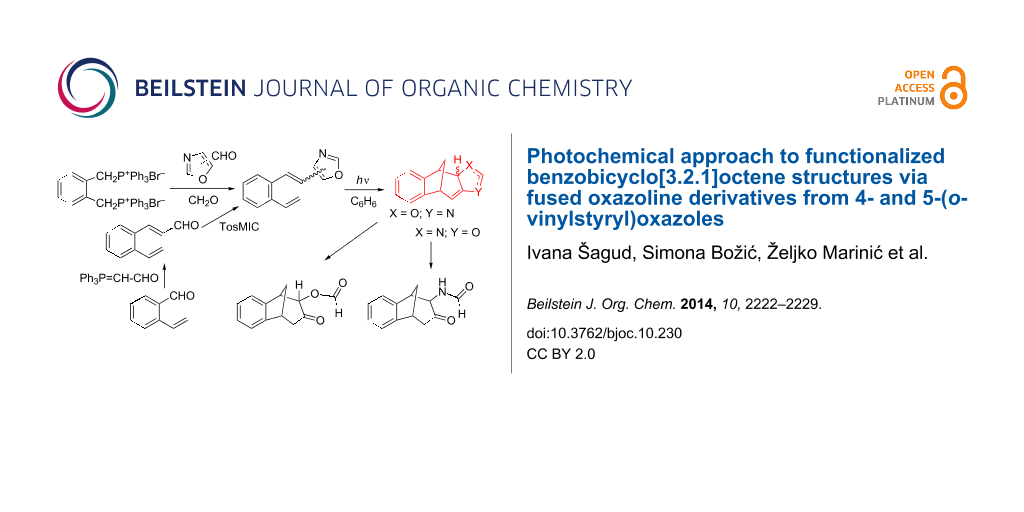
Abstract
Novel cis/trans-4- and cis/trans-5-(2-vinylstyryl)oxazoles have been synthesized by Wittig reactions from the diphosphonium salt of α,α’-o-xylene dibromide, formaldehyde and 4- and 5-oxazolecarbaldehydes, respectively. In contrast, trans-5-(2-vinylstyryl)oxazole has been synthesized by the van Leusen reaction from trans-3-(2-vinylphenyl)acrylaldehyde which is prepared from o-vinylbenzaldehyde and (formylmethylene)triphenylphosphorane. The 4- and 5-(2-vinylstyryl)oxazoles afford, by photochemical intramolecular cycloaddition, diverse fused oxazoline-benzobicyclo[3.2.1]octadienes, which are identified and characterized by spectroscopic methods. The photoproducts formed are relatively unstable and spontaneously or on silica gel undergo oxazoline ring opening followed by formation of formiato- or formamido-benzobicyclo[3.2.1]octenone derivatives. On irradiation of 4-(2-vinylstyryl)oxazole small quantities of electrocyclization product, 4-(1,2-dihydronaphthalen-2-yl)oxazole, are isolated and spectroscopically characterized.

Graphical Abstract
Introduction
The bicyclo[3.2.1]octane skeleton is the basic framework of numerous important biologically active natural compounds or their metabolites [1]. Properly functionalized bicyclo[3.2.1]octanes have proved as useful reactive intermediates in stereoselective transformations making these derivatives powerful building blocks in organic synthetic strategies [2]. Various methodologies and new synthetic approaches for their preparation and reactivity have been reviewed [3]. Continuing our long-standing interest for photochemical intramolecular cycloaddition reactions of various β-heteroaryl-o-divinylbenzenes, furans [4-6], thiophenes [6-8], pyroles [9,10] and sydnones [11-13], as routes to polycyclic compounds, we turned our attention to oxazole derivatives. The oxazole structure is commonly found in natural products and pharmaceuticals [14-17] and is applied in useful reagents and intermediates in organic synthesis [18-25]. There are examples of oxazole photochemical intermolecular cycloadditions [26-32], but to the best of our knowledge, there are no examples of intramolecular photocycloaddition. We describe herein, the synthesis of new 4- and 5-(2-vinylstyryl)oxazoles (1, 2) and their intramolecular photocycloaddition to diverse fused tetracyclic oxazoline compounds which further spontaneously or during the work-up procedure hydrolyze to benzobicyclo[3.2.1]octenone derivatives. This is a new method for the synthesis of functionalized benzobicyclo[3.2.1]octenes.
