Abstract
Highly efficient CO2 absorption was realized through formation of zwitterionic adducts, combining synthetic strategies to ionic liquids (ILs) and coordination. The essence of our strategy is to make use of multidentate cation coordination between Li+ and an organic base. Also PEG-functionalized organic bases were employed to enhance the CO2-philicity. The ILs were reacted with CO2 to form the zwitterionic adduct. Coordination effects between various lithium salts and neutral ligands, as well as the CO2 capacity of the chelated ILs obtained were investigated. For example, the CO2 capacity of PEG150MeBu2N increased steadily from 0.10 to 0.66 (mol CO2 absorbed per mol of base) through the formation of zwitterionic adducts being stabilized by Li+.
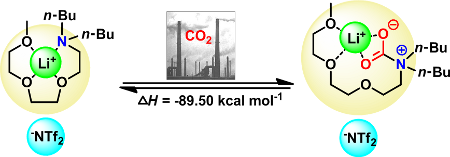
Graphical Abstract
Introduction
Carbon capture and sequestration (CCS) from flue gas formed by combustion of fossil fuel is a critical part of efforts directed towards the stabilization of atmospheric greenhouse gas levels [1]. In recent years, there has been intense research worldwide aimed at the development of various processes and technologies for efficient CO2 capture. These efforts include the development of liquid and solid absorbents and membranes [2-7].
Ionic liquids (ILs), which have attractive properties such as negligible vapor pressure, a wide liquid temperature ranges, good thermal stability, high ionic conductivity, and versatile solvation properties [8-11], can be designed for task-specific applications through the smart choice of the respective cations and/or anions. Application fields include green solvents for synthesis [9,12-15], efficient catalysts in organic synthesis [2,16,17], media for advanced separation [18,19], novel electrolytes for energy applications [20,21], and efficient absorbents for gas separation [2,22-24]. In particular, amino-functionalized IL [APBIm][BF4] (1-aminopropyl-3-butylimidazolium tetrafluoroborate) and ILs being composed of amino acid (AA) anions and phosphonium or ammonium cations were developed for efficient CO2 chemisorption [23,25-30]. Binary absorbents derived from superbases together with various non-volatile weak proton donors such as hydroxy-functionalized ILs, imidazolium ILs, fluorinated alcohol, imidazole and phenol, were also found to be efficient liquid absorbents allowing for reversible CO2 chemisorption [31-35]. In general, two absorbent molecules are involved to react with one CO2 molecule generating ammonium carbamate (Scheme 1a) or ammonium alkyl formate (Scheme 1b). Hence, increasing the 1:2 (CO2:absorbent molecule) stoichiometry for the CO2 capacity to 1:1 is an essential prerequisite for a breakthrough in absorption techniques [23]. In this respect, task-specifically designed absorbents have been successfully synthesized from AAs and applied for 1:1 CO2 capture through a carbamic acid formation pathway (Scheme 1a, step 1). Notably, equimolar CO2 absorption was obtained using task-specific ionic liquids (TSILs) with the phosphonium cation containing long alkyl chains and anions derived from AAs (prolinate and methioninate) [36], or AA salts with bulky N-substituents in polyethylene glycol (PEG) solution [37]. However, procedures for the preparation of ILs usually include complicated purification procedures or the use of volatile organic solvents (e.g., toluene, acetonitrile). Recently, Wang et al. developed novel alkanolamine-based ILs through multi-dentate cation coordination between alkanolamine and Li+ for reversible CO2 capture, by simple mixing of equimolar amounts of alkanolamines with LiNTf2 [38]. The strong complexation of alkali metal cations by crown ethers could be used to achieve equimolar CO2 absorption in systems containing crown ethers and easily available alkali metal salts of amino acids, resulting in the respective carbamates (onium salts) [39].
![[1860-5397-10-204-i1]](/bjoc/content/inline/1860-5397-10-204-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of CO2 with amino-group containing absorbents (a), base/proton donor binary system (b) or strong organic base (c).
Scheme 1: Reactions of CO2 with amino-group containing absorbents (a), base/proton donor binary system (b) or...
As previously reported, strong amidine and guanidine bases such as 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) can form the base-CO2 zwitterionic adduct in a 1:1 manner under strictly anhydrous conditions (Scheme 1c) [40,41]. Herein, we present such a method combining the formation of ILs and coordination to achieve equimolar CO2 capture through zwitterionic adduct formation. The essence of our strategy is to make use of the multisite coordination interaction between Li+ and organic bases or PEG-functionalized organic bases. The readily prepared ILs were reacted then with CO2 to form intramolecular zwitterionic adducts.
Results and Discussion
Taking PEG150MeTMG as a neutral ligand (Table 1), coordination effects of various lithium salts were investigated (Table S1, Supporting Information File 1). Only LiSO3CF3 and LiNTf2 formed complexes with PEG150MeTMG through multisite coordination. Subsequently, different neutral ligands with alkyl chains or PEG chain were selected to evaluate the effect of chelating with LiNTf2 on the physicochemical properties of the resulting ILs as well as the CO2 capacities (Table 1).
