007, 9, 4583–4586. doi:10.1021/ol702094d
Return to citation in text: [1] -
Kuninobu, Y.; Seiki, T.; Kanamaru, S.; Nishina, Y.; Takai, K. Org. Lett. 2010, 12, 5287–5289. doi:10.1021/ol102349r
Return to citation in text: [1] -
Li, Y.; Wu, Y.; Liu, P.; Prostran, Z.; Gardner, S.; Ong, B. S. Chem. Mater. 2007, 19, 418–423. doi:10.1021/cm062378n
Return to citation in text: [1] -
Lee, S. H.; Kim, S. H.; Choo, D. J.; Jang, J. Org. Electron. 2010, 11, 1268–1272. doi:10.1016/j.orgel.2010.03.018
Return to citation in text: [1] -
Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z
Return to citation in text: [1] -
Zade, S. S.; Bendikov, M. J. Phys. Org. Chem. 2012, 25, 452–461. doi:10.1002/poc.1941
Return to citation in text: [1] -
Biermann, D.; Schmidt, W. J. Am. Chem. Soc. 1980, 102, 3163–3173. doi:10.1021/ja00529a046
Return to citation in text: [1] -
Bénard, C. P.; Geng, Z.; Heuft, M. A.; VanCrey, K.; Fallis, A. G. J. Org. Chem. 2007, 72, 7229–7236. doi:10.1021/jo0709807
Return to citation in text: [1] -
Anthony, J. E.; Eaton, D. L.; Parkin, S. R. Org. Lett. 2002, 4, 15–18. doi:10.1021/ol0167356
Return to citation in text: [1] [2] [3] -
Etschel, S. H.; Waterloo, A. R.; Margraf, J. T.; Amin, A. Y.; Hampel, F.; Jäger, C. M.; Clark, T.; Halik, M.; Tykwinski, R. R. Chem. Commun. 2013, 49, 6725–6727. doi:10.1039/c3cc43270j
Return to citation in text: [1] [2] [3] -
Zhang, J.; Pawle, R. H.; Haas, T. E.; Thomas, S. W., III. Chem.–Eur. J. 2014, 20, 5880–5884. doi:10.1002/chem.201402003
Return to citation in text: [1] -
Kim, Y.-E.; Park, J.-W. Mol. Cryst. Liq. Cryst. 2006, 444, 137–143. doi:10.1080/15421400500365318
Return to citation in text: [1] -
Zhang, X.; Jiang, X.; Luo, J.; Chi, C.; Chen, H.; Wu, J. Chem.–Eur. J. 2010, 16, 464–468. doi:10.1002/chem.200902675
Return to citation in text: [1] [2] -
Lehnherr, D.; McDonald, R.; Tykwinski, R. R. Org. Lett. 2008, 10, 4163–4166. doi:10.1021/ol801464k
Return to citation in text: [1] [2] [3] -
Waterloo, A. R.; Kunakom, S.; Hampel, F.; Tykwinski, R. R. Macromol. Chem. Phys. 2012, 213, 1020–1032. doi:10.1002/macp.201100622
Return to citation in text: [1] [2] -
Boudebous, A.; Constable, E. C.; Housecroft, C. E.; Neuburger, M.; Schaffner, S. Acta Crystallogr., Sect. C 2006, 62, o243–o245. doi:10.1107/S0108270106008262
Return to citation in text: [1] -
Vets, N.; Smet, M.; Dehaen, W. Synlett 2005, 217–222. doi:10.1055/s-2004-836055
Return to citation in text: [1] -
Maulding, D. R.; Roberts, B. G. J. Org. Chem. 1969, 34, 1734–1736. doi:10.1021/jo01258a045
Return to citation in text: [1] [2] -
Platt, A. D.; Day, J.; Subramanian, S.; Anthony, J. E.; Ostroverkhova, O. J. Phys. Chem. C 2009, 113, 14006–14014. doi:10.1021/jp904021p