Results and Discussion
cis/trans-Isomers of 4- and 5-oxazole derivatives (1, 2) were synthesized by Wittig reactions from the diphosphonium salt of α,α’-o-xylene dibromide, formaldehyde and oxazole-4- and 5-carbaldehydes (3, 4), respectively, in absolute ethanol with sodium ethoxide as a base (Scheme 1).
![[1860-5397-10-230-i1]](/bjoc/content/inline/1860-5397-10-230-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 4- (1) and 5-(2-vinylstyryl)oxazoles (2).
Scheme 1: Synthesis of 4- (1) and 5-(2-vinylstyryl)oxazoles (2).
The procedure of this multicomponent reaction is slightly modified, compared to the described method [33], in order to optimize the yields. The yield of 4-oxazole derivative 1 was 50% whereas the best result found for the 5-oxazole derivative 2 was 22%. The required oxazole-4/5-carbaldehydes (3, 4) [34-36] were prepared from commercially available esters by DIBALH reduction following the procedure [34] for oxazole-4-carbaldehyde (3). The crude products obtained were used in the next step of the synthesis without purification because of their volatility. Reduction of ethyl oxazole-4-carboxylate proceeds completely whereas the crude reaction sample of the oxazole-5-carbaldehyde (4) contains 10% of unreacted ester. The unreacted ester is difficult to separate by column chromatography from the trans-isomer 2. It could be removed from the reaction mixture by mild basic hydrolysis [37].
To avoid the use of volatile oxazole-5-carbaldehyde (4) we developed a new synthetic route to 5-(2-vinylstyryl)oxazole (2) in which the oxazole ring is formed at the end of the reaction sequence (Scheme 1). An oxazole ring substituted in the 5-position can be synthesized from the corresponding aldehydes using van Leusen’s reagent, tosyl methyl isocyanide (TosMIC) [38,39]. For the preparation of 5-(2-vinylstyryl)oxazole (2) by this method 3-(2-vinylphenyl)acrylaldehyde (6) was needed. This new o-substituted phenylacrylaldehyde 6 was prepared using (formylmethylene)triphenylphosphorane by a Wittig reaction from o-vinylbenzaldehyde (5). The yield of desired product 6 is lower, compared to the yields of previously prepared β-heteroarylacrylaldehydes [23]. This can be explained by the diminished nucleophilic attack of the reagent to the carbonyl moiety due to the steric hindrance of the o-vinyl group in 5 and continued competitive reaction of the carbonyl from the formed o-vinylphenylacrylaldehyde 6 with (formylmethylene)triphenylphosphorane to give 5-(2-vinylphenyl)penta-2,4-dienal (7) as byproduct. Under optimal reaction conditions (see Supporting Information File 1) 32% trans-3-(2-vinylphenyl)prop-2-enal (6) is obtained in addition with 5% trans,trans-5-(2-vinylphenyl)penta-2,4-dienal (7) as a contaminant, alongside with a large amount of resinous material. The required o-vinylbenzaldehyde (5) [40] was synthesized from 2-bromostyrene and used without purification. As the starting aldehyde 6 for the reaction with TosMIC was in trans configuration the 5-(2-vinylstyryl)oxazole (2) obtained retained the trans configuration. This is clearly seen from the coupling constants of the ethylene protons (J = 16 Hz) in the 1H NMR. All new compounds for further experiments, cis/trans-1, cis/trans-2 and trans-6 are isolated by column chromatography on silica gel in moderate yields (22–50%) and characterized by spectroscopic methods (see Supporting Information File 1).
The irradiation experiments have been performed in a Rayonet reactor (>300 nm, rt, using up to 16 lamps each with a power of 8 W) with petroleum ether, acetonitrile or benzene as a solvent. Benzene gave the cleanest reaction profile and was used as the solvent in further preparative experiments. The 1H NMR spectra of the crude photomixtures showed complete conversion after 3–5 hours of irradiation. In the case of cis/trans-4-(2-vinylstyryl)oxazoles 1 two dominant products 8a (74%) and 8b (20%) were observed in the 1H NMR spectra and a small quantity of 9 (6%) was observed as well (Scheme 2).
