Abstract
In this work we present for the first time the synthesis of novel 5-hydroxymethylcytosine (5hmC) and 5-formylcytosine (5fC) derivatives that can be used as tools in the emerging field of epigenetics for deciphering chemical biology of TET-mediated processes.

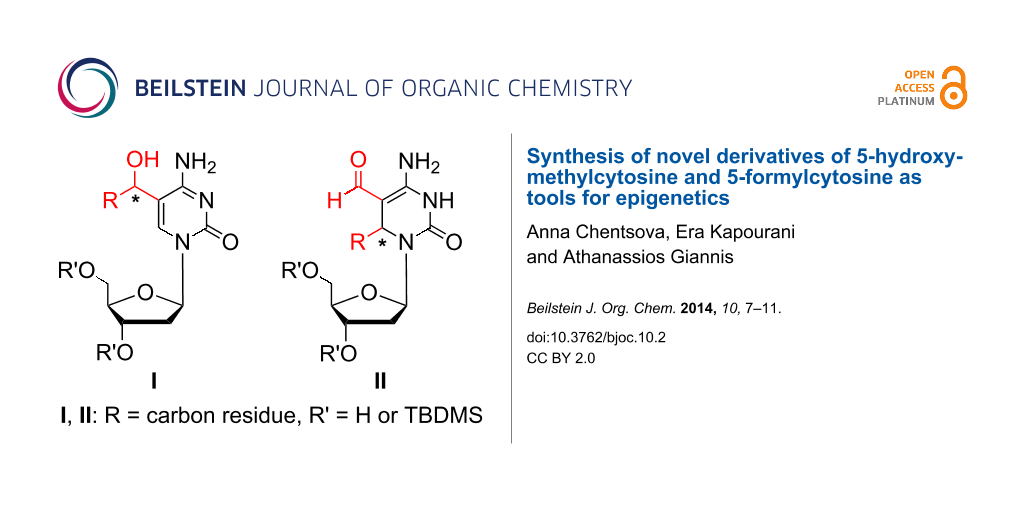
Graphical Abstract
Introduction
Epigenetic modifications play a crucial role in cell differentiation and cell development [1]. They control gene expression through several mechanisms such as non-coding RNAs, histone modifications (acetylation, methylation, phosphorylation, etc.) [2], and DNA methylation [3-7]. The latter takes place at the C-5 position of the cytosine moiety in CpG islands establishing the so called 5th base: 5-methylcytosine (5mC), a well-known epigenetic mark that correlates with gene silencing [8]. Recently, conversion of the 5mC moiety to 5-hydroxymethylcytosine (5hmC) and to higher oxidation products such as 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) by the action of ten-eleven-translocation (TET) enzymes was discovered [9-13]. The TET proteins are identified as 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenases [10,14]. Whereas the DNA methylation is a densely studied field, its reverse process has not yet been deciphered. In trying to understand DNA demethylation several mechanisms involving new cytosine-modified bases as intermediates have been proposed (Scheme 1). (1) The most widely accepted pathway includes iterative oxidation of 5mC catalyzed by TET enzymes followed by removal of 5fC and 5caC by thymine DNA glycosylase (TDG). Excision of 5fC and 5caC generates an abasic site, which is further repaired resulting in replacement of 5mC with unmodified cytosine (C) [15-18]. (2) The second alternative scenario still remains controversial [19,20]. It links the oxidative action of TET enzymes, the subsequent deamination of 5hmC to 5-hydroxymethyluracil (5hmU) by cytidine deaminases AID (activation-induced cytidine deaminase) or APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide) with the base excision repair (BER) machinery [15-17,21]. (3) Among other putative demethylation mechanisms is the direct dehydroxymethylation of 5hmC to cytosine by action of DNA methyltransferases (DNMT). This enzymatic process was observed in vitro, whether it also works in vivo is yet to be elucidated [15,22]. (4) Lastly, decarboxylation of the 5caC by an unknown decarboxylase excluding action of BER should also be considered [15,23]. This variety of demethylation pathways might indicate that different tissues utilize different demethylation pathways [1,24].
![[1860-5397-10-2-i1]](/bjoc/content/inline/1860-5397-10-2-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed steps for DNA demethylation (for details see text).
Scheme 1: Proposed steps for DNA demethylation (for details see text).