Table 1: Stability (ΔHf) of the cations derived from coordination of Li+ in LiNTf2 with various neutral ligands and CO2 capacity of the derived ionic liquidsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-204-i2.svg?max-width=637&scale=1.0)
|
|||
| Entry | Ionic liquid | ΔHf/kcal mol−1 b | CO2 capacityc |
|---|---|---|---|
| 1 | [PEG150MeLi][NTf2] | −100.94 | 0.09 (0.9%) |
| 2 | [OctImLi][NTf2] | −56.49 | 0.11 (1.0%) |
| 3 | [PEG150MeImLi][NTf2] | −91.17 | 0.16 (1.4%) |
| 4 | [PEG150MeNH2Li][NTf2] | −89.39 | 0.45 (4.4%) |
| 5 | [TMGLi][NTf2] | −41.59 | 0.65 (7.1%) |
| 6 | [OctTMGLi][NTf2] | −47.79 | 0.80 (6.8%) |
| 7 | [PEG150MeTMGLi][NTf2] | −106.56 | 0.89 (7.1%) |
| 8 | OctBu2N/LiNTf2 | – | – |
| 9 | PEG150MeBu2N | – | 0.10 (1.6%) |
| 10 | [PEG150MeBu2NLi][NTf2] | −96.26 | 0.66 (5.2%) |
| 11 | [PEG150MeBu2NLi][SO3CF3] | −96.26 | 0.61 (6.2%) |
| 12 | [DBULi][NTf2] | −60.22 | 0.50 (5.0%) |
| 13 | [DBNLi][NTf2] | −60.50 | 0.75 (8.0%) |
aIonic liquids were prepared by mixing of a neutral ligand with LiNTf2 in 1:1 molar ratio. CO2 absorption was carried out at 25 °C and absorption equilibrium was reached within 20 min. bEnergy of the gas phase reaction between the neutral ligand and Li+, was calculated with DFT, using the B3PW91 functional with the 6-311++G (d,p) basis set as implemented in the Gaussian 09 program package. cMol of CO2 captured per mol of ionic liquid. Results in bracket were grams of CO2 absorbed per gram of absorbent.
Typical optimized structures of cations derived from chelation between neutral ligands and Li+ are shown in Figure 1. The geometry optimizations were carried out by performing DFT calculations. Four O atoms in PEG150Me chelate with Li+ in a quasi-crown ether manner. For neutral ligands with PEG chain (PEG150MeIm, PEG150MeNH2, PEG150MeTMG and PEG150MeBu2N), all the O and N atoms coordinate with Li+ in a quasi-aza-crown ether fashion. OctIm, OctTMG, TMG, DBU and DBN generate complexes whereby Li+ is bound only to N atoms. In contrast, the coordination ability of the N atom in OctBu2N is not strong enough to form a homogeneous chelated IL with LiNTf2.
![[1860-5397-10-204-1]](/bjoc/content/figures/1860-5397-10-204-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Typical optimized structures of complex cations derived from chelation between Li+ and neutral ligands. H: white, C: grey, O: red, N: blue, Li: purple. Bond lengths are in Å.
Figure 1: Typical optimized structures of complex cations derived from chelation between Li+ and neutral liga...
Calculation of the energy of the gas phase reaction between neutral ligands and Li+ gave a value for the enthalpy change in the range of −41.59 to −106.56 kcal mol−1 (Table 1, entries 1–11), indicating that the formation of chelated ILs is feasible.
In addition, PEG-functionalization of the organic base enhanced the complexation ability. For example, ΔHf decreased from −56.49 kal mol−1 (OctIm) to −91.17 kcal mol−1 (PEG150MeIm) for imidazole (Table 1, entry 2 vs 3), from −47.79 kcal mol−1 (OctTMG) to −106.56 kcal mol−1 (PEG150MeTMG) for guanidine (Table 1, entry 6 vs 7), and from no complexation (OctBu2N) to −96.26 kal mol−1 (PEG150MeBu2N) for tertiary amine (Table 1, entry 8 vs 9).
The structures of the chelated ILs were confirmed by thermogravimetric analysis (TGA), NMR (see Supporting Information File 1, Figure S2), in situ FTIR under CO2 pressure and mass spectrometry. The thermal stability of the chelated ILs was strongly increased compared to the corresponding neutral ligands. The decomposition temperature increased by 90 °C and 60 °C for PEG150MeTMG and PEG150MeBu2N, respectively, after coordination with LiNTf2 (Figure 2a).
![[1860-5397-10-204-2]](/bjoc/content/figures/1860-5397-10-204-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Comparison of the thermal stability between the neutral ligands and the corresponding chelated ionic liquids after coordinating with LiNTf2, being detected by TGA; (b) 1H NMR (CDCl3, 400 MHz) spectrum of PEG150MeTMG and PEG150MeBu2N before and after reacting with lithium salts (LiNTf2 and LiSO3CF3).
Figure 2: (a) Comparison of the thermal stability between the neutral ligands and the corresponding chelated ...