![[1860-5397-10-230-i2]](/bjoc/content/inline/1860-5397-10-230-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Irradiation of 4- (1) and 5-(2-vinylstyryl)oxazoles (2) (crude reaction mixtures).
Scheme 2: Irradiation of 4- (1) and 5-(2-vinylstyryl)oxazoles (2) (crude reaction mixtures).
Whereas in the case of cis/trans-5-(2-vinylstyryl)oxazoles (2) one major product 10 (75%) and minor product 11 (25%) along with a lot of small quantities of unidentified side products (Scheme 2) were observed. Irradiation of compounds 1 and 2 in NMR tubes dissolved in deuterated benzene and recording the spectra at timed intervals demonstrated that 11 is not the photochemical product as can be clearly seen in Figure 1.
![[1860-5397-10-230-1]](/bjoc/content/figures/1860-5397-10-230-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Part of 1H NMR spectra in C6D6 of the crude photomixtures after 200 min (300 nm, rt ) of irradiation of cis/trans-1 (a) and cis/trans-2 (b).
Figure 1: Part of 1H NMR spectra in C6D6 of the crude photomixtures after 200 min (300 nm, rt ) of irradiatio...
After complete conversion of the starting compound 2 only compound 10 was present in the 1H NMR spectrum in contrast to photochemical conversion of compound 1 in which the same mixture of three products (8a, 8b and 9) is obtained as in preparative experiments.
The structure of photoproduct 10 was completely determined using COSY, NOESY and HSQC techniques (see Supporting Information File 1). Aromatic protons of 10 are at 6.9–7.3 ppm and the proton on the oxazoline moiety is a singlet at 6.2 ppm. The specific aliphatic protons HA–HF of the bicyclic skeleton (Figure 1) show a similar pattern as the previously described benzofuran intermediate [41]. In the 13C NMR spectrum there are 5 signals in the region from 108 to 40 ppm. The doublet at 108 ppm indicates the structure with sp2-hybridized carbon (CH(A)) and the triplet at 44 ppm indicates the existence of one geminal carbon atom. The tetracyclic oxazoline stereoisomer rel-(2S)-10 undergoes spontaneously oxazoline ring-opening to 11 during the solvent evaporation after the irradiation and therefore the identification of this compound had to be done immediately after the work-up procedure. The formation of tricyclic formamido derivative rel-(9S)-11 can be explained by the addition of water to the exo-double bond of the bicyclic skeleton or the C=N double bond and further oxazoline ring-opening (Scheme 3).
![[1860-5397-10-230-i3]](/bjoc/content/inline/1860-5397-10-230-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible mechanisms of oxazoline ring-opening in photoproduct 10.
Scheme 3: Plausible mechanisms of oxazoline ring-opening in photoproduct 10.
Formamido derivative rel-(9S)-11 is completely characterized by spectroscopic methods. In its IR spectrum there are signals at 3334 cm−1 of the NH group, and strong signals at 1722 and 1683 cm−1 of two carbonyl groups. The formamido proton in the 1H NMR spectrum appears at 8.29 ppm and the proton of the NH group as a broad singlet at 6.29 ppm. Specific signals for the aliphatic protons HA–HG of the benzobicyclo[3.2.1]octenone structure are present at higher field from 4.8 to 2.3 ppm as expected (Figure 2: (b)). From the NOESY spectrum was evident that the HA is oriented towards the methano bridge.
![[1860-5397-10-230-2]](/bjoc/content/figures/1860-5397-10-230-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectra in C6D6 of rel-(9S)-12a (a) and rel-(9S)-11 (b).
Figure 2: 1H NMR spectra in C6D6 of rel-(9S)-12a (a) and rel-(9S)-11 (b).
The diastereomeric fused tetracyclic oxazolines rel-(2S)-8a and rel-(2R)-8b (Scheme 2, Figure 1), that were obtained on irradiation of cis/trans-4-(2-vinylstyryl)oxazole (1), are more stable than 10 but not sufficiently so that they can be separated chromatographically on silica gel. The major diastereomer 8a is isolated mixing the diastereomeric mixture for several hours in dry ether with some quantity of silica gel. The minor diastereomer 8b is identified and characterized in the NMR spectra of the photomixtures. The difference in structures of 8a and 8b is in the orientation of HB (8a) or Hb (8b) protons. In the NOESY spectrum of the diastereomeric mixture the interaction between HB and HF protons can be seen which is a clear proof that the HB is facing the methano bridge in the major diastereomer 8a. There is no interaction between Hb and Hf protons in the minor isomer 8b suggesting that Hb proton is opposite to the methano-bridge. The diastereomer in which the HB is oriented towards the methano bridge is the main product in photochemical reactions of either 4- or 5-(2-vinylstyryl)oxazole.