While DNA methylation is usually associated with gene repression [8,25], active demethylation seems to allow cells to unblock silenced genes aiming at epigenetic reprogramming of their genetic material [26]. Current accepted models propose that 5hmC could be involved in epigenetic modulation of gene activity. In fact, 5hmC was discovered also in embryonic stem cells and seems to play a decisive role in their self-renewal process [27]. Interestingly, the levels of 5hmC in several cancer types are strongly reduced relative to the corresponding normal tissue around the tumor [28]. To gain deeper insights into the chemical biology of DNA demethylation pathways further exploration of the TET-mediated processes is necessary. Analogues of 5hmC with substituents preventing formation of 5fC and 5caC species could serve as useful tools for ongoing investigations in this emerging field.
Results and Discussion
Herein, we describe the synthesis of compounds with the general formula I which represents modified cytidine analogues bearing a secondary alcohol at position C-5 of cytosine. Additionally, a synthesis of 3,6-dihydrodeoxycytidine derivatives of general formula II is presented (Figure 1).
![[1860-5397-10-2-1]](/bjoc/content/figures/1860-5397-10-2-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of the synthesized compounds.
Figure 1: Structures of the synthesized compounds.
We chose the known aldehyde 1 [29] (prepared from commercially available 2’-deoxycytidine) as a starting material for the envisioned transformations (Scheme 2). To the best of our knowledge, the addition of organometallic compounds (organolithium and organomagnesium, etc.) to aldehyde 1 is not described in the literature. Compound 1 was readily converted to 5hmC analogues 2a–e by treatment with various Grignard reagents (methylmagnesium bromide, THF, 0 °C → room temperature, or vinylmagnesium bromide, THF, 0 °C → room temperature) and organolithium reagents (lithium (trimethylsilyl)acetylide, THF, −40 °C → −20 °C or lithium phenylacetylide, THF, −78 °C → −50 °C) (Scheme 2). These alcohols were obtained as a mixture of diastereomers in yields ranging from 43% to 96% (Table 1). Compound 2b was isolated in moderate yield of 43% due to the cleavage of the TMS-group during the reaction resulting in formation of derivative 2e with a yield of 26%. The obtained derivatives 2a–d were further treated with Olah’s reagent and pyridine in EtOAc at room temperature or HF·triethylamine complex [30] in DCM at 0 °C to afford the deprotected 2’-deoxycytidine analogues 3a–d as mixtures of diastereomers in yields of 60–75%.
![[1860-5397-10-2-i2]](/bjoc/content/inline/1860-5397-10-2-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the 2'-deoxycytidine analogues.
Scheme 2: Synthesis of the 2'-deoxycytidine analogues.
Table 1: Yields and ratios of diastereomeric alcohols 2a–e and 3a–d.
| Entry | 2/3 | Yield [%] | Ratioa |
|---|---|---|---|
| 1 | 2a, R = methyl | 96 | 1.1:1 |
| 2 | 2b, R = (TMS)ethynyl | 43 | 1.9:1 |
| 3 | 2c, R = phenylethynyl | 68 | 1.2:1 |
| 4 | 2d, R = vinyl | 77 | 1.1:1 |
| 5 | 2e, R = ethynyl | 26 | 1.2:1 |
| 6 | 3a, R = methyl | 75 | n.d. |
| 7 | 3b, R = ethynyl | 60 | n.d. |
| 8 | 3c, R = phenylethynyl | 72 | 1.1:1 |
| 9 | 3d, R = vinyl | 73 | n.d. |
aDetermined by 1H NMR; n.d. = not determined.
Next, we synthesized the N-4-protected cytidine derivatives 4 and 5 by treatment of aldehyde 1 with β,β,β-trichloro-tert-butoxycarbonyl chloride (TCBocCl) [31] in the presence of pyridine in DCM (Scheme 3). The reaction of 4 with Grignard (methylmagnesium bromide, THF, 0 °C → room temperature, or vinylmagnesium bromide, THF, 0 °C → room temperature) and organolithium reagents (lithium (trimethylsilyl)acetylide, THF, −60 °C → −50 °C or lithium phenylacetylide, THF, −78 °C) afforded derivatives 6a–c and carbamates 7a–c as mixtures of diastereomers (Table 2). It should be mentioned that upon storage at room temperature derivatives 6a–c undergo slow intramolecular cyclization to the corresponding carbamates 7a–c. The reaction of aldehyde 4 and vinylmagnesium bromide yielded directly carbamate 7d.