In the 1H NMR spectrum, the proton signals of the four methyl groups in guanidine of PEG150MeTMG shifted from 2.63–2.72 ppm to 2.96 ppm after reacting with equimolar amounts of LiNTf2 (Figure 2b). The corresponding anion CF3SO3−, the twelve protons moving only from 2.63–2.72 ppm to 2.71–2.75 ppm, indicate that there is only little influence of the anion on the nature of the coordinative bond. In the case of tertiary amines (e.g., PEG150MeBu2N), all the protons shifted downfield after forming the chelated IL with LiNTf2. Changing the anion to CF3SO3− had negligible influence on the coordination ability. All the neutral ligands shown in Table 1 gave a downfield shift in the 1H NMR spectrum after chelating with Li+, in accordance with the electron density decreasing through coordination. Furthermore, the formation of chelated ILs was also verified by ESI-MS (m/z = 268.27 for [PEG150MeTMGLi]+ and m/z = 282.32 for [PEG150MeBu2NLi]+) (Figure S3, see Supporting Information File 1). In the in situ FTIR spectrum, the C=N absorption band of the neutral ligand PEG150MeTMG was shifted from 1616 cm−1 to 1592 cm−1 after reacting with equimolar amounts of LiNTf2 as depicted in Figure 3a.
![[1860-5397-10-204-3]](/bjoc/content/figures/1860-5397-10-204-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: In situ FTIR spectra of neutral ligands and the corresponding chelated ionic liquids after reaction with LiNTf2, as well as the reaction mixture after CO2 absorption in the absence or presence of water.
Figure 3: In situ FTIR spectra of neutral ligands and the corresponding chelated ionic liquids after reaction...
The effect of various chelated ILs on CO2 absorption was subsequently examined using LiNTf2 as coordinating reagent. The CO2 absorption capacity, defined as mol of CO2 captured per mol of IL, was estimated from the weight increase of the reaction mixture. As shown in Table 1, ILs of weak basicity, such as [PEG150MeLi][NTf2], [OctImLi][NTf2] and [PEG150MeImLi][NTf2] showed a poor CO2 sorption capacity, implying that only physical interactions with CO2 were present (Table 1, entries 1–3). The primary amine-functionalized IL [PEG150MeNH2Li][NTf2] gave rise to CO2 uptake approaching 1:2 stoichiometry (Table 1, entry 4) as expected from the proposed mechanism for the formation of ammonium carbamate as shown in Scheme 1a [38]. Notably, the CO2 capacity of guanidine-functionalized ILs increased in the order of [TMGLi][NTf2] (0.65) < [OctTMGLi][NTf2] (0.80) < [PEG150MeTMGLi][NTf2] (0.89 mol CO2 absorbed per mol of base) (Table 1, entries 5–7), indicating that the CO2-philic nature of the PEG chain facilitates CO2 sorption. Generally, anhydrous tertiary amines absorb CO2 only under high CO2 pressures to form instable zwitterionic alkylcarbonate salts (Table 1, entry 9) [42]. [PEG150MeBu2NLi][NTf2] and [PEG150MeBu2NLi][SO3CF3] were able to rapidly reach 0.66 and 0.61 CO2 capacity, respectively. Thus, tertiary amino-functionalized ILs with multidentate cation coordination have a much better performance probably due to the formation of a zwitterionic adduct being stabilized by Li+ (Table 1, entries 10 and 11). Indeed, the CO2 absorption capacity of [PEG150MeBu2NLi][NTf2] increased steadily from 0.10 to 0.66 mol CO2 absorbed per mol of base when the molar ratio of LiNTf2/PEG150MeBu2N was varied from 0 to 1. When the molar ratio was increased to 1.5, no further promotion of the CO2 capacity was observed (Figure 4). In contrast, when PEG150MeTMG was employed as the neutral ligand, the CO2 capacity decreased from 1.66 to 0.89 mol CO2 absorbed per mol of base as the molar ratio of LiNTf2/PEG150MeTMG was increased from 0 to 1, probably due to decreased basicity of guanidine after coordinating with Li+. At last, ILs [DBULi][NTf2] (0.50) and [DBNLi][NTf2] (0.75 mol CO2 absorbed per mol of base) had a CO2 capacity below 1:1 stoichiometry expected from the proposed mechanism (Scheme 1c), owing to highly increased viscosity after CO2 absorption (Table 1, entries 12 and 13). Compared on a weight basis, a CO2 capacity of 5.0 wt % to 8.0 wt % was obtained with ILs from neutral ligands/LiNTf2 (Table 1, entries 5–7 and 10–13). This is much higher than for the conventional IL 1-hexyl-3-methylimidazolium hexafluorophosphate (0.0881 wt %) [43], and comparable to amino-functionalized imidazolium-based IL (7.4 wt %) [23] and ILs derived from amino acids [24]. Hence, our IL system has the potential to be utilized for industrialized CO2 absorption processes.
![[1860-5397-10-204-4]](/bjoc/content/figures/1860-5397-10-204-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Influence of the ratio of LiNTf2/neutral ligands (PEG150MeTMG and PEG150MeBu2N) on the CO2 capacity of the coordinating mixtures.
Figure 4: Influence of the ratio of LiNTf2/neutral ligands (PEG150MeTMG and PEG150MeBu2N) on the CO2 capacity...
