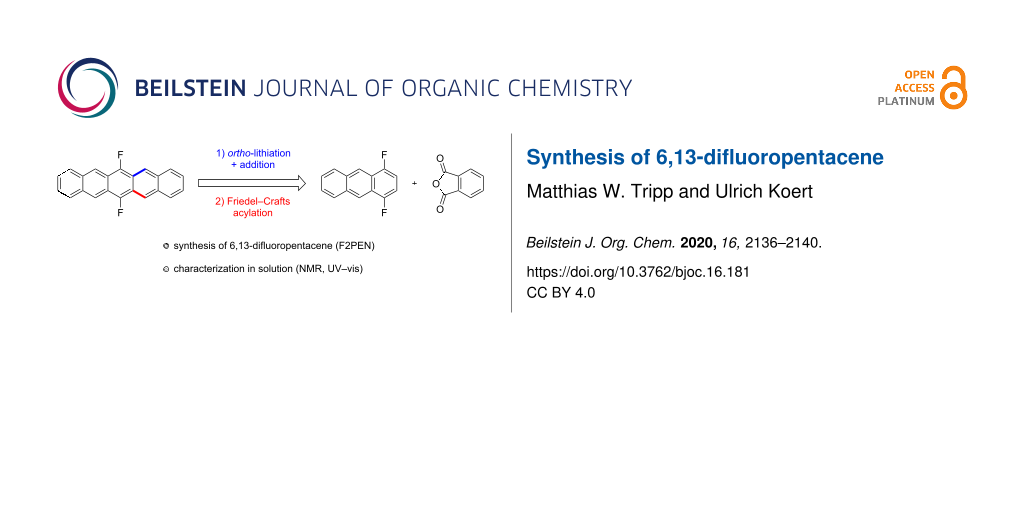
Abstract
6,13-Difluoropentacene was synthesized from 1,4-difluorobenzene. Friedel–Crafts annulation of the latter with phthalic anhydride and subsequent reduction of the anthraquinone gave 1,4-difluoroanthracene. After ortho-lithiation and reaction with phthalic anhydride a carboxylic acid was obtained whose Friedel–Crafts acylation and subsequent reductive removal of the oxygen-functionalities resulted in the formation of the target compound. The HOMO–LUMO gap of 6,13-difluoropentacene was determined via UV–vis spectroscopy and compared to other fluorinated pentacenes.

Graphical Abstract
Introduction
Pentacenes are a prototype in the field of organic semiconductors due to their expanded π-systems and low HOMO–LUMO gaps with applications in different molecular-based organic electronics like OFETs, OLEDs or organic photovoltaics [1-3]. An advantage of organic molecular electronics over inorganic alternatives is the versatility in design of new molecular materials by derivatization of known compounds [4]. Since the performance of these devices is strongly dependent on the optoelectronic solid-state properties of the pentacenes, predictable control of the electronic structure is desirable. A common strategy to influence the electronic properties of pentacenes, without significant effect on the molecular shape, is fluorination [5]. Suzuki et al. showed in 2004 the strong effect of perfluorination on the electronic properties [6]. While unsubstituted pentacene (1, PEN, Figure 1) is known to be a p-type semiconductor, the perfluorinated counterpart perfluoropentacene (2, PFP) showed an n-type behavior. In their synthesis and structural as well as electronic characterization of F4PEN 3, Bettinger et al. showed that the electronic properties of pentacenes are not a simple linear function of the degree of fluorination [7,8]. They reported that the fluorination in the 2,3,9,10-positions resulted in a larger HOMO–LUMO gap compared to PEN, while PFP showed a smaller gap than PEN. Another example for a partially fluorinated pentacene is the F6PEN 4 [9]. Its unilateral substitution pattern leads to a disctinct electronic structure and a criss-cross packing motif, unlike the herringbone structure of 1–3.
![[1860-5397-16-181-1]](/bjoc/content/figures/1860-5397-16-181-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of pentacene and fluorinated pentacenes.