![[1860-5397-10-2-i3]](/bjoc/content/inline/1860-5397-10-2-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions of TCBoc-protected aldehydes 4 and 5 with organometallic reagents.
Scheme 3: Reactions of TCBoc-protected aldehydes 4 and 5 with organometallic reagents.
Table 2: Yields and ratios of diastereomers 6a–c, 7a–d, 8a–e and 9a–d.
| Entry | 6/7/8/9 | Yield [%] | Ratioa |
|---|---|---|---|
| 1 | 6a, R = methyl | 28 | n.d |
| 2 | 6b, R = (TMS)ethynyl | 42 | 2:1 |
| 3 | 6c, R = phenylethynyl | 42 | 1.6:1 |
| 4 | 7a, R = methyl | 35 | 1.9:1 |
| 5 | 7b, R = (TMS)ethynyl | 30 | 1.1:1 |
| 6 | 7c, R = phenylethynyl | 30 | 1.4:1 |
| 7 | 7d, R = vinyl | 69 | 2.3:1 |
| 8 | 8a, R = methyl, R1 = H | 38 | 2.4:1 |
| 9 | 8b, R = (TMS)ethynyl, R1 = H | 80 | 3.2:1b |
| 10 | 8c, R = phenylethynyl, R1 = H | 71 | 1.1:1 |
| 11 | 8d, R = vinyl, R1 = H | 37 | 2.6:1 |
| 12 | 8e, R = vinyl, R1 = TCBoc | 17 | 5.7:1 |
| 13 | 9a, R = methyl | 40 | 2.4:1 |
| 14 | 9b, R = (TMS)ethynyl | 77 | – |
| 15 | 9c, R = phenylethynyl | 44 | 1:1 |
| 16 | 9d, R = vinyl | 61 | 2.6:1 |
aDetermined by 1H NMR; bpure epimers were isolated by HPLC; n.d. = not determined.
Surprisingly, the reaction of derivative 5 bearing a N-(TCBoc)2 group with organometallic compounds (methylmagnesium bromide, THF, 0 °C → room temperature, lithium (trimethylsilyl)acetylide, THF, −50 °C, lithium phenylacetylide, THF, −78 °C → −50 °C, vinylmagnesium bromide, THF, 0 °C) afforded 3,6-dihydrodeoxycytidine derivatives 8a–e as mixtures of diastereomers. In case of 8b the diastereomers were separated by HPLC. Products arising from addition of the organometallic reagents to the aldehyde group (1,2-addition) were not observed. The formation of compounds 8a–d can be explained assuming a Michael-type reaction of aldehyde 5 with organometallic reagents, subsequent isomerisation of the double bond followed by removal of one TCBoc group during the reaction and work-up as shown in Scheme 4.
![[1860-5397-10-2-i4]](/bjoc/content/inline/1860-5397-10-2-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the formation of 3,6-dihydrodeoxycytidine derivatives 8a–d (M = Li, Mg).
Scheme 4: Proposed mechanism for the formation of 3,6-dihydrodeoxycytidine derivatives 8a–d (M = Li, Mg).
Finally, cleavage of the TCBoc group was achieved by the action of the 10% Cd–Pb couple [32] on compounds 8a–d in THF and 1.0 M aq NH4OAc to provide derivatives 9a–d (Scheme 3).
Conclusion
In summary, the reaction of 5fC derivatives 1, 4, and 5 with organometallic reagents (RMgBr, RLi) was investigated and enabled the synthesis of novel derivatives of 5-hydroxymethylcytosine and 5-formylcytosine: whereas aldehydes 1 and 4 afforded cytosine derivatives 2a–e, 6a–c and 7a–d, the reaction of derivative 5 yielded 3,6-dihydrodeoxycytidine derivatives 8a–d which subsequently after removal of the TCBoc group afforded derivatives 9a–d. These new nucleobase modified 2’-deoxycytidine analogues can be used in the synthesis [29,33,34] of modified DNA oligomers for further studies of the TET-mediated processes which are of great importance in the emerging field of epigenetics. In addition they could find application as novel antivirals and/or as antimetabolites [35,36]. The majority of the obtained compounds contain functionalized side chains thus allowing further manipulations.