The formation of the photoproducts 8 and 10 can be explained by intramolecular cycloaddition and formation of resonance stabilized biradicals A/A’ followed by the 1,6-ring closure (Scheme 4).
![[1860-5397-10-230-i4]](/bjoc/content/inline/1860-5397-10-230-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism of the formation of polycyclic compounds (8–10).
Scheme 4: Mechanism of the formation of polycyclic compounds (8–10).
An 1,3-H shift, as in furan and thiophene derivatives [6], and rearomatization to fused oxazole derivatives B/B’ is not detected. The 1,6-ring closure of the biradicals A/A’ occurs stereoselectively giving the major products rel-(2S)-8a/rel-(2S)-10 in which the hydrogen on C-2 is oriented toward the methano bridge. The formation of dihydronaphthalene derivative 9, found only on irradiation of 1, is explained by 6π electrocyclization of the benzodivinyl moiety to intermediate C followed by 1,5-H shift and rearomatization of the benzene ring. Analogue electrocyclization was not detected in phenyl or furyl-substituted o-divinylbenzenes but instead stilbene-like 6π electrocyclization and formation of 1-vinylphenanthrene [42] or 6-vinylnaphtho[2,1-b]thiophene [6] occured, respectively.
During chromatography on silica gel the tricyclic formiato derivative rel-(9S)-12 is formed from 8 (Scheme 5).
![[1860-5397-10-230-i5]](/bjoc/content/inline/1860-5397-10-230-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reactions of the photochemical product 8 with EtOH, MeOD and H2O/silica gel.
Scheme 5: Reactions of the photochemical product 8 with EtOH, MeOD and H2O/silica gel.
In the 1H NMR spectrum the main diastereomer rel-(9S)-12a shows the signal of the formiato proton in the low field at 8.2 ppm. The signals from 5.6 to 2.3 ppm are assigned to HA–HG protons of the bicyclic skeleton. Using COSY and NOESY techniques all corresponding interactions are found. The signals of two carbonyl groups located at 200.8 (C=O) and 159.3 (CH'=O) ppm in the 13C NMR spectrum were confirmed in the IR spectrum with the bands at 1740 and 1714 cm–1. No signal for a NH proton was found in the 1H NMR (Figure 2: (a)) nor the NH band in the IR spectrum. Its HRMS also confirmed the compound without the presence of nitrogen. The NOE interaction between protons HA and HG proved that the HA proton is facing to the methano bridge and that in the open structure rel-(9S)-12a HA retained the same orientation as it had in the closed product 8a. 1H NMR spectra of rel-(9S)-12a and rel-(9R)-12b are similar and have comparable interactions of protons in the COSY spectra. Unlike the NOESY spectrum of rel-(9S)-12a with HA at 5.61 ppm in interaction with HG, the Ha proton at 5.07 ppm of rel-(9R)-12b has no interaction with Hg.
The photoproduct 8 is more stable than 10. The stability can be attributed to the existence of conjugated double bonds present in the structure. The structure of 8 in which the exo-double bond is in conjugation with the oxazoline double bond is also confirmed by isolation of adducts 13 and 14 (Scheme 5).
When the crude photomixture, containing 8a, 8b and 9, was dissolved in absolute ethanol and left in a refrigerator at 13 °C over the weekend the adduct 13 was formed as a main product. The same procedure with MeOD showed the incorporation of deuterium in the bicyclo[3.2.1]octadiene moiety and a methoxy group on the N=C oxazoline bond by 1,4-addition or more likely by addition to the N=C bond followed by keto–enol tautomerization giving 14. The adducts are spectroscopically completely identified (see Supporting Information File 1). The spectra of alcohol adducts show that alongside with (2S)-13/14 there were traces of 9 and traces of what we suspect to be (2R)-13/14. Products 13/14 easily undergo ring opening on silica gel giving the same formiato derivative 12. Plausible mechanism of the ring opening of oxazoline derivative 8 might be as outlined in Scheme 6.
