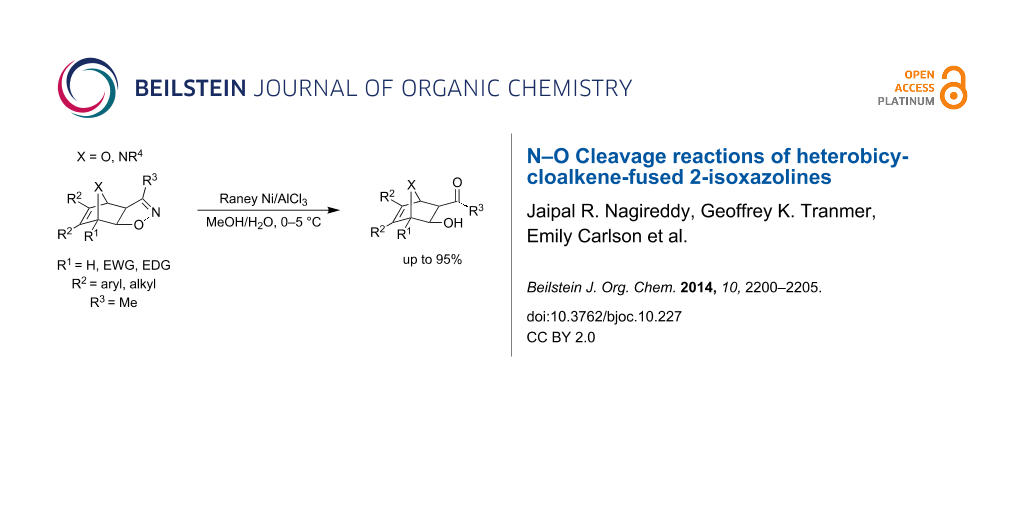
Abstract
Transition metal-mediated N–O bond cleavage reactions of heterobicycloalkene-fused 3-methyl-2-isoxazolines were investigated. Optimal cleavage conditions were found with Raney nickel/AlCl3 mediation in aqueous methanol. The reaction provided a diverse collection of novel heterobicycle-fused β-hydroxyketones with good to excellent yields (66–95%) and without the need for chromatographic purification.

Graphical Abstract
Introduction
2-Isoxazolines 1 are practical precursors to countless structural motifs found in nature. Various carbonyl compounds 2, β-hydroxyimines 3, β-hydroxynitriles 4, β-aminoketones 5, γ-aminoalcohols 6 and oximes 7 can all be readily attained through ring cleavage of 2-isoxazolines using suitable reagents for each transformation (Scheme 1) [1-7]. In recent years, there has been considerable interest in applying this isoxazoline-cleavage methodology to the preparation of biologically active natural products, including novel antibacterial, antifungal and anticancer treatments [8-11]. Some common methods of reductive N–O bond cleavage in isoxazolines include hydrogenolysis using Raney nickel, reduction by LiAlH4, TiCl3, SmI2, or treatment with Mo(CO)6 [12-19]. Although cleavage reactions of 2-isoxazolines have been extensively studied, little is still known regarding cleavage reactions of 2-isoxazolines fused to bicyclic compounds.
![[1860-5397-10-227-i1]](/bjoc/content/inline/1860-5397-10-227-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cleavage reactions of 2-isoxazoline 1.
Scheme 1: Cleavage reactions of 2-isoxazoline 1.
We have previously shown that the Mo(CO)6-mediated cleavage of carbobicycle-fused 2-isoxazolines converts both exo- and endo stereoisomers of 8 into the corresponding single isomer of cyclopentene 9 (Table 1) [20].
Table 1: Molybdenum-mediated cleavage of carbobicycle-fused isoxazolines.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-227-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | R | Isoxazoline | Product | Yield (%)a |
|---|---|---|---|---|
| 1 | Ph | exo-8a | 9a | 66 |
| 2 | Ph | endo-8a | 9a | 70 |
| 3 | Me | exo-8b | 9b | 61 |
| 4 | Me | endo-8b | 9b | 62 |
aIsolated yield after column chromatography.