Figure 1: Structures of pentacene and fluorinated pentacenes.
Here, the synthesis and characterization of 6,13-difluoropentacene (5, F2PEN) is reported. Since the central ring of pentacenes is known to be a weak spot (because of light-induced dimerization or oxidation) [2] the effects of fluorination in these positions are of interest.
Results and Discussion
6,13-Disubstituted pentacenes are often synthesized starting from 6,13-pentacenedione [2,3,10-12]. Initial attempts to prepare 6,13-difluoropentacene (5) along this route failed [13]. Therefore, an alternative route that commences with the central ring already carrying the two fluorine substituents was investigated. The retrosynthetic analysis for this strategy is shown in Scheme 1. The formation of the C5,5a-bond colored in red could be accomplished by an intramolecular Friedel–Crafts type acylation with the acylium-cation intermediate 6. The corresponding carboxylic acid precursor could be prepared by reaction of the anthracenyllithium 7 with phthalic anhydride (8). Intermediate 7 could be accessed by ortho-lithiation of anthracene 9. The synthesis of 9 by two consecutive Friedel–Crafts acylation reactions and reduction of the resulting anthraquinone could start from 8 and 1,4-difluorobenzene (10).
![[1860-5397-16-181-i1]](/bjoc/content/inline/1860-5397-16-181-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of F2PEN 5.
Scheme 1: Retrosynthetic analysis of F2PEN 5.
Starting point of the synthesis was the conversion of 1,4-difluorobenzene (10) to 1,4-difluoroanthraquinone (11, Scheme 2) [14]. The first Friedel-Crafts acylation of phthalic anhydride was achieved using AlCl3 in boiling 1,4-difluorobenzene (10). The second Friedel–Crafts acylation is then performed in polyphosphoric acid at 140 °C, giving anthraquinone 11 in 53% yield over two steps. Reduction of 11 to the anthracene 9 proceeded smoothly using zinc powder in 1,4-dioxane and aqueous ammonia under copper catalysis [15]. To achieve good yields, it was crucial to perform the reaction in a sealed pressure tube to prevent ammonia from degassing.
![[1860-5397-16-181-i2]](/bjoc/content/inline/1860-5397-16-181-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Different bases, temperatures and reaction times were screened to optimize the ortho-lithiation of 9. The best results were achieved using LDA as a base at –78 °C for 15 min and then rapidly adding the lithiated species 7 to phthalic anhydride in THF precooled to –100 °C [16]. Using these conditions, the carboxylic acid 12 was isolated in 82% yield.
The subsequent Friedel–Crafts acylation proofed to be challenging. Using reagents like PCl5 resulted in a quantitative substitution of the fluorine substituents in the corresponding pentacenequinone by chloride. This side reaction could be avoided using polyphosphoric acid as Friedel–Crafts reagent. The pentacenequinone showed very low solubility, which complicated a chromatographic isolation. However, the crude 1H NMR showed sufficiently clean conversion, hence the subsequent reduction step using NaBH4 to diol 13 was performed without further purification of the quinone.
The low yield for the formation of 13 seems to be an intrinsic instability of its ortho-fluorobenzylic alcohol moiety. Moreover, the compound quantitatively decomposes to 6,13-pentacenequinone 15 in CD2Cl2 solution at room temperature within 2 h. A possible reaction pathway for this degradation in the presence of moisture is an intermolecular attack of water, leading to a fluorohydrine 14 (Scheme 3). In absence of water, this reaction might take place in an intramolecular manner. The fluorohydrine 14 then rapidly decomposes to 6,13-pentacenequinone (15) after elimination of HF.
![[1860-5397-16-181-i3]](/bjoc/content/inline/1860-5397-16-181-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Decomposition of diol 13 in solution.
Scheme 3: Decomposition of diol 13 in solution.