Supporting Information
| Supporting Information File 1: Experimental details and analytical data of all synthesized compounds. | ||
| Format: PDF | Size: 10.0 MB | Download |
References
-
Branco, M. R.; Ficz, G.; Reik, W. Nat. Rev. Genet. 2012, 13, 7–13. doi:10.1038/nrg3080
Return to citation in text: [1] [2] -
Biel, M.; Wascholowski, V.; Giannis, A. Angew. Chem., Int. Ed. 2005, 44, 3186–3216. doi:10.1002/anie.200461346
Return to citation in text: [1] -
Esteller, M. Genes Cancer 2011, 2, 604–606. doi:10.1177/1947601911423096
Return to citation in text: [1] -
Fuks, F. Curr. Opin. Genet. Dev. 2005, 15, 490–495. doi:10.1016/j.gde.2005.08.002
Return to citation in text: [1] -
Dawson, M. A.; Kouzarides, T. Cell 2012, 150, 12–27. doi:10.1016/j.cell.2012.06.013
Return to citation in text: [1] -
Dawson, M. A.; Kouzarides, T.; Huntly, B. J. P. N. Engl. J. Med. 2012, 367, 647–657. doi:10.1056/NEJMra1112635
Return to citation in text: [1] -
Hamm, C. A.; Costa, F. F. Drug Discovery Today 2011, 16, 626–635. doi:10.1016/j.drudis.2011.04.007
Return to citation in text: [1] -
Klose, R. J.; Bird, A. P. Trends Biochem. Sci. 2006, 31, 89–97. doi:10.1016/j.tibs.2005.12.008
Return to citation in text: [1] [2] -
Kriaucionis, S.; Heintz, N. Science 2009, 324, 929–930. doi:10.1126/science.1169786
Return to citation in text: [1] -
Tahiliani, M.; Koh, K. P.; Shen, Y.; Pastor, W. A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L. M.; Liu, D. R.; Aravind, L.; Rao, A. Science 2009, 324, 930–935. doi:10.1126/science.1170116
Return to citation in text: [1] [2] -
He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; Sun, Y.; Li, X.; Dai, Q.; Song, C.-X.; Zhang, K.; He, C.; Xu, G.-L. Science 2011, 333, 1303–1307. doi:10.1126/science.1210944
Return to citation in text: [1] -
Ito, S.; Shen, L.; Dai, Q.; Wu, S. C.; Collins, L. B.; Swenberg, J. A.; He, C.; Zhang, Y. Science 2011, 333, 1300–1303. doi:10.1126/science.1210597
Return to citation in text: [1] -
Pfaffeneder, T.; Hackner, B.; Truß, M.; Münzel, M.; Müller, M.; Deiml, C. A.; Hagemeier, C.; Carell, T. Angew. Chem., Int. Ed. 2011, 50, 7008–7012. doi:10.1002/anie.201103899
Return to citation in text: [1] -
Rose, N. R.; McDonough, M. A.; King, O. N. F.; Kawamura, A.; Schofield, C. J. Chem. Soc. Rev. 2011, 40, 4364–4397. doi:10.1039/c0cs00203h
See for an excellent review on 2-oxoglutarate dependend oxygenases.