Since then, we have been interested in the outcomes of cleavage reactions of 2-isoxazolines fused to heterobicyclic compounds 10 or 11, which we were able to prepare in a highly regio- and stereoselective manner [21]. Selective cleavage of the N–O bond or simultaneous cleavage of both N–O and bridge C–C bonds of these isoxazolines could provide novel routes to the aforementioned functionalities attached to heterobicyclic ring systems such as β-hydroxycarbonyl derivatives 12 and γ-aminoalcohols 13, as well as carbonyl compounds including phthalan 14 and isoindoline derivatives 15 (R2 = Ar; Scheme 2).
![[1860-5397-10-227-i2]](/bjoc/content/inline/1860-5397-10-227-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Potential modes of cleavage for heterobicycloalkene-fused isoxazolines 10 or 11.
Scheme 2: Potential modes of cleavage for heterobicycloalkene-fused isoxazolines 10 or 11.
Results and Discussion
For our investigation of heterobicycloalkene-fused 2-isoxazoline cleavage, we initially planned to follow our former Mo(CO)6-promoted approach. We elected substrate 10a (Scheme 2, R’ = Me, R = X = Y = H) for early optimization trials. The molybdenum-mediated conditions, however, were unsuccessful at both the previously optimized temperature of 80 °C (which led to extensive degradation) and at lower temperatures of 45–60 °C (where only 10a was recovered). We then adopted the methodology of Simoni and coworkers, treating 10a with refluxing Mo(CO)6 in acetonitrile [18]. Unfortunately, in our studies this showed no noticeable improvement, as the reaction did not proceed at room temperature, and at 45–60 °C we observed a series of spots by TLC which were not separable by column chromatography. This prompted us to seek other reagents capable of inducing N–O cleavage. We subsequently attempted hydrogenation using Pd/C with various solvent systems (MeOH, MeOH/AcOH, AcOH/H2O) as well as hydrogen sources (H2, ammonium formate) [22], as well as the combination of Pd(OH)2 with H2. However, none of these palladium-mediated attempts resulted in a clean transformation. Trials involving zinc dust in aqueous AcOH, as well as iron and ammonium chloride in aqueous EtOH at various temperatures also failed [23,24]. Finally, we turned to nickel-mediated reactions.
As before, our early trials with Raney nickel/H2 in MeOH and Raney nickel in AcOH showed no signs of progress. However, under conditions of Raney nickel/AlCl3 in a 5:1 MeOH/H2O mixture [25,26], we observed a clean transformation and the product readily precipitated out of solution following work-up, allowing us to bypass chromatographic purification. The product so obtained was found to be β-hydroxyketone 16a (Table 2, entry 1).
Once the optimal conditions were found, various symmetrical heterobicycle-fused 2-isoxazolines were opened by Raney-Ni/AlCl3 mediation. These results are summarized in Table 2. Compared to the parent compound 10a which gave an appreciable 93% yield of the corresponding β-hydroxyketone (Table 2, entry 1), replacement of two of the arene hydrogens for bromine atoms in 10b resulted in a lower tendency toward N–O bond cleavage (Table 2, entry 2). In addition, while both 6,7- and 5,8-dimethoxy-substituted compounds 10c and 10d gave excellent yields of 87% and 94%, respectively (Table 2, entries 4 and 5), the 5,8-dimethyl derivative 10e was slightly lower yielding, providing 76% isolated product (Table 2, entry 6). Overall, isoxazolines 10a–e all showed good conversion to their β-hydroxyketones. We then proceeded to expand the reaction scope to include unsymmetrical substrates, namely those with bridgehead (C1) substituents.
Table 2: Effects of arene substitution on Raney Ni/AlCl3-mediated cleavage of symmetrical 7-oxabenzonorbornadiene-fused 3-methyl-2-isoxazolines.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-227-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | X | Y | Isoxazoline | Product | Yield (%)a |
|---|---|---|---|---|---|
| 1 | H | H | 10a | 16a | 93 |
| 2 | H | Br | 10b | 16b | 71 |
| 3 | H | OMe | 10c | 16c | 87 |
| 4 | OMe | H | 10d | 16d | 94 |
| 5 | Me | H | 10e | 16e | 76 |
aIsolated yield.