![[1860-5397-10-230-i6]](/bjoc/content/inline/1860-5397-10-230-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Plausible mechanisms of oxazoline ring opening in photoproduct 10 and formation of 12.
Scheme 6: Plausible mechanisms of oxazoline ring opening in photoproduct 10 and formation of 12.
Conclusion
In summary, photochemical fused oxazoline-benzobicyclo[3.2.1]octadiene products 8 and 10 are formed by photochemical intramolecular cycloaddition of 4- (1) and 5-(2-vinylstyryl)oxazoles (2), respectively. Product 10 spontaneously undergoes ring opening and formation of benzobicyclo[3.2.1]octenone derivative 11. Diastereomers 8 are stable under non-acidic conditions allowing the isolation of the main diastereomer 8a. They are easily hydrolyzed under mildly acidic conditions (silica gel) to functionalized benzobicyclo[3.2.1]octenone derivatives 12. Related benzobicyclo[3.2.1]octen-3-ones have been prepared by the method of Lansbury from chloroallylindene [43-45], by carbene reaction from benzonorbornadiene [46,47] or by intramolecular insertion of the vinyl group into a carbon–carbon single bond using organometallic catalysts [48]. Herein we have reported a new simple method for the synthesis of functionalized benzobicyclo[3.2.1]octene derivatives using light as a traceless reagent [49].
Supporting Information
| Supporting Information File 1: Experimental part, NMR and IR spectra. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Snyder, S. A.; Gollner, A.; Chiriac, M. I. Nature 2011, 474, 461–466. doi:10.1038/nature10197
Return to citation in text: [1] -
Filippini, M.-H.; Rodriguez, J. Chem. Rev. 1999, 99, 27–76. doi:10.1021/cr970029u
Return to citation in text: [1] -
Presset, M.; Coquerel, Y.; Rodriguez, J. Chem. Rev. 2013, 113, 525–595. doi:10.1021/cr200364p
Return to citation in text: [1] -
Škorić, I.; Basarić, N.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Chem. – Eur. J. 2005, 11, 543–551. doi:10.1002/chem.200401005
Return to citation in text: [1] -
Šindler-Kulyk, M.; Špoljarić, L.; Marinić, Ž. Heterocycles 1989, 29, 679–682. doi:10.3987/COM-89-4629
Return to citation in text: [1] -
Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062
Return to citation in text: [1] [2] [3] [4] -
Vuk, D.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron 2012, 68, 6873–6880. doi:10.1016/j.tet.2012.06.019
Return to citation in text: [1] -
Vuk, D.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Croat. Chem. Acta 2012, 85, 425–434. doi:10.5562/cca2120
Return to citation in text: [1] -
Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2006, 71, 9382–9392. doi:10.1021/jo061435t
Return to citation in text: [1] -
Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2003, 68, 7524–7527. doi:10.1021/jo0346454
Return to citation in text: [1] -
Butković, K.; Basarić, N.; Lovreković, K.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron Lett. 2004, 45, 9057–9060. doi:10.1016/j.tetlet.2004.10.036
Return to citation in text: [1] -
Butković, K.; Vuk, D.; Marinić, Ž.; Penić, J.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9356–9362. doi:10.1016/j.tet.2010.10.013
Return to citation in text: [1] -
Butković, K.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Beilstein J. Org. Chem. 2011, 7, 1663–1670. doi:10.3762/bjoc.7.196
Return to citation in text: [1] -
Palmer, D. C. Oxazoles: Synthesis, Reactions, and Spectroscopy. In The Chemistry of heterocyclic compounds, Part A; Taylor, E. C.; Wipf, P., Eds.; John Wiley and Sons Inc.: New Jersey, 2003; pp 94–118.