Return to citation in text: [1] -
Ostroverkhova, O.; Shcherbyna, S.; Cooke, D. G.; Egerton, R. F.; Hegmann, F. A.; Tykwinski, R. R.; Parkin, S. R.; Anthony, J. E. J. Appl. Phys. 2005, 98, 033701. doi:10.1063/1.1949711
Return to citation in text: [1] -
Lee, K. O.; Gan, T. T. Chem. Phys. Lett. 1977, 51, 120–124. doi:10.1016/0009-2614(77)85368-2
Return to citation in text: [1] -
Mattheus, C. C.; Dros, A. B.; Baas, J.; Oostergetel, G. T.; Meetsma, A.; de Boer, J. L.; Palstra, T. T. M. Synth. Met. 2003, 138, 475–481. doi:10.1016/S0379-6779(02)00467-8
Return to citation in text: [1] -
Mattheus, C. C.; Dros, A. B.; Baas, J.; Meetsma, A.; de Boer, J. L.; Palstra, T. T. M. Acta Crystallogr., Sect. C 2001, 57, 939–941. doi:10.1107/S010827010100703X
Return to citation in text: [1] -
Siegrist, T.; Kloc, C.; Schön, J. H.; Batlogg, B.; Haddon, R. C.; Berg, S.; Thomas, G. A. Angew. Chem., Int. Ed. 2001, 40, 1732–1736. doi:10.1002/1521-3773(20010504)40:9<1732::AID-ANIE17320>3.0.CO;2-7
Return to citation in text: [1] -
Mattheus, C. C.; de Wijs, G. A.; de Groot, R. A.; Palstra, T. T. M. J. Am. Chem. Soc. 2003, 125, 6323–6330. doi:10.1021/ja0211499
Return to citation in text: [1] -
Campbell, R. B.; Robertson, J. M.; Trotter, J. Acta Crystallogr. 1962, 15, 289–290. doi:10.1107/S0365110X62000699
Return to citation in text: [1] -
Anthony, J. E.; Subramanian, S.; Parkin, S. R.; Park, S. K.; Jackson, T. N. J. Mater. Chem. 2009, 19, 7984–7989. doi:10.1039/b911560a
Return to citation in text: [1] -
Kim, D. H.; Lee, D. Y.; Lee, H. S.; Lee, W. H.; Kim, Y. H.; Han, J. I.; Cho, K. Adv. Mater. 2007, 19, 678–682. doi:10.1002/adma.200601259
Return to citation in text: [1] -
Lim, Y.-F.; Shu, Y.; Parkin, S. R.; Anthony, J. E.; Malliaras, G. G. J. Mater. Chem. 2009, 19, 3049–3056. doi:10.1039/b818693f
Return to citation in text: [1]
| 43. | Mattheus, C. C.; Dros, A. B.; Baas, J.; Oostergetel, G. T.; Meetsma, A.; de Boer, J. L.; Palstra, T. T. M. Synth. Met. 2003, 138, 475–481. doi:10.1016/S0379-6779(02)00467-8 |
| 44. | Mattheus, C. C.; Dros, A. B.; Baas, J.; Meetsma, A.; de Boer, J. L.; Palstra, T. T. M. Acta Crystallogr., Sect. C 2001, 57, 939–941. doi:10.1107/S010827010100703X |
| 45. | Siegrist, T.; Kloc, C.; Schön, J. H.; Batlogg, B.; Haddon, R. C.; Berg, S.; Thomas, G. A. Angew. Chem., Int. Ed. 2001, 40, 1732–1736. doi:10.1002/1521-3773(20010504)40:9<1732::AID-ANIE17320>3.0.CO;2-7 |
| 46. | Mattheus, C. C.; de Wijs, G. A.; de Groot, R. A.; Palstra, T. T. M. J. Am. Chem. Soc. 2003, 125, 6323–6330. doi:10.1021/ja0211499 |
| 47. | Campbell, R. B.; Robertson, J. M.; Trotter, J. Acta Crystallogr. 1962, 15, 289–290. doi:10.1107/S0365110X62000699 |