The results of isoxazoline cleavage on unsymmetrical oxabicyclic alkene-fused systems are summarized in Table 3. Due to decreased solubility in aqueous methanol, the C1-substituted compounds 10f–k were reacted in a 5:5:2 THF/MeOH/H2O solvent system. Relative to the unsubstituted parent 10a (Table 3, entry 1), reactions of isoxazolines 10f and 10g bearing alkyl substituents in the C1 position were still reasonably good (Table 3, entries 2 and 3), and even with electron-withdrawing C1 substituents in compounds 10h–j, the yields did not suffer to any extreme (Table 3, entries 4–6). In addition, the transformation to β-hydroxyketone succeeded with compound 10k bearing both arene and C1 substituents to furnish 16k (Table 3, entry 7) whose structure was confirmed by X-ray crystallographic analysis [27].
Table 3: Effects of bridgehead substitution on Raney Ni/AlCl3 mediated cleavage of 7-oxabenzonorbornadiene-fused 3-methyl-2-isoxazolines.
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-227-i8.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | X | Y | Isoxazoline | Product | Yield (%)a |
|---|---|---|---|---|---|---|
| 1b | H | H | H | 10a | 16a | 93 |
| 2 | Me | H | H | 10f | 16f | 80 |
| 3 | Et | H | H | 10g | 16g | 95 |
| 4 | CH2OH | H | H | 10h | 16h | 85 |
| 5 | C(O)CH3 | H | H | 10i | 16i | 69 |
| 6 | COOMe | H | H | 10j | 16j | 85 |
| 7 | Me | OMe | H | 10k | 16k | 86 |
aIsolated yield. b5:1 MeOH/H2O solvent system.
Finally, we applied this N–O bond-cleavage methodology to several other heterobicycle-fused 2-isoxazolines to verify its compatibility with similar structures (Scheme 3). As with isoxazolines 10, each of these compounds (17, 18 and 19) was prepared in our laboratories by a combination of Diels–Alder cycloaddition and 1,3-dipolar cycloaddition reactions [21]. We found that the Raney nickel-mediated cleavage took place readily on 17 to yield a β-hydroxyketo-functionalized 7-oxanorbornane 20 (Scheme 3), and was also successful with isoxazolines 18a and 18b, affording N-phenylmaleimide-fused 21a and 21b (where the solvent ratio was modified to account for the lower solubility of 18a/18b in aqueous methanol). Furthermore, the reaction was also successful in the conversion of 7-azanorbornadiene-fused 19 to 22, suggesting the potential utility of the isoxazoline-cleavage reaction with other azabicyclic systems.
![[1860-5397-10-227-i3]](/bjoc/content/inline/1860-5397-10-227-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Raney Ni/AlCl3-mediated cleavage of 2-isoxazolines with various fused heterobicyclic frameworks.
Scheme 3: Raney Ni/AlCl3-mediated cleavage of 2-isoxazolines with various fused heterobicyclic frameworks.
In our initial report of molybdenum-mediated cleavage of carbobicycloalkene-fused isoxazolines 8, we proposed a general mechanism of the transformation to attached ring system 9 (Scheme 4). As the carbobicyclic framework contains no metal-coordinatable atom, we assumed that the cleavage reaction proceeds with initial coordination of the nitrogen atom to molybdenum, as suggeseted by Simoni and coworkers [18].
![[1860-5397-10-227-i4]](/bjoc/content/inline/1860-5397-10-227-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for Mo-mediated cleavage of carbobicycle-fused 2-isoxazoline.
Scheme 4: Proposed mechanism for Mo-mediated cleavage of carbobicycle-fused 2-isoxazoline.