Return to citation in text: [1] -
Yeh, V. S. C. Tetrahedron 2004, 60, 11995–12042. doi:10.1016/j.tet.2004.10.001
Return to citation in text: [1] -
Jin, Z. Nat. Prod. Rep. 2011, 28, 1143–1191. doi:10.1039/c0np00074d
Return to citation in text: [1] -
Jin, Z. Nat. Prod. Rep. 2013, 30, 869–915. doi:10.1039/c3np70006b
Return to citation in text: [1] -
Zhang, J.; Coqueron, P.-Y.; Vors, J.-P.; Ciufolini, M. A. Org. Lett. 2010, 12, 3942–3945. doi:10.1021/ol101704r
Return to citation in text: [1] -
Zhang, J.; Ciufolini, M. A. Org. Lett. 2011, 13, 390–393. doi:10.1021/ol102678j
Return to citation in text: [1] -
Zhang, J.; Ciufolini, M. A. Org. Lett. 2009, 11, 2389–2392. doi:10.1021/ol900455m
Return to citation in text: [1] -
Cardwell, K. S.; Hermitage, S. A.; Sjolin, A. Tetrahedron Lett. 2000, 41, 4239–4242. doi:10.1016/S0040-4039(00)00573-6
Return to citation in text: [1] -
Swaminathan, S.; Singh, A. K.; Li, W.-S.; Venit, J. J.; Natalie, K. J., Jr.; Simpson, J. H.; Weaver, R. E.; Silverberg, L. J. Tetrahedron Lett. 1998, 39, 4769–4772. doi:10.1016/S0040-4039(98)00904-6
Return to citation in text: [1] -
Šagud, I.; Faraguna, F.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2011, 76, 2904–2908. doi:10.1021/jo1025942
Return to citation in text: [1] [2] -
Webb, M. R.; Addie, M. S.; Crawforth, C. M.; Dale, J. W.; Franci, X.; Pizzonero, M.; Donald, C.; Taylor, R. J. K. Tetrahedron 2008, 64, 4778–4791. doi:10.1016/j.tet.2008.01.116
Return to citation in text: [1] -
Hoffman, T. J.; Rigby, J. H.; Arseniyadis, S.; Cossy, J. J. Org. Chem. 2008, 73, 2400–2403. doi:10.1021/jo702305g
Return to citation in text: [1] -
Griesbeck, A. G.; Fiege, M.; Lex, J. J. Chem. Commun. 2000, 589–590. doi:10.1039/b000578i
Return to citation in text: [1] -
Weuthen, M.; Scharf, H.-D.; Runsink, J. Chem. Ber. 1987, 120, 1023–1026. doi:10.1002/cber.19871200623
Return to citation in text: [1] -
Griesbeck, A. G.; Bondock, S. Can. J. Chem. 2003, 81, 555–559. doi:10.1139/v03-029
Return to citation in text: [1] -
Bondock, S.; Griesback, A. G. Monatsh. Chem. 2006, 137, 765–777. doi:10.1007/s00706-006-0474-4
Return to citation in text: [1] -
Griesbeck, A. G.; Franke, M.; Neudörfl, J.; Kotaka, H. Beilstein J. Org. Chem. 2011, 7, 127–134. doi:10.3762/bjoc.7.18
Return to citation in text: [1] -
Zhang, Y.; Wang, L.; Zhang, M.; Fun, H.-K.; Xu, J.-H. Org. Lett. 2004, 26, 4893–4895. doi:10.1021/ol048028t
Return to citation in text: [1] -
Wang, L.; Huang, Y.-C.; Liu, Y.; Fun, H.-K.; Zhang, Y.; Xu, J.-H. J. Org. Chem. 2010, 75, 7757–7768. doi:10.1021/jo101764f
Return to citation in text: [1] -
Šindler-Kulyk, M.; Stiplošek, Z.; Vojnović, D.; Metelko, B.; Marinić, Ž. Heterocycles 1991, 32, 2357–2363. doi:10.3987/COM-91-5846
Return to citation in text: [1] -
Reeves, J. T.; Song, J. J.; Tan, Z.; Lee, H.; Yee, N. K.; Senanayake, C. H. Org. Lett. 2007, 9, 1875–1878. doi:10.1021/ol070340q
Return to citation in text: [1] [2] -
Mulder, R. J.; Shafer, C. M.; Molinski, T. F. J. Org. Chem. 1999, 64, 4995–4998. doi:10.1021/jo9906328
Return to citation in text: [1] -
Addie, M. S.; Taylor, R. J. K. J. Chem. Soc., Perkin Trans. 1 2000, 527–531. doi:10.1039/a908987j
Return to citation in text: [1] -