Return to citation in text: [1] -
Pastor, W. A.; Aravind, L.; Rao, A. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. doi:10.1038/nrm3589
Return to citation in text: [1] [2] [3] [4] -
Fu, Y.; He, C. Curr. Opin. Chem. Biol. 2012, 16, 516–524. doi:10.1016/j.cbpa.2012.10.002
Return to citation in text: [1] [2] -
Carell, T.; Brandmayr, C.; Hienzsch, A.; Müller, M.; Pearson, D.; Reiter, V.; Thoma, I.; Thumbs, P.; Wagner, M. Angew. Chem., Int. Ed. 2012, 51, 7110–7131. doi:10.1002/anie.201201193
Return to citation in text: [1] [2] -
Pfeifer, G. P.; Kadam, S.; Jin, S. G. Epigenet. Chromatin 2013, 6, 10. doi:10.1186/1756-8935-6-10
Return to citation in text: [1] -
Guo, J. U.; Su, Y.; Zhong, C.; Ming, G.-l.; Song, H. Cell 2011, 145, 423–434. doi:10.1016/j.cell.2011.03.022
Return to citation in text: [1] -
Nabel, C. S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H. L.; Stivers, J. T.; Zhang, Y.; Kohli, R. M. Nat. Chem. Biol. 2012, 8, 751–758. doi:10.1038/nchembio.1042
Return to citation in text: [1] -
Bhutani, N.; Burns, D. M.; Blau, H. M. Cell 2011, 146, 866–872. doi:10.1016/j.cell.2011.08.042
Return to citation in text: [1] -
Chen, C.-C.; Wang, K.-Y.; Shen, C.-K. J. J. Biol. Chem. 2012, 287, 33116–33121. doi:10.1074/jbc.C112.406975
Return to citation in text: [1] -
Schiesser, S.; Hackner, B.; Pfaffeneder, T.; Müller, M.; Hagemeier, C.; Truss, M.; Carell, T. Angew. Chem., Int. Ed. 2012, 51, 6516–6520. doi:10.1002/anie.201202583
Return to citation in text: [1] -
Auclair, G.; Weber, M. Biochimie 2012, 94, 2202–2211. doi:10.1016/j.biochi.2012.05.016
Return to citation in text: [1] -
Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A. J.; Socci, N. D.; Scandura, J. M. PLoS One 2011, 6, e14524. doi:10.1371/journal.pone.0014524
Return to citation in text: [1] -
Münzel, M.; Lischke, U.; Stathis, D.; Pfaffeneder, T.; Gnerlich, F. A.; Deiml, C. A.; Koch, S. C.; Karaghiosoff, K.; Carell, T. Chem.–Eur. J. 2011, 17, 13782–13788. doi:10.1002/chem.201102782
Return to citation in text: [1] -
Ito, S.; D'Alessio, A. C.; Taranova, O. V.; Hong, K.; Sowers, L. C.; Zhang, Y. Nature 2010, 466, 1129–1133. doi:10.1038/nature09303
Return to citation in text: [1] -
Jin, S.-G.; Jiang, Y.; Qiu, R.; Rauch, T. A.; Wang, Y.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G. P. Cancer Res. 2011, 71, 7360–7365. doi:10.1158/0008-5472.CAN-11-2023
Return to citation in text: [1] -
Münzel, M.; Globisch, D.; Trindler, C.; Carell, T. Org. Lett. 2010, 12, 5671–5673. doi:10.1021/ol102408t
Return to citation in text: [1] [2] -
Münzel, M.; Globisch, D.; Brückl, T.; Wagner, M.; Welzmiller, V.; Michalakis, S.; Müller, M.; Biel, M.; Carell, T. Angew. Chem., Int. Ed. 2010, 49, 5375–5377. doi:10.1002/anie.201002033
Return to citation in text: [1] -
Schneiderwind, R. G. K.; Ugi, I. Tetrahedron 1983, 39, 2207–2210. doi:10.1016/S0040-4020(01)91939-8
Return to citation in text: [1] -
Dong, Q.; Anderson, C. E.; Ciufolini, M. A. Tetrahedron Lett. 1995, 36, 5681–5682. doi:10.1016/0040-4039(95)01122-X
Return to citation in text: [1] -
Steigenberger, B.; Schiesser, S.; Hackner, B.; Brandmayr, C.; Laube, S. K.; Steinbacher, J.; Pfaffeneder, T.; Carell, T. Org. Lett. 2013, 15, 366–369. doi:10.1021/ol3033219
Return to citation in text: [1] -