In comparison, the β-hydroxyketones 16 generated in the current investigation suggest both reductive N–O cleavage and imine hydrolysis occurring in a single pot. Moreover, there is no indication of a retro-aldol cleavage. Since molybdenum catalysis was ineffective with the heterobicycloalkene-fused isoxazolines, it is possible that molybdenum preferentially coordinates to the heteroatom of the bicycloalkene rather than to the nitrogen of the isoxazoline, reducing its tendency toward N–O cleavage. Furthermore, since the reaction did not proceed in the absence of either AlCl3 or nickel, coordination of both metals appears to be an essential step in the formation of 16. Under the optimized conditions, we suggest that initial binding of Lewis acidic aluminum to the bridge heteroatom of 10 leaves its isoxazoline nitrogen free to interact with the Raney nickel (Scheme 5, 23). The N–O bond is subsequently broken to afford 24, and it is conceivable at this stage that the added coordination of AlCl3 to the alkoxy oxygen of 24 prevents retro-aldol cleavage from taking place. Finally, in a manner analogous to our former proposal, hydrolysis ensues to produce 16 (Scheme 4).
![[1860-5397-10-227-i5]](/bjoc/content/inline/1860-5397-10-227-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for Raney nickel-mediated formation of β-hydroxyketones from heterobicycloalkene-fused 2-isoxazolines.
Scheme 5: Proposed mechanism for Raney nickel-mediated formation of β-hydroxyketones from heterobicycloalkene...
Conclusion
To conclude, we have demonstrated the first examples of Raney nickel/AlCl3-mediated cleavage reactions of 2-isoxazoline rings fused to oxabicyclic and azabicyclic frameworks, which show novel reactivity compared to our former report of carbobicyclic analogues. The transformation involves N–O bond cleavage of the 2-isoxazoline followed by imine hydrolysis, leading to β-hydroxyketone products with moderate to excellent yields. We have investigated the efficacy of various cleavage conditions and found that although none of Mo(CO)6, Zn/AcOH, Fe/NH4Cl, Pd-C/H2, Pd(OH)2/H2 or Raney nickel/AcOH/H2 is suitable for cleavage of 2-isoxazolines fused to heterobicyclic frameworks, the combination of Raney nickel/AlCl3 is a suitable mediator for N–O bond cleavage in a variety of heterobicycloalkene-fused 2-isoxazoline systems.
References
-
Nitta, M.; Kobayashi, T. J. Chem. Soc., Perkin Trans. 1 1984, 2103–2108. doi:10.1039/P19840002103
Return to citation in text: [1] -
Curran, D. P.; Fenk, C. J. Tetrahedron Lett. 1986, 27, 4865–4868. doi:10.1016/S0040-4039(00)85083-2
Return to citation in text: [1] -
Pohjakallio, A.; Pihko, P. M.; Liu, J. J. Org. Chem. 2010, 75, 6712–6715. doi:10.1021/jo1013788
Return to citation in text: [1] -
Gelabert, L. I.; Fascio, M. L.; D’Accorso, N. B. J. Heterocycl. Chem. 2003, 40, 341–344. doi:10.1002/jhet.5570400223
Return to citation in text: [1] -
Kozikowski, A. P. Acc. Chem. Res. 1984, 17, 410–416. doi:10.1021/ar00108a001
Return to citation in text: [1] -
Burri, K. F.; Cardone, R. A.; Chen, W. Y.; Rosen, P. J. Am. Chem. Soc. 1978, 100, 7069–7071. doi:10.1021/ja00490a052
Return to citation in text: [1] -