Young, G. L.; Smith, S. A.; Taylor, R. J. K. Tetrahedron Lett. 2004, 45, 3797–3801. doi:10.1016/j.tetlet.2004.03.083
Return to citation in text: [1] -
van Leusen, A. M.; Hoogenboom, B. E.; Siderius, H. Tetrahedron Lett. 1972, 13, 2369–2372. doi:10.1016/S0040-4039(01)85305-3
Return to citation in text: [1] -
Saikachi, H.; Kitagawa, T.; Sasaki, H.; van Leusen, A. M. Chem. Pharm. Bull. 1979, 27, 793–796. doi:10.1248/cpb.27.793
Return to citation in text: [1] -
Dale, W. J.; Starr, L.; Strobel, C. W. J. Org. Chem. 1961, 26, 2225–2227. doi:10.1021/jo01351a016
Return to citation in text: [1] -
Škorić, I.; Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. Heterocycles 2001, 55, 1889–1896. doi:10.3987/COM-01-9314
Return to citation in text: [1] -
Šindler-Kulyk, M.; Laarhoven, W. H. Recl. Trav. Chim. Pays-Bas 1979, 98, 187–191. doi:10.1002/recl.19790980410
Return to citation in text: [1] -
Lansbury, P. T.; Nienhouse, E. J. J. Am. Chem. Soc. 1966, 88, 4290–4291. doi:10.1021/ja00970a052
Return to citation in text: [1] -
Lansbury, P. T.; Nienhouse, E. J.; Scharf, D. J.; Hilfiker, F. R. J. Am. Chem. Soc. 1970, 92, 5649–5657. doi:10.1021/ja00722a018
Return to citation in text: [1] -
Wilt, J. W.; Rasmussen, R. R. J. Org. Chem. 1975, 40, 1031–1036. doi:10.1021/jo00896a007
Return to citation in text: [1] -
Kitahonoki, K.; Takano, Y.; Matsura, A.; Kotera, K. Tetrahedron 1969, 25, 335–353. doi:10.1016/S0040-4020(01)82628-4
Return to citation in text: [1] -
Balci, M.; Harmandar, M. Tetrahedron Lett. 1984, 25, 237–240. doi:10.1016/S0040-4039(00)99849-6
Return to citation in text: [1] -
Murakami, M.; Itahashi, T.; Ito, Y. J. Am. Chem. Soc. 2002, 124, 13976–13977. doi:10.1021/ja021062n
Return to citation in text: [1] -
Hoffmann, N. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. doi:10.1039/c2pp25074h
Return to citation in text: [1]
| 1. | Snyder, S. A.; Gollner, A.; Chiriac, M. I. Nature 2011, 474, 461–466. doi:10.1038/nature10197 |
| 6. | Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062 |
| 7. | Vuk, D.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron 2012, 68, 6873–6880. doi:10.1016/j.tet.2012.06.019 |
| 8. | Vuk, D.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Croat. Chem. Acta 2012, 85, 425–434. doi:10.5562/cca2120 |
| 38. | van Leusen, A. M.; Hoogenboom, B. E.; Siderius, H. Tetrahedron Lett. 1972, 13, 2369–2372. doi:10.1016/S0040-4039(01)85305-3 |
| 39. | Saikachi, H.; Kitagawa, T.; Sasaki, H.; van Leusen, A. M. Chem. Pharm. Bull. 1979, 27, 793–796. doi:10.1248/cpb.27.793 |
| 4. | Škorić, I.; Basarić, N.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Chem. – Eur. J. 2005, 11, 543–551. doi:10.1002/chem.200401005 |
| 5. | Šindler-Kulyk, M.; Špoljarić, L.; Marinić, Ž. Heterocycles 1989, 29, 679–682. doi:10.3987/COM-89-4629 |
| 6. | Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062 |
| 23. | Šagud, I.; Faraguna, F.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2011, 76, 2904–2908. doi:10.1021/jo1025942 |
| 3. | Presset, M.; Coquerel, Y.; Rodriguez, J. Chem. Rev. 2013, 113, 525–595. doi:10.1021/cr200364p |
| 34. | Reeves, J. T.; Song, J. J.; Tan, Z.; Lee, H.; Yee, N. K.; Senanayake, C. H. Org. Lett. 2007, 9, 1875–1878. doi:10.1