The final aromatization of diol 13 to the target molecule 5 proceeded smoothly in 74% yield using SnCl2 in 1,4-dioxane and aqueous HCl [11,12]. F2PEN 5 can be stored under inertgas atmosphere at −20 °C for a month without noticeable decomposition. However, in solution under ambient atmosphere and sunlight decomposition takes place quickly, which is indicated by decolorization of a purple solution in CH2Cl2 within 3 min. 1H NMR analysis showed 6,13-pentacenequinone (15) as the degradation product. The rate of degradation in degassed C6D6 was slow enough to obtain a clean 1H NMR spectrum (see Supporing Information File 1) [17].
Next, the HOMO–LUMO gap of 5 was investigated via UV–vis spectroscopy in solution (Figure 2). The λmax was determined to be at 597 nm, which corresponds to a HOMO–LUMO gap of ΔE = 2.08 eV. Comparison with the values of PEN (ΔE = 2.13 eV) [9] and PFP (ΔE = 1.99 eV) [9] shows that the electronic structure of F2PEN 5 lies in between these two. While fluorine substituents in 2,3,9,10-positions (F4PEN 3) result in a larger HOMO–LUMO gap compared to PEN [7,8], substitution in the 6,13-positions leads to smaller band-gaps, similar to the unilaterally substituted F6PEN 4 [9].
![[1860-5397-16-181-2]](/bjoc/content/figures/1860-5397-16-181-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: UV–vis spectrum of F2PEN 5 in CH2Cl2.
Figure 2: UV–vis spectrum of F2PEN 5 in CH2Cl2.
Conclusion
Starting from 1,4-difluorobenzene a synthetic route to 6,13-difluoropentacene was established (7% over seven steps). Key steps are Friedel–Crafts acylation reactions and an ortho-directed lithiation of 1,4-difluoroanthracene. This strategy could be applicable for the synthesis of differently substituted 6,13-difluoropentacenes as well.
Supporting Information
| Supporting Information File 1: Experimental details, spectroscopic and analytical data of all new compounds. | ||
| Format: PDF | Size: 1.4 MB | Download |
References
-
Ruiz, R.; Choudhary, D.; Nickel, B.; Toccoli, T.; Chang, K.-C.; Mayer, A. C.; Clancy, P.; Blakely, J. M.; Headrick, R. L.; Iannotta, S.; Malliaras, G. G. Chem. Mater. 2004, 16, 4497–4508. doi:10.1021/cm049563q
Return to citation in text: [1] -
Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045
Return to citation in text: [1] [2] [3] -
Bunz, U. H. F. Acc. Chem. Res. 2015, 48, 1676–1686. doi:10.1021/acs.accounts.5b00118
Return to citation in text: [1] [2] -
Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689–6735. doi:10.1021/cr9002984
Return to citation in text: [1] -
Tang, M. L.; Bao, Z. Chem. Mater. 2011, 23, 446–455. doi:10.1021/cm102182x
Return to citation in text: [1] -
Sakamoto, Y.; Suzuki, T.; Kobayashi, M.; Gao, Y.; Fukai, Y.; Inoue, Y.; Sato, F.; Tokito, S. J. Am. Chem. Soc. 2004, 126, 8138–8140. doi:10.1021/ja0476258
Return to citation in text: [1] -
Shen, B.; Geiger, T.; Einholz, R.; Reicherter, F.; Schundelmeier, S.; Maichle-Mössmer, C.; Speiser, B.; Bettinger, H. F. J. Org. Chem. 2018, 83, 3149–3158. doi:10.1021/acs.joc.7b03241