The in situ FTIR spectrum of [PEG150MeTMGLi][NTf2] and [PEG150MeBu2NLi][NTf2] before and after reaction with CO2 are shown in Figure 3. For [PEG150MeTMGLi][NTf2] as absorbent, the C=N stretching band at 1592 cm−1 shifted to 1622 cm−1 after reaction with CO2. A characteristic peak centered at 1685 cm−1 is assigned to the stretching vibration of the carbonyl group in the zwitterionic alkyl carbamate, which is quite different from that in HCO3− (1588 cm−1). For the reaction of [PEG150MeBu2NLi][NTf2] with CO2, a new band at 1635 cm−1 was assigned to the stretching vibration of the C=O bond in the zwitterionic product. In addition, a distinct broad band at around 2118 cm−1 corresponding to the ammonium cation was observed in the presence of water.
To gain deeper insight into the reaction mechanism of CO2 absorption with chelated ILs, DFT calculations were carried out. We performed geometry and energy optimizations for the free [BaseLi]+ cation, free CO2 and the [BaseLi]+ + CO2 complex. As shown in Figure 5, the chelated ILs react with CO2 through nucleophilic attack by the N atom, to form the zwitterionic adducts, which are stabilized through coordination with Li+. In addition, formation of the zwitterionic alkyl carbamate is calculated to be associated with an enthalpy changes of −102.36 kcal mol−1 and −89.50 kcal mol−1 for [PEG150MeTMGLi]+/CO2 and [PEG150MeBu2NLi]+/CO2, respectively, indicating that the absorption process is thermodynamically favourable.
![[1860-5397-10-204-5]](/bjoc/content/figures/1860-5397-10-204-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: The quantum chemistry calculations (enthalpy changes) of the reaction between CO2 and [PEG150MeTMGLi]+ (a) or [PEG150MeBu2NLi]+ (b); H: white, C: grey, O: red, N: blue, Li: purple. Bond lengths are in Å.
Figure 5: The quantum chemistry calculations (enthalpy changes) of the reaction between CO2 and [PEG150MeTMGL...
Conclusion
In summary, efficient CO2 capture was achieved through formation of zwitterionic adducts with readily synthesized chelated ILs. Multisite coordination interaction between the Li+ cation and organic bases or PEG-functionalized organic bases is thought to be responsible for forming the amidine, guanidine or tertiary amine-functionalized ILs, after reaction with CO2 to form zwitterionic adducts in a 1:1 manner. Coordination effects between various lithium salts and neutral ligands, and the CO2 capacity of the obtained chelated ILs were investigated. Indeed, the thermal stability and the CO2 capacity of the neutral ligands (e.g., PEG150MeBu2N) was highly increased after coordination with lithium salts to form chelated ILs (e.g., [PEG150MeBu2NLi][NTf2]).
Experimental
Materials
All reagents used in this work were purchased from Sigma-Aldrich and used without further purification. CO2 with a purity of 99.999% was obtained commercially.
Experimental methods
1H NMR spectra were recorded on a Brucker 400 spectrometer in CDCl3. Residual CHCl3 (7.26 ppm) was used as internal reference. 13C NMR spectra were recorded at 100.6 MHz in CDCl3. Residual CHCl3 (77.0 ppm) was used as internal reference. In situ FTIR spectra were collected on a Mettler Toledo React IR ic10, which was equipped with a diamond ATR probe, using an ic IR analysis system. The probe was placed into the absorption mixture. Spectra were collected in situ during CO2 absorption, while the mixture was stirred continuously using a magnetic stir bar. ESI-MS spectra were recorded on a Thermo Finnigan LCQ Advantage spectrometer in ESI mode at a spray voltage of 4.8 kV.
General procedure for CO2 absorption
The CO2 absorption procedure was analogous to the CO2/SO2 absorption procedure we had reported before [35,44]. In a typical procedure, the CO2 capture was carried out in a 10 mL Schlenk flask. The absorbents were charged into the reactor at room temperature. Then, the air in the flask was replaced by passing CO2 through a needle, which was inserted into the bottom of the flask. The absorption was conducted at 25 °C with a CO2 flow rate of 0.1 L/min. The amount of CO2 absorbed was determined by following the weight of the mixture with an Analytical Balance. Data points were taken with an accuracy of ±0.0001 g every five minutes. Absorption/desorption was determined for at least three cycles.
Supporting Information
| Supporting Information File 1: General experimental methods, synthesis and characterization of the neutral ligands, lithium salts and the corresponding chelated ionic liquids. | ||
| Format: PDF | Size: 3.6 MB | Download |
Acknowledgements
We are grateful to the National Natural Science Foundation of China (No. 21172125, 21121002), the ‘‘111’’ Project of Ministry of Education of China (Project No. B06005), and Specialized Research Fund for the Doctoral Program of Higher Education (20130031110013) for financial support.