Dai, Q.; He, C. Org. Lett. 2011, 13, 3446–3449. doi:10.1021/ol201189n
Return to citation in text: [1] -
Kifli, N.; De Clercq, E.; Balzarini, J.; Simons, C. Bioorg. Med. Chem. 2004, 12, 4245–4252. doi:10.1016/j.bmc.2004.05.017
Return to citation in text: [1] -
Matsuda, A.; Sasaki, T. Cancer Sci. 2004, 95, 105–111. doi:10.1111/j.1349-7006.2004.tb03189.x
Return to citation in text: [1]
| 31. | Schneiderwind, R. G. K.; Ugi, I. Tetrahedron 1983, 39, 2207–2210. doi:10.1016/S0040-4020(01)91939-8 |
| 29. | Münzel, M.; Globisch, D.; Trindler, C.; Carell, T. Org. Lett. 2010, 12, 5671–5673. doi:10.1021/ol102408t |
| 30. | Münzel, M.; Globisch, D.; Brückl, T.; Wagner, M.; Welzmiller, V.; Michalakis, S.; Müller, M.; Biel, M.; Carell, T. Angew. Chem., Int. Ed. 2010, 49, 5375–5377. doi:10.1002/anie.201002033 |
| 1. | Branco, M. R.; Ficz, G.; Reik, W. Nat. Rev. Genet. 2012, 13, 7–13. doi:10.1038/nrg3080 |
| 9. | Kriaucionis, S.; Heintz, N. Science 2009, 324, 929–930. doi:10.1126/science.1169786 |
| 10. | Tahiliani, M.; Koh, K. P.; Shen, Y.; Pastor, W. A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L. M.; Liu, D. R.; Aravind, L.; Rao, A. Science 2009, 324, 930–935. doi:10.1126/science.1170116 |
| 11. | He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; Sun, Y.; Li, X.; Dai, Q.; Song, C.-X.; Zhang, K.; He, C.; Xu, G.-L. Science 2011, 333, 1303–1307. doi:10.1126/science.1210944 |
| 12. | Ito, S.; Shen, L.; Dai, Q.; Wu, S. C.; Collins, L. B.; Swenberg, J. A.; He, C.; Zhang, Y. Science 2011, 333, 1300–1303. doi:10.1126/science.1210597 |
| 13. | Pfaffeneder, T.; Hackner, B.; Truß, M.; Münzel, M.; Müller, M.; Deiml, C. A.; Hagemeier, C.; Carell, T. Angew. Chem., Int. Ed. 2011, 50, 7008–7012. doi:10.1002/anie.201103899 |
| 27. | Ito, S.; D'Alessio, A. C.; Taranova, O. V.; Hong, K.; Sowers, L. C.; Zhang, Y. Nature 2010, 466, 1129–1133. doi:10.1038/nature09303 |
| 8. | Klose, R. J.; Bird, A. P. Trends Biochem. Sci. 2006, 31, 89–97. doi:10.1016/j.tibs.2005.12.008 |
| 28. | Jin, S.-G.; Jiang, Y.; Qiu, R.; Rauch, T. A.; Wang, Y.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G. P. Cancer Res. 2011, 71, 7360–7365. doi:10.1158/0008-5472.CAN-11-2023 |
| 3. | Esteller, M. Genes Cancer 2011, 2, 604–606. doi:10.1177/1947601911423096 |
| 4. | Fuks, F. Curr. Opin. Genet. Dev. 2005, 15, 490–495. doi:10.1016/j.gde.2005.08.002 |
| 5. | Dawson, M. A.; Kouzarides, T. Cell 2012, 150, 12–27. doi:10.1016/j.cell.2012.06.013 |
| 6. | Dawson, M. A.; Kouzarides, T.; Huntly, B. J. P. N. Engl. J. Med. 2012, 367, 647–657. doi:10.1056/NEJMra1112635 |
| 7. | Hamm, C. A.; Costa, F. F. Drug Discovery Today 2011, 16, 626–635. doi:10.1016/j.drudis.2011.04.007 |
| 8. | Klose, R. J.; Bird, A. P. Trends Biochem. Sci. 2006, 31, 89–97. doi:10.1016/j.tibs.2005.12.008 |
| 25. | Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A. J.; Socci, N. D.; Scandura, J. M. PLoS One 2011, 6, e14524. doi:10.1371/journal.pone.0014524 |
| 2. | Biel, M.; Wascholowski, V.; Giannis, A. Angew. Chem., Int. Ed. 2005, 44, 3186–3216. doi:10.1002/anie.200461346 |
| 26. | Münzel, M.; Lischke, U.; Stathis, D.; Pfaffeneder, T.; Gnerlich, F. A.; Deiml, C. A.; Koch, S. C.; Karaghiosoff, K.; Carell, T. Chem.–Eur. J. 2011, 17, 13782–13788. doi:10.1002/chem.201102782 |