Return to citation in text: [1] [2] -
Geiger, T.; Schundelmeier, S.; Hummel, T.; Ströbele, M.; Leis, W.; Seitz, M.; Zeiser, C.; Moretti, L.; Maiuri, M.; Cerullo, G.; Broch, K.; Vahland, J.; Leo, K.; Maichle-Mössmer, C.; Speiser, B.; Bettinger, H. F. Chem. – Eur. J. 2020, 26, 3420–3434. doi:10.1002/chem.201905843
Return to citation in text: [1] [2] -
Hofmann, P. E.; Tripp, M. W.; Bischof, D.; Grell, Y.; Schiller, A. L. C.; Breuer, T.; Ivlev, S. I.; Witte, G.; Koert, U. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202006489
Return to citation in text: [1] [2] [3] [4] -
Tykwinski, R. R. Acc. Chem. Res. 2019, 52, 2056–2069. doi:10.1021/acs.accounts.9b00216
Return to citation in text: [1] -
Schwaben, J.; Münster, N.; Klues, M.; Breuer, T.; Hofmann, P.; Harms, K.; Witte, G.; Koert, U. Chem. – Eur. J. 2015, 21, 13758–13771. doi:10.1002/chem.201501399
Return to citation in text: [1] [2] -
Schwaben, J.; Münster, N.; Breuer, T.; Klues, M.; Harms, K.; Witte, G.; Koert, U. Eur. J. Org. Chem. 2013, 1639–1643. doi:10.1002/ejoc.201201714
Return to citation in text: [1] [2] -
Preparations of alkyl-disubstituted 6,13-difluoropentacenes via the corresponding pentacenequinone are reported in a patent: Okamoto, K; Jpn. Kokai Tokkyo Koho, JP 2008024684, 07.02.2008.
Return to citation in text: [1] -
Krapcho, A. P.; Getahun, Z. Synth. Commun. 1985, 15, 907–910. doi:10.1080/00397918508063889
Return to citation in text: [1] -
Wang, J.; Leung, L. M. Dyes Pigm. 2013, 99, 105–115. doi:10.1016/j.dyepig.2013.04.021
Return to citation in text: [1] -
Parham, W. E.; Piccirilli, R. M. J. Org. Chem. 1976, 41, 1268–1269. doi:10.1021/jo00869a046
Return to citation in text: [1] -
Because of the low solubility and decomposition in solution, no 13C NMR spectrum could be obtained.
Return to citation in text: [1]
| 7. | Shen, B.; Geiger, T.; Einholz, R.; Reicherter, F.; Schundelmeier, S.; Maichle-Mössmer, C.; Speiser, B.; Bettinger, H. F. J. Org. Chem. 2018, 83, 3149–3158. doi:10.1021/acs.joc.7b03241 |
| 8. | Geiger, T.; Schundelmeier, S.; Hummel, T.; Ströbele, M.; Leis, W.; Seitz, M.; Zeiser, C.; Moretti, L.; Maiuri, M.; Cerullo, G.; Broch, K.; Vahland, J.; Leo, K.; Maichle-Mössmer, C.; Speiser, B.; Bettinger, H. F. Chem. – Eur. J. 2020, 26, 3420–3434. doi:10.1002/chem.201905843 |
| 9. | Hofmann, P. E.; Tripp, M. W.; Bischof, D.; Grell, Y.; Schiller, A. L. C.; Breuer, T.; Ivlev, S. I.; Witte, G.; Koert, U. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202006489 |
| 1. | Ruiz, R.; Choudhary, D.; Nickel, B.; Toccoli, T.; Chang, K.-C.; Mayer, A. C.; Clancy, P.; Blakely, J. M.; Headrick, R. L.; Iannotta, S.; Malliaras, G. G. Chem. Mater. 2004, 16, 4497–4508. doi:10.1021/cm049563q |
| 2. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 3. | Bunz, U. H. F. Acc. Chem. Res. 2015, 48, 1676–1686. doi:10.1021/acs.accounts.5b00118 |