References
-
Raupach, M. R.; Marland, G.; Ciais, P.; Le Quéré, C.; Canadell, J. G.; Klepper, G.; Field, C. B. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 10288–10293. doi:10.1073/pnas.0700609104
Return to citation in text: [1] -
Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N. RSC Adv. 2011, 1, 545–567. doi:10.1039/c1ra00307k
Return to citation in text: [1] [2] [3] -
Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Energy Environ. Sci. 2012, 5, 6602–6639. doi:10.1039/C2EE02774G
Return to citation in text: [1] -
D'Alessandro, D. M.; Smit, B.; Long, J. R. Angew. Chem., Int. Ed. 2010, 49, 6058–6082. doi:10.1002/anie.201000431
Return to citation in text: [1] -
Choi, S.; Drese, J. H.; Jones, C. W. ChemSusChem 2009, 2, 796–854. doi:10.1002/cssc.200900036
Return to citation in text: [1] -
Wang, Q.; Luo, J.; Zhong, Z.; Borgna, A. Energy Environ. Sci. 2011, 4, 42–55. doi:10.1039/c0ee00064g
Return to citation in text: [1] -
Li, L.; Zhao, N.; Wei, W.; Sun, Y. Fuel 2013, 108, 112–130. doi:10.1016/j.fuel.2011.08.022
Return to citation in text: [1] -
Welton, T. Chem. Rev. 1999, 99, 2071–2084. doi:10.1021/cr980032t
Return to citation in text: [1] -
Wasserscheid, P.; Keim, W. Angew. Chem., Int. Ed. 2000, 39, 3772–3789. doi:10.1002/1521-3773(20001103)39:21<3772::AID-ANIE3772>3.0.CO;2-5
Return to citation in text: [1] [2] -
Petkovic, M.; Seddon, K. R.; Rebelo, L. P. N.; Pereira, C. S. Chem. Soc. Rev. 2011, 40, 1383–1403. doi:10.1039/c004968a
Return to citation in text: [1] -
Plechkova, N. V.; Seddon, K. R. Chem. Soc. Rev. 2008, 37, 123–150. doi:10.1039/b006677j
Return to citation in text: [1] -
Ma, Z.; Yu, J.; Dai, S. Adv. Mater. 2010, 22, 261–285. doi:10.1002/adma.200900603
Return to citation in text: [1] -
Antonietti, M.; Kuang, D.; Smarsly, B.; Zhou, Y. Angew. Chem., Int. Ed. 2004, 43, 4988–4992. doi:10.1002/anie.200460091
Return to citation in text: [1] -
Dupont, J.; de Souza, R. F.; Suarez, P. A. Z. Chem. Rev. 2002, 102, 3667–3692. doi:10.1021/cr010338r
Return to citation in text: [1] -
Zakrzewska, M. E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Chem. Rev. 2011, 111, 397–417. doi:10.1021/cr100171a
Return to citation in text: [1] -
Miao, C.-X.; He, L.-N.; Wang, J.-Q.; Wang, J.-L. Adv. Synth. Catal. 2009, 351, 2209–2216. doi:10.1002/adsc.200900285
Return to citation in text: [1] -
Zhang, Q.; Zhang, S.; Deng, Y. Green Chem. 2011, 13, 2619–2637. doi:10.1039/c1gc15334j
Return to citation in text: [1] -
Han, X.; Armstrong, D. W. Acc. Chem. Res. 2007, 40, 1079–1086. doi:10.1021/ar700044y
Return to citation in text: [1] -
Berthod, A.; Ruiz-Ángel, M. J.; Carda-Broch, S. J. Chromatogr., A 2008, 1184, 6–18. doi:10.1016/j.chroma.2007.11.109
Return to citation in text: [1] -
Armand, M.; Endres, F.; MacFarlane, D. R.; Ohno, H.; Scrosati, B. Nat. Mater. 2009, 8, 621–629. doi:10.1038/nmat2448
Return to citation in text: [1] -
MacFarlane, D. R.; Forsyth, M.; Howlett, P. C.; Pringle, J. M.; Sun, J.; Annat, G.; Neil, W.; Izgorodina, E. I. Acc. Chem. Res. 2007, 40, 1165–1173. doi:10.1021/ar7000952
Return to citation in text: [1] -
Ramdin, M.; de Loos, T. W.; Vlugt, T. J. H. Ind. Eng. Chem. Res. 2012, 51, 8149–8177. doi:10.1021/ie3003705
Return to citation in text: [1] -
Bates, E. D.; Mayton, R. D.; Ntai, I.; Davis, J. H., Jr. J. Am. Chem. Soc. 2002, 124, 926–927. doi:10.1021/ja017593d
Return to citation in text: [1] [2] [3] [4] -