| 15. | Pastor, W. A.; Aravind, L.; Rao, A. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. doi:10.1038/nrm3589 |
| 16. | Fu, Y.; He, C. Curr. Opin. Chem. Biol. 2012, 16, 516–524. doi:10.1016/j.cbpa.2012.10.002 |
| 17. | Carell, T.; Brandmayr, C.; Hienzsch, A.; Müller, M.; Pearson, D.; Reiter, V.; Thoma, I.; Thumbs, P.; Wagner, M. Angew. Chem., Int. Ed. 2012, 51, 7110–7131. doi:10.1002/anie.201201193 |
| 21. | Bhutani, N.; Burns, D. M.; Blau, H. M. Cell 2011, 146, 866–872. doi:10.1016/j.cell.2011.08.042 |
| 15. | Pastor, W. A.; Aravind, L.; Rao, A. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. doi:10.1038/nrm3589 |
| 23. | Schiesser, S.; Hackner, B.; Pfaffeneder, T.; Müller, M.; Hagemeier, C.; Truss, M.; Carell, T. Angew. Chem., Int. Ed. 2012, 51, 6516–6520. doi:10.1002/anie.201202583 |
| 35. | Kifli, N.; De Clercq, E.; Balzarini, J.; Simons, C. Bioorg. Med. Chem. 2004, 12, 4245–4252. doi:10.1016/j.bmc.2004.05.017 |
| 36. | Matsuda, A.; Sasaki, T. Cancer Sci. 2004, 95, 105–111. doi:10.1111/j.1349-7006.2004.tb03189.x |
| 19. | Guo, J. U.; Su, Y.; Zhong, C.; Ming, G.-l.; Song, H. Cell 2011, 145, 423–434. doi:10.1016/j.cell.2011.03.022 |
| 20. | Nabel, C. S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H. L.; Stivers, J. T.; Zhang, Y.; Kohli, R. M. Nat. Chem. Biol. 2012, 8, 751–758. doi:10.1038/nchembio.1042 |
| 1. | Branco, M. R.; Ficz, G.; Reik, W. Nat. Rev. Genet. 2012, 13, 7–13. doi:10.1038/nrg3080 |
| 24. | Auclair, G.; Weber, M. Biochimie 2012, 94, 2202–2211. doi:10.1016/j.biochi.2012.05.016 |
| 15. | Pastor, W. A.; Aravind, L.; Rao, A. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. doi:10.1038/nrm3589 |
| 16. | Fu, Y.; He, C. Curr. Opin. Chem. Biol. 2012, 16, 516–524. doi:10.1016/j.cbpa.2012.10.002 |
| 17. | Carell, T.; Brandmayr, C.; Hienzsch, A.; Müller, M.; Pearson, D.; Reiter, V.; Thoma, I.; Thumbs, P.; Wagner, M. Angew. Chem., Int. Ed. 2012, 51, 7110–7131. doi:10.1002/anie.201201193 |
| 18. | Pfeifer, G. P.; Kadam, S.; Jin, S. G. Epigenet. Chromatin 2013, 6, 10. doi:10.1186/1756-8935-6-10 |
| 32. | Dong, Q.; Anderson, C. E.; Ciufolini, M. A. Tetrahedron Lett. 1995, 36, 5681–5682. doi:10.1016/0040-4039(95)01122-X |
| 10. | Tahiliani, M.; Koh, K. P.; Shen, Y.; Pastor, W. A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L. M.; Liu, D. R.; Aravind, L.; Rao, A. Science 2009, 324, 930–935. doi:10.1126/science.1170116 |
| 14. |
Rose, N. R.; McDonough, M. A.; King, O. N. F.; Kawamura, A.; Schofield, C. J. Chem. Soc. Rev. 2011, 40, 4364–4397. doi:10.1039/c0cs00203h
See for an excellent review on 2-oxoglutarate dependend oxygenases. |
| 15. | Pastor, W. A.; Aravind, L.; Rao, A. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. doi:10.1038/nrm3589 |
| 22. | Chen, C.-C.; Wang, K.-Y.; Shen, C.-K. J. J. Biol. Chem. 2012, 287, 33116–33121. doi:10.1074/jbc.C112.406975 |
| 29. | Münzel, M.; Globisch, D.; Trindler, C.; Carell, T. Org. Lett. 2010, 12, 5671–5673. doi:10.1021/ol102408t |
| 33. | Steigenberger, B.; Schiesser, S.; Hackner, B.; Brandmayr, C.; Laube, S. K.; Steinbacher, J.; Pfaffeneder, T.; Carell, T. Org. Lett. 2013, 15, 366–369. doi:10.1021/ol3033219 |
| 34. | Dai, Q.; He, C. Org. Lett. 2011, 13, 3446–3449. doi:10.1021/ol201189n |
© 2014 Chentsova et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)