Nagireddy, J. R.; Raheem, M.-A.; Haner, J.; Tam, W. Curr. Org. Synth. 2011, 8, 659–700. doi:10.2174/157017911796957348
Return to citation in text: [1] -
Bull, J. A.; Balskus, E. P.; Horan, R. A. J.; Langner, M.; Ley, S. V. Chem.–Eur. J. 2007, 13, 5515–5538. doi:10.1002/chem.200700033
Return to citation in text: [1] -
Das, B.; Satyakumar, A.; Ravikanth, B.; Rao, B. V.; Raju, T. V.; Siddhardha, B.; Murty, U. S. J. Heterocycl. Chem. 2013, 50, 430–434. doi:10.1002/jhet.982
Return to citation in text: [1] -
Bode, J. W.; Carreira, E. M. J. Am. Chem. Soc. 2001, 123, 3611–3612. doi:10.1021/ja0155635
Return to citation in text: [1] -
Litvinovskaya, R. P.; Koval’, N. V.; Khripach, V. A. Chem. Heterocycl. Compd. 2000, 36, 190–194. doi:10.1007/BF02283551
Return to citation in text: [1] -
Curran, D. P. J. Am. Chem. Soc. 1983, 105, 5826–5833. doi:10.1021/ja00356a021
Return to citation in text: [1] -
Jiang, B.; Liu, Y.; Zhou, W.-s. J. Org. Chem. 2000, 65, 6231–6236. doi:10.1021/jo000059o
Return to citation in text: [1] -
Barco, A.; Benetti, S.; De Risi, C.; Pollini, G. P.; Zanirato, V. Tetrahedron 1995, 51, 7721–7726. doi:10.1016/0040-4020(95)00392-L
Return to citation in text: [1] -
Andersen, S. H.; Sharma, K. K.; Torssell, K. B. G. Tetrahedron 1983, 39, 2241–2245. doi:10.1016/S0040-4020(01)91946-5
Return to citation in text: [1] -
Jäger, V.; Grund, H. Angew. Chem., Int. Ed. Engl. 1976, 15, 50–51. doi:10.1002/anie.197600501
Return to citation in text: [1] -
Bode, J. W.; Carreira, E. M. Org. Lett. 2001, 3, 1587–1590. doi:10.1021/ol015885d
Return to citation in text: [1] -
Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Simoni, D. Synthesis 1987, 276–278. doi:10.1055/s-1987-27915
Return to citation in text: [1] [2] [3] -
Baraldi, P. G.; Bigoni, A.; Cacciari, B.; Caldari, C.; Manfredini, S.; Spalluto, G. Synthesis 1994, 1158–1162. doi:10.1055/s-1994-25663
Return to citation in text: [1] -
Tranmer, G. K.; Tam, W. Org. Lett. 2002, 4, 4101–4104. doi:10.1021/ol026846k
Return to citation in text: [1] -
Nagireddy, J. R.; Carlson, E.; Tam, W. Can. J. Chem. 2014, 92, 635–639. doi:10.1139/cjc-2014-0140
Return to citation in text: [1] [2] -
Asaoka, M.; Mukuta, T.; Takei, H. Tetrahedron Lett. 1981, 22, 735–738. doi:10.1016/0040-4039(81)80137-2
Return to citation in text: [1] -
King, G. S.; Magnus, P. D.; Rzepa, H. S. J. Chem. Soc., Perkin Trans. 1 1972, 437–443. doi:10.1039/P19720000437
Return to citation in text: [1] -
Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c
Return to citation in text: [1] -
Kozikowski, A. P.; Adamczyk, M. Tetrahedron Lett. 1982, 23, 3123–3126. doi:10.1016/S0040-4039(00)88575-5
Return to citation in text: [1] -
Bondar, N. F.; Isaenya, L. P.; Skupskaya, R. V.; Lakhvich, F. A. Russ. J. Org. Chem. 2003, 39, 1095–1103.
Return to citation in text: [1] -
Lough, A. J.; Nagireddy, J. R.; Tam, W. Acta Crystallogr., Sect. E 2014, 70, o545. doi:10.1107/S1600536814007752
Return to citation in text: [1]
| 1. | Nitta, M.; Kobayashi, T. J. Chem. Soc., Perkin Trans. 1 1984, 2103–2108. doi:10.1039/P19840002103 |
| 2. | Curran, D. P.; Fenk, C. J. Tetrahedron Lett. 1986, 27, 4865–4868. doi:10.1016/S0040-4039(00)85083-2 |