Zhang, X.; Zhang, X.; Dong, H.; Zhao, Z.; Zhang, S.; Huang, Y. Energy Environ. Sci. 2012, 5, 6668–6681. doi:10.1039/c2ee21152a
Return to citation in text: [1] [2] -
Zhang, J.; Zhang, S.; Dong, K.; Zhang, Y.; Shen, Y.; Lv, X. Chem. – Eur. J. 2006, 12, 4021–4026. doi:10.1002/chem.200501015
Return to citation in text: [1] -
Jiang, Y.-Y.; Wang, G.-N.; Zhou, Z.; Wu, Y.-T.; Geng, J.; Zhang, Z.-B. Chem. Commun. 2008, 505–507. doi:10.1039/b713648j
Return to citation in text: [1] -
Zhang, Y.; Zhang, S.; Lu, X.; Zhou, Q.; Fan, W.; Zhang, X. Chem. – Eur. J. 2009, 15, 3003–3011. doi:10.1002/chem.200801184
Return to citation in text: [1] -
Yu, H.; Wu, Y.-T.; Jiang, Y.-Y.; Zhou, Z.; Zhang, Z.-B. New J. Chem. 2009, 33, 2385–2390. doi:10.1039/b9nj00330d
Return to citation in text: [1] -
Li, X.; Hou, M.; Zhang, Z.; Han, B.; Yang, G.; Wang, X.; Zou, L. Green Chem. 2008, 10, 879–884. doi:10.1039/b801948g
Return to citation in text: [1] -
Luo, X. Y.; Ding, F.; Lin, W. J.; Qi, Y. Q.; Li, H. R.; Wang, C. M. J. Phys. Chem. Lett. 2014, 5, 381–386. doi:10.1021/jz402531n
Return to citation in text: [1] -
Wang, C.; Mahurin, S. M.; Luo, H.; Baker, G. A.; Li, H.; Dai, S. Green Chem. 2010, 12, 870–874. doi:10.1039/b927514b
Return to citation in text: [1] -
Wang, C.; Luo, H.; Luo, X.; Li, H.; Dai, S. Green Chem. 2010, 12, 2019–2023. doi:10.1039/c0gc00070a
Return to citation in text: [1] -
Wang, C.; Luo, H.; Jiang, D.-e.; Li, H.; Dai, S. Angew. Chem., Int. Ed. 2010, 49, 5978–5981. doi:10.1002/anie.201002641
Return to citation in text: [1] -
Wang, C.; Luo, X.; Luo, H.; Jiang, D.-e.; Li, H.; Dai, S. Angew. Chem., Int. Ed. 2011, 50, 4918–4922. doi:10.1002/anie.201008151
Return to citation in text: [1] -
Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Li, B.; Yu, B. Energy Environ. Sci. 2011, 4, 3971–3975. doi:10.1039/C1EE02156G
Return to citation in text: [1] [2] -
Gurkan, B. E.; de la Fuente, J. C.; Mindrup, E. M.; Ficke, L. E.; Goodrich, B. F.; Price, E. A.; Schneider, W. F.; Brennecke, J. F. J. Am. Chem. Soc. 2010, 132, 2116–2117. doi:10.1021/ja909305t
Return to citation in text: [1] -
Liu, A.-H.; Ma, R.; Song, C.; Yang, Z.-Z.; Yu, A.; Cai, Y.; He, L.-N.; Zhao, Y.-N.; Yu, B.; Song, Q.-W. Angew. Chem., Int. Ed. 2012, 51, 11306–11310. doi:10.1002/anie.201205362
Return to citation in text: [1] -
Wang, C.; Guo, Y.; Zhu, X.; Cui, G.; Li, H.; Dai, S. Chem. Commun. 2012, 48, 6526–6528. doi:10.1039/c2cc32365f
Return to citation in text: [1] [2] -
Yang, Z.-Z.; Jiang, D.-e.; Zhu, X.; Tian, C.; Brown, S.; Do-Thanh, C.-L.; He, L.-N.; Dai, S. Green Chem. 2014, 16, 253–258. doi:10.1039/c3gc41513a
Return to citation in text: [1] -
Das Neves Gomes, C.; Jacquet, O.; Villiers, C.; Thuéry, P.; Ephritikhine, M.; Cantat, T. Angew. Chem., Int. Ed. 2012, 51, 187–190. doi:10.1002/anie.201105516
Return to citation in text: [1] -
Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035
Return to citation in text: [1] -
Rainbolt, J. E.; Koech, P. K.; Yonker, C. R.; Zheng, F.; Main, D.; Weaver, M. L.; Linehan, J. C.; Heldebrant, D. J. Energy Environ. Sci. 2011, 4, 480–484. doi:10.1039/c0ee00506a
Return to citation in text: [1] -
Blanchard, L. A.; Gu, Z.; Brennecke, J. F. J. Phys. Chem. B 2001, 105, 2437–2444. doi:10.1021/jp003309d
Return to citation in text: [1] -
Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Yu, B. Environ. Sci. Technol. 2013, 47, 1598–1605. doi:10.1021/es304147q
Return to citation in text: [1]
| 43. | Blanchard, L. A.; Gu, Z.; Brennecke, J. F. J. Phys. Chem. B 2001, 105, 2437–2444. doi:10.1021/jp003309d |