| 1. | Figueira-Duarte, T. M.; Müllen, K. Chem. Rev. 2011, 111, 7260–7314. doi:10.1021/cr100428a |
| 12. | Lehnherr, D.; Tykwinski, R. R. Aust. J. Chem. 2011, 64, 919–929. doi:10.1071/CH11169 |
| 13. | Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z |
| 14. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 15. | Bendikov, M.; Wudl, F.; Perepichka, D. F. Chem. Rev. 2004, 104, 4891–4946. doi:10.1021/cr030666m |
| 21. | Lehnherr, D.; Gao, J.; Hegmann, F. A.; Tykwinski, R. R. Org. Lett. 2008, 10, 4779–4782. doi:10.1021/ol801886h |
| 35. | Lehnherr, D.; McDonald, R.; Tykwinski, R. R. Org. Lett. 2008, 10, 4163–4166. doi:10.1021/ol801464k |
| 36. | Waterloo, A. R.; Kunakom, S.; Hampel, F.; Tykwinski, R. R. Macromol. Chem. Phys. 2012, 213, 1020–1032. doi:10.1002/macp.201100622 |
| 37. | Boudebous, A.; Constable, E. C.; Housecroft, C. E.; Neuburger, M.; Schaffner, S. Acta Crystallogr., Sect. C 2006, 62, o243–o245. doi:10.1107/S0108270106008262 |
| 38. | Vets, N.; Smet, M.; Dehaen, W. Synlett 2005, 217–222. doi:10.1055/s-2004-836055 |
| 4. | Beaujuge, P. M.; Fréchet, J. M. J. J. Am. Chem. Soc. 2011, 133, 20009–20029. doi:10.1021/ja2073643 |
| 5. | Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Chem. Rev. 2009, 109, 5868–5923. doi:10.1021/cr900182s |
| 6. | Mártin, N.; Sánchez, L.; Herranz, M. Á.; Illescas, B.; Guldi, D. M. Acc. Chem. Res. 2007, 40, 1015–1024. doi:10.1021/ar700026t |
| 7. | Dang, M. T.; Hirsch, L.; Wantz, G.; Wuest, J. D. Chem. Rev. 2013, 113, 3734–3765. doi:10.1021/cr300005u |
| 8. | Arias, A. C.; MacKenzie, J. D.; McCulloch, I.; Rivnay, J.; Salleo, A. Chem. Rev. 2010, 110, 3–24. doi:10.1021/cr900150b |
| 9. | Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689–6735. doi:10.1021/cr9002984 |
| 10. | Mei, J.; Diao, Y.; Appleton, A. L.; Fang, L.; Bao, Z. J. Am. Chem. Soc. 2013, 135, 6724–6746. doi:10.1021/ja400881n |
| 11. | Lehnherr, D.; Tykwinski, R. R. Materials 2010, 3, 2772–2800. doi:10.3390/ma3042772 |
| 17. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Tykwinski, R. R. Angew. Chem., Int. Ed. 2010, 49, 6190–6194. doi:10.1002/anie.201000555 |
| 18. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179 |
| 34. | Zhang, X.; Jiang, X.; Luo, J.; Chi, C.; Chen, H.; Wu, J. Chem.–Eur. J. 2010, 16, 464–468. doi:10.1002/chem.200902675 |
| 3. | Lucas, B.; Trigaud, T.; Videlot-Ackermann, C. Polym. Int. 2012, 61, 374–389. doi:10.1002/pi.3213 |
| 18. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179 |
| 31. | Etschel, S. H.; Waterloo, A. R.; Margraf, J. T.; Amin, A. Y.; Hampel, F.; Jäger, C. M.; Clark, T.; Halik, M.; Tykwinski, R. R. Chem. Commun. 2013, 49, 6725–6727. doi:10.1039/c3cc43270j |
| 31. | Etschel, S. H.; Waterloo, A. R.; Margraf, J. T.; Amin, A. Y.; Hampel, F.; Jäger, C. M.; Clark, T.; Halik, M.; Tykwinski, R. R. Chem. Commun. 2013, 49, 6725–6727. doi:10.1039/c3cc43270j |
| 2. | Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689–6735. doi:10.1021/cr9002984 |
| 32. | Zhang, J.; Pawle, R. H.; Haas, T. E.; Thomas, S. W., III. Chem.–Eur. J. 2014, 20, 5880–5884. doi:10.1002/chem.201402003 |
| 33. | Kim, Y.-E.; Park, J.-W. Mol. Cryst. Liq. Cryst. 2006, 444, 137–143. doi:10.1080/15421400500365318 |
| 34. | Zhang, X.; Jiang, X.; Luo, J.; Chi, C.; Chen, H.; Wu, J. Chem.–Eur. J. 2010, 16, 464–468. doi:10.1002/chem.200902675 |
| 17. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Tykwinski, R. R. Angew. Chem., Int. Ed. 2010, 49, 6190–6194. doi:10.1002/anie.201000555 |
| 18. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179 |
| 19. | Lehnherr, D.; Gao, J.; Hegmann, F. A.; Tykwinski, R. R. J. Org. Chem. 2009, 74, 5017–5024. doi:10.1021/jo9007089 |
| 20. | Lehnherr, D.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Tetrahedron 2008, 64, 11449–11461. doi:10.1016/j.tet.2008.09.041 |
| 21. | Lehnherr, D.; Gao, J.; Hegmann, F. A.; Tykwinski, R. R. Org. Lett. 2008, 10, 4779–4782. doi:10.1021/ol801886h |
| 22. | Lehnherr, D.; Tykwinski, R. R. Org. Lett. 2007, 9, 4583–4586. doi:10.1021/ol702094d |
| 23. | Kuninobu, Y.; Seiki, T.; Kanamaru, S.; Nishina, Y.; Takai, K. Org. Lett. 2010, 12, 5287–5289. doi:10.1021/ol102349r |
| 24. | Li, Y.; Wu, Y.; Liu, P.; Prostran, Z.; Gardner, S.; Ong, B. S. Chem. Mater. 2007, 19, 418–423. doi:10.1021/cm062378n |
| 25. | Lee, S. H.; Kim, S. H.; Choo, D. J.; Jang, J. Org. Electron. 2010, 11, 1268–1272. doi:10.1016/j.orgel.2010.03.018 |
| 27. | Zade, S. S.; Bendikov, M. J. Phys. Org. Chem. 2012, 25, 452–461. doi:10.1002/poc.1941 |
| 28. | Biermann, D.; Schmidt, W. J. Am. Chem. Soc. 1980, 102, 3163–3173. doi:10.1021/ja00529a046 |
| 29. | Bénard, C. P.; Geng, Z.; Heuft, M. A.; VanCrey, K.; Fallis, A. G. J. Org. Chem. 2007, 72, 7229–7236. doi:10.1021/jo0709807 |
| 49. | Kim, D. H.; Lee, D. Y.; Lee, H. S.; Lee, W. H.; Kim, Y. H.; Han, J. I.; Cho, K. Adv. Mater. 2007, 19, 678–682. doi:10.1002/adma.200601259 |
| 13. | Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z |
| 14. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 30. | Anthony, J. E.; Eaton, D. L.; Parkin, S. R. Org. Lett. 2002, 4, 15–18. doi:10.1021/ol0167356 |
| 50. | Lim, Y.-F.; Shu, Y.; Parkin, S. R.; Anthony, J. E.; Malliaras, G. G. J. Mater. Chem. 2009, 19, 3049–3056. doi:10.1039/b818693f |
| 16. | Anthony, J. E.; Brooks, J. S.; Eaton, D. L.; Parkin, S. R. J. Am. Chem. Soc. 2001, 123, 9482–9483. doi:10.1021/ja0162459 |
| 48. | Anthony, J. E.; Subramanian, S.; Parkin, S. R.; Park, S. K.; Jackson, T. N. J. Mater. Chem. 2009, 19, 7984–7989. doi:10.1039/b911560a |
| 13. | Anthony, J. E. Chem. Rev. 2006, 106, 5028–5048. doi:10.1021/cr050966z |
| 14. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 12. | Lehnherr, D.; Tykwinski, R. R. Aust. J. Chem. 2011, 64, 919–929. doi:10.1071/CH11169 |
| 26. | Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Chem. Rev. 2012, 112, 2208–2267. doi:10.1021/cr100380z |
| 30. | Anthony, J. E.; Eaton, D. L.; Parkin, S. R. Org. Lett. 2002, 4, 15–18. doi:10.1021/ol0167356 |
| 11. | Lehnherr, D.; Tykwinski, R. R. Materials 2010, 3, 2772–2800. doi:10.3390/ma3042772 |
| 30. | Anthony, J. E.; Eaton, D. L.; Parkin, S. R. Org. Lett. 2002, 4, 15–18. doi:10.1021/ol0167356 |
| 31. | Etschel, S. H.; Waterloo, A. R.; Margraf, J. T.; Amin, A. Y.; Hampel, F.; Jäger, C. M.; Clark, T.; Halik, M.; Tykwinski, R. R. Chem. Commun. 2013, 49, 6725–6727. doi:10.1039/c3cc43270j |
| 36. | Waterloo, A. R.; Kunakom, S.; Hampel, F.; Tykwinski, R. R. Macromol. Chem. Phys. 2012, 213, 1020–1032. doi:10.1002/macp.201100622 |
| 39. | Maulding, D. R.; Roberts, B. G. J. Org. Chem. 1969, 34, 1734–1736. doi:10.1021/jo01258a045 |
| 18. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179 |
| 18. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179 |
| 35. | Lehnherr, D.; McDonald, R.; Tykwinski, R. R. Org. Lett. 2008, 10, 4163–4166. doi:10.1021/ol801464k |
| 17. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Tykwinski, R. R. Angew. Chem., Int. Ed. 2010, 49, 6190–6194. doi:10.1002/anie.201000555 |
| 18. | Lehnherr, D.; Murray, A. H.; McDonald, R.; Ferguson, M. J.; Tykwinski, R. R. Chem.–Eur. J. 2009, 15, 12580–12584. doi:10.1002/chem.200902179 |
| 41. | Ostroverkhova, O.; Shcherbyna, S.; Cooke, D. G.; Egerton, R. F.; Hegmann, F. A.; Tykwinski, R. R.; Parkin, S. R.; Anthony, J. E. J. Appl. Phys. 2005, 98, 033701. doi:10.1063/1.1949711 |
| 42. | Lee, K. O.; Gan, T. T. Chem. Phys. Lett. 1977, 51, 120–124. doi:10.1016/0009-2614(77)85368-2 |
| 11. | Lehnherr, D.; Tykwinski, R. R. Materials 2010, 3, 2772–2800. doi:10.3390/ma3042772 |
| 11. | Lehnherr, D.; Tykwinski, R. R. Materials 2010, 3, 2772–2800. doi:10.3390/ma3042772 |
| 35. | Lehnherr, D.; McDonald, R.; Tykwinski, R. R. Org. Lett. 2008, 10, 4163–4166. doi:10.1021/ol801464k |
| 40. | Platt, A. D.; Day, J.; Subramanian, S.; Anthony, J. E.; Ostroverkhova, O. J. Phys. Chem. C 2009, 113, 14006–14014. doi:10.1021/jp904021p |
| 39. | Maulding, D. R.; Roberts, B. G. J. Org. Chem. 1969, 34, 1734–1736. doi:10.1021/jo01258a045 |
© 2014 Waterloo et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)