| 3. | Pohjakallio, A.; Pihko, P. M.; Liu, J. J. Org. Chem. 2010, 75, 6712–6715. doi:10.1021/jo1013788 |
| 4. | Gelabert, L. I.; Fascio, M. L.; D’Accorso, N. B. J. Heterocycl. Chem. 2003, 40, 341–344. doi:10.1002/jhet.5570400223 |
| 5. | Kozikowski, A. P. Acc. Chem. Res. 1984, 17, 410–416. doi:10.1021/ar00108a001 |
| 6. | Burri, K. F.; Cardone, R. A.; Chen, W. Y.; Rosen, P. J. Am. Chem. Soc. 1978, 100, 7069–7071. doi:10.1021/ja00490a052 |
| 7. | Nagireddy, J. R.; Raheem, M.-A.; Haner, J.; Tam, W. Curr. Org. Synth. 2011, 8, 659–700. doi:10.2174/157017911796957348 |
| 21. | Nagireddy, J. R.; Carlson, E.; Tam, W. Can. J. Chem. 2014, 92, 635–639. doi:10.1139/cjc-2014-0140 |
| 12. | Curran, D. P. J. Am. Chem. Soc. 1983, 105, 5826–5833. doi:10.1021/ja00356a021 |
| 13. | Jiang, B.; Liu, Y.; Zhou, W.-s. J. Org. Chem. 2000, 65, 6231–6236. doi:10.1021/jo000059o |
| 14. | Barco, A.; Benetti, S.; De Risi, C.; Pollini, G. P.; Zanirato, V. Tetrahedron 1995, 51, 7721–7726. doi:10.1016/0040-4020(95)00392-L |
| 15. | Andersen, S. H.; Sharma, K. K.; Torssell, K. B. G. Tetrahedron 1983, 39, 2241–2245. doi:10.1016/S0040-4020(01)91946-5 |
| 16. | Jäger, V.; Grund, H. Angew. Chem., Int. Ed. Engl. 1976, 15, 50–51. doi:10.1002/anie.197600501 |
| 17. | Bode, J. W.; Carreira, E. M. Org. Lett. 2001, 3, 1587–1590. doi:10.1021/ol015885d |
| 18. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Simoni, D. Synthesis 1987, 276–278. doi:10.1055/s-1987-27915 |
| 19. | Baraldi, P. G.; Bigoni, A.; Cacciari, B.; Caldari, C.; Manfredini, S.; Spalluto, G. Synthesis 1994, 1158–1162. doi:10.1055/s-1994-25663 |
| 8. | Bull, J. A.; Balskus, E. P.; Horan, R. A. J.; Langner, M.; Ley, S. V. Chem.–Eur. J. 2007, 13, 5515–5538. doi:10.1002/chem.200700033 |
| 9. | Das, B.; Satyakumar, A.; Ravikanth, B.; Rao, B. V.; Raju, T. V.; Siddhardha, B.; Murty, U. S. J. Heterocycl. Chem. 2013, 50, 430–434. doi:10.1002/jhet.982 |
| 10. | Bode, J. W.; Carreira, E. M. J. Am. Chem. Soc. 2001, 123, 3611–3612. doi:10.1021/ja0155635 |
| 11. | Litvinovskaya, R. P.; Koval’, N. V.; Khripach, V. A. Chem. Heterocycl. Compd. 2000, 36, 190–194. doi:10.1007/BF02283551 |
| 25. | Kozikowski, A. P.; Adamczyk, M. Tetrahedron Lett. 1982, 23, 3123–3126. doi:10.1016/S0040-4039(00)88575-5 |
| 26. | Bondar, N. F.; Isaenya, L. P.; Skupskaya, R. V.; Lakhvich, F. A. Russ. J. Org. Chem. 2003, 39, 1095–1103. |
| 21. | Nagireddy, J. R.; Carlson, E.; Tam, W. Can. J. Chem. 2014, 92, 635–639. doi:10.1139/cjc-2014-0140 |
| 23. | King, G. S.; Magnus, P. D.; Rzepa, H. S. J. Chem. Soc., Perkin Trans. 1 1972, 437–443. doi:10.1039/P19720000437 |
| 24. | Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181–9183. doi:10.1021/jo801831c |
| 18. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Simoni, D. Synthesis 1987, 276–278. doi:10.1055/s-1987-27915 |
| 22. | Asaoka, M.; Mukuta, T.; Takei, H. Tetrahedron Lett. 1981, 22, 735–738. doi:10.1016/0040-4039(81)80137-2 |
| 18. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Simoni, D. Synthesis 1987, 276–278. doi:10.1055/s-1987-27915 |
| 27. | Lough, A. J.; Nagireddy, J. R.; Tam, W. Acta Crystallogr., Sect. E 2014, 70, o545. doi:10.1107/S1600536814007752 |
© 2014 Nagireddy et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)