| 38. | Wang, C.; Guo, Y.; Zhu, X.; Cui, G.; Li, H.; Dai, S. Chem. Commun. 2012, 48, 6526–6528. doi:10.1039/c2cc32365f |
| 42. | Rainbolt, J. E.; Koech, P. K.; Yonker, C. R.; Zheng, F.; Main, D.; Weaver, M. L.; Linehan, J. C.; Heldebrant, D. J. Energy Environ. Sci. 2011, 4, 480–484. doi:10.1039/c0ee00506a |
| 1. | Raupach, M. R.; Marland, G.; Ciais, P.; Le Quéré, C.; Canadell, J. G.; Klepper, G.; Field, C. B. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 10288–10293. doi:10.1073/pnas.0700609104 |
| 2. | Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N. RSC Adv. 2011, 1, 545–567. doi:10.1039/c1ra00307k |
| 16. | Miao, C.-X.; He, L.-N.; Wang, J.-Q.; Wang, J.-L. Adv. Synth. Catal. 2009, 351, 2209–2216. doi:10.1002/adsc.200900285 |
| 17. | Zhang, Q.; Zhang, S.; Deng, Y. Green Chem. 2011, 13, 2619–2637. doi:10.1039/c1gc15334j |
| 39. | Yang, Z.-Z.; Jiang, D.-e.; Zhu, X.; Tian, C.; Brown, S.; Do-Thanh, C.-L.; He, L.-N.; Dai, S. Green Chem. 2014, 16, 253–258. doi:10.1039/c3gc41513a |
| 9. | Wasserscheid, P.; Keim, W. Angew. Chem., Int. Ed. 2000, 39, 3772–3789. doi:10.1002/1521-3773(20001103)39:21<3772::AID-ANIE3772>3.0.CO;2-5 |
| 12. | Ma, Z.; Yu, J.; Dai, S. Adv. Mater. 2010, 22, 261–285. doi:10.1002/adma.200900603 |
| 13. | Antonietti, M.; Kuang, D.; Smarsly, B.; Zhou, Y. Angew. Chem., Int. Ed. 2004, 43, 4988–4992. doi:10.1002/anie.200460091 |
| 14. | Dupont, J.; de Souza, R. F.; Suarez, P. A. Z. Chem. Rev. 2002, 102, 3667–3692. doi:10.1021/cr010338r |
| 15. | Zakrzewska, M. E.; Bogel-Łukasik, E.; Bogel-Łukasik, R. Chem. Rev. 2011, 111, 397–417. doi:10.1021/cr100171a |
| 40. | Das Neves Gomes, C.; Jacquet, O.; Villiers, C.; Thuéry, P.; Ephritikhine, M.; Cantat, T. Angew. Chem., Int. Ed. 2012, 51, 187–190. doi:10.1002/anie.201105516 |
| 41. | Villiers, C.; Dognon, J.-P.; Pollet, R.; Thuéry, P.; Ephritikhine, M. Angew. Chem., Int. Ed. 2010, 49, 3465–3468. doi:10.1002/anie.201001035 |
| 8. | Welton, T. Chem. Rev. 1999, 99, 2071–2084. doi:10.1021/cr980032t |
| 9. | Wasserscheid, P.; Keim, W. Angew. Chem., Int. Ed. 2000, 39, 3772–3789. doi:10.1002/1521-3773(20001103)39:21<3772::AID-ANIE3772>3.0.CO;2-5 |
| 10. | Petkovic, M.; Seddon, K. R.; Rebelo, L. P. N.; Pereira, C. S. Chem. Soc. Rev. 2011, 40, 1383–1403. doi:10.1039/c004968a |
| 11. | Plechkova, N. V.; Seddon, K. R. Chem. Soc. Rev. 2008, 37, 123–150. doi:10.1039/b006677j |
| 37. | Liu, A.-H.; Ma, R.; Song, C.; Yang, Z.-Z.; Yu, A.; Cai, Y.; He, L.-N.; Zhao, Y.-N.; Yu, B.; Song, Q.-W. Angew. Chem., Int. Ed. 2012, 51, 11306–11310. doi:10.1002/anie.201205362 |
| 2. | Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N. RSC Adv. 2011, 1, 545–567. doi:10.1039/c1ra00307k |
| 3. | Yang, Z.-Z.; He, L.-N.; Gao, J.; Liu, A.-H.; Yu, B. Energy Environ. Sci. 2012, 5, 6602–6639. doi:10.1039/C2EE02774G |
| 4. | D'Alessandro, D. M.; Smit, B.; Long, J. R. Angew. Chem., Int. Ed. 2010, 49, 6058–6082. doi:10.1002/anie.201000431 |
| 5. | Choi, S.; Drese, J. H.; Jones, C. W. ChemSusChem 2009, 2, 796–854. doi:10.1002/cssc.200900036 |
| 6. | Wang, Q.; Luo, J.; Zhong, Z.; Borgna, A. Energy Environ. Sci. 2011, 4, 42–55. doi:10.1039/c0ee00064g |
| 7. | Li, L.; Zhao, N.; Wei, W.; Sun, Y. Fuel 2013, 108, 112–130. doi:10.1016/j.fuel.2011.08.022 |
| 38. | Wang, C.; Guo, Y.; Zhu, X.; Cui, G.; Li, H.; Dai, S. Chem. Commun. 2012, 48, 6526–6528. doi:10.1039/c2cc32365f |