| 7. | Shen, B.; Geiger, T.; Einholz, R.; Reicherter, F.; Schundelmeier, S.; Maichle-Mössmer, C.; Speiser, B.; Bettinger, H. F. J. Org. Chem. 2018, 83, 3149–3158. doi:10.1021/acs.joc.7b03241 |
| 8. | Geiger, T.; Schundelmeier, S.; Hummel, T.; Ströbele, M.; Leis, W.; Seitz, M.; Zeiser, C.; Moretti, L.; Maiuri, M.; Cerullo, G.; Broch, K.; Vahland, J.; Leo, K.; Maichle-Mössmer, C.; Speiser, B.; Bettinger, H. F. Chem. – Eur. J. 2020, 26, 3420–3434. doi:10.1002/chem.201905843 |
| 9. | Hofmann, P. E.; Tripp, M. W.; Bischof, D.; Grell, Y.; Schiller, A. L. C.; Breuer, T.; Ivlev, S. I.; Witte, G.; Koert, U. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202006489 |
| 6. | Sakamoto, Y.; Suzuki, T.; Kobayashi, M.; Gao, Y.; Fukai, Y.; Inoue, Y.; Sato, F.; Tokito, S. J. Am. Chem. Soc. 2004, 126, 8138–8140. doi:10.1021/ja0476258 |
| 9. | Hofmann, P. E.; Tripp, M. W.; Bischof, D.; Grell, Y.; Schiller, A. L. C.; Breuer, T.; Ivlev, S. I.; Witte, G.; Koert, U. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202006489 |
| 11. | Schwaben, J.; Münster, N.; Klues, M.; Breuer, T.; Hofmann, P.; Harms, K.; Witte, G.; Koert, U. Chem. – Eur. J. 2015, 21, 13758–13771. doi:10.1002/chem.201501399 |
| 12. | Schwaben, J.; Münster, N.; Breuer, T.; Klues, M.; Harms, K.; Witte, G.; Koert, U. Eur. J. Org. Chem. 2013, 1639–1643. doi:10.1002/ejoc.201201714 |
| 4. | Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689–6735. doi:10.1021/cr9002984 |
| 17. | Because of the low solubility and decomposition in solution, no 13C NMR spectrum could be obtained. |
| 13. | Preparations of alkyl-disubstituted 6,13-difluoropentacenes via the corresponding pentacenequinone are reported in a patent: Okamoto, K; Jpn. Kokai Tokkyo Koho, JP 2008024684, 07.02.2008. |
| 15. | Wang, J.; Leung, L. M. Dyes Pigm. 2013, 99, 105–115. doi:10.1016/j.dyepig.2013.04.021 |
| 2. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 3. | Bunz, U. H. F. Acc. Chem. Res. 2015, 48, 1676–1686. doi:10.1021/acs.accounts.5b00118 |
| 10. | Tykwinski, R. R. Acc. Chem. Res. 2019, 52, 2056–2069. doi:10.1021/acs.accounts.9b00216 |
| 11. | Schwaben, J.; Münster, N.; Klues, M.; Breuer, T.; Hofmann, P.; Harms, K.; Witte, G.; Koert, U. Chem. – Eur. J. 2015, 21, 13758–13771. doi:10.1002/chem.201501399 |
| 12. | Schwaben, J.; Münster, N.; Breuer, T.; Klues, M.; Harms, K.; Witte, G.; Koert, U. Eur. J. Org. Chem. 2013, 1639–1643. doi:10.1002/ejoc.201201714 |
| 16. | Parham, W. E.; Piccirilli, R. M. J. Org. Chem. 1976, 41, 1268–1269. doi:10.1021/jo00869a046 |
| 2. | Anthony, J. E. Angew. Chem., Int. Ed. 2008, 47, 452–483. doi:10.1002/anie.200604045 |
| 9. | Hofmann, P. E.; Tripp, M. W.; Bischof, D.; Grell, Y.; Schiller, A. L. C.; Breuer, T.; Ivlev, S. I.; Witte, G.; Koert, U. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202006489 |
| 14. | Krapcho, A. P.; Getahun, Z. Synth. Commun. 1985, 15, 907–910. doi:10.1080/00397918508063889 |
© 2020 Tripp and Koert; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)