| 23. | Bates, E. D.; Mayton, R. D.; Ntai, I.; Davis, J. H., Jr. J. Am. Chem. Soc. 2002, 124, 926–927. doi:10.1021/ja017593d |
| 25. | Zhang, J.; Zhang, S.; Dong, K.; Zhang, Y.; Shen, Y.; Lv, X. Chem. – Eur. J. 2006, 12, 4021–4026. doi:10.1002/chem.200501015 |
| 26. | Jiang, Y.-Y.; Wang, G.-N.; Zhou, Z.; Wu, Y.-T.; Geng, J.; Zhang, Z.-B. Chem. Commun. 2008, 505–507. doi:10.1039/b713648j |
| 27. | Zhang, Y.; Zhang, S.; Lu, X.; Zhou, Q.; Fan, W.; Zhang, X. Chem. – Eur. J. 2009, 15, 3003–3011. doi:10.1002/chem.200801184 |
| 28. | Yu, H.; Wu, Y.-T.; Jiang, Y.-Y.; Zhou, Z.; Zhang, Z.-B. New J. Chem. 2009, 33, 2385–2390. doi:10.1039/b9nj00330d |
| 29. | Li, X.; Hou, M.; Zhang, Z.; Han, B.; Yang, G.; Wang, X.; Zou, L. Green Chem. 2008, 10, 879–884. doi:10.1039/b801948g |
| 30. | Luo, X. Y.; Ding, F.; Lin, W. J.; Qi, Y. Q.; Li, H. R.; Wang, C. M. J. Phys. Chem. Lett. 2014, 5, 381–386. doi:10.1021/jz402531n |
| 23. | Bates, E. D.; Mayton, R. D.; Ntai, I.; Davis, J. H., Jr. J. Am. Chem. Soc. 2002, 124, 926–927. doi:10.1021/ja017593d |
| 35. | Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Li, B.; Yu, B. Energy Environ. Sci. 2011, 4, 3971–3975. doi:10.1039/C1EE02156G |
| 44. | Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Yu, B. Environ. Sci. Technol. 2013, 47, 1598–1605. doi:10.1021/es304147q |
| 2. | Yang, Z.-Z.; Zhao, Y.-N.; He, L.-N. RSC Adv. 2011, 1, 545–567. doi:10.1039/c1ra00307k |
| 22. | Ramdin, M.; de Loos, T. W.; Vlugt, T. J. H. Ind. Eng. Chem. Res. 2012, 51, 8149–8177. doi:10.1021/ie3003705 |
| 23. | Bates, E. D.; Mayton, R. D.; Ntai, I.; Davis, J. H., Jr. J. Am. Chem. Soc. 2002, 124, 926–927. doi:10.1021/ja017593d |
| 24. | Zhang, X.; Zhang, X.; Dong, H.; Zhao, Z.; Zhang, S.; Huang, Y. Energy Environ. Sci. 2012, 5, 6668–6681. doi:10.1039/c2ee21152a |
| 36. | Gurkan, B. E.; de la Fuente, J. C.; Mindrup, E. M.; Ficke, L. E.; Goodrich, B. F.; Price, E. A.; Schneider, W. F.; Brennecke, J. F. J. Am. Chem. Soc. 2010, 132, 2116–2117. doi:10.1021/ja909305t |
| 20. | Armand, M.; Endres, F.; MacFarlane, D. R.; Ohno, H.; Scrosati, B. Nat. Mater. 2009, 8, 621–629. doi:10.1038/nmat2448 |
| 21. | MacFarlane, D. R.; Forsyth, M.; Howlett, P. C.; Pringle, J. M.; Sun, J.; Annat, G.; Neil, W.; Izgorodina, E. I. Acc. Chem. Res. 2007, 40, 1165–1173. doi:10.1021/ar7000952 |
| 23. | Bates, E. D.; Mayton, R. D.; Ntai, I.; Davis, J. H., Jr. J. Am. Chem. Soc. 2002, 124, 926–927. doi:10.1021/ja017593d |
| 18. | Han, X.; Armstrong, D. W. Acc. Chem. Res. 2007, 40, 1079–1086. doi:10.1021/ar700044y |
| 19. | Berthod, A.; Ruiz-Ángel, M. J.; Carda-Broch, S. J. Chromatogr., A 2008, 1184, 6–18. doi:10.1016/j.chroma.2007.11.109 |
| 31. | Wang, C.; Mahurin, S. M.; Luo, H.; Baker, G. A.; Li, H.; Dai, S. Green Chem. 2010, 12, 870–874. doi:10.1039/b927514b |
| 32. | Wang, C.; Luo, H.; Luo, X.; Li, H.; Dai, S. Green Chem. 2010, 12, 2019–2023. doi:10.1039/c0gc00070a |
| 33. | Wang, C.; Luo, H.; Jiang, D.-e.; Li, H.; Dai, S. Angew. Chem., Int. Ed. 2010, 49, 5978–5981. doi:10.1002/anie.201002641 |
| 34. | Wang, C.; Luo, X.; Luo, H.; Jiang, D.-e.; Li, H.; Dai, S. Angew. Chem., Int. Ed. 2011, 50, 4918–4922. doi:10.1002/anie.201008151 |
| 35. | Yang, Z.-Z.; He, L.-N.; Zhao, Y.-N.; Li, B.; Yu, B. Energy Environ. Sci. 2011, 4, 3971–3975. doi:10.1039/C1EE02156G |
| 24. | Zhang, X.; Zhang, X.; Dong, H.; Zhao, Z.; Zhang, S.; Huang, Y. Energy Environ. Sci. 2012, 5, 6668–6681. doi:10.1039/c2ee21152a |
© 2014 Yang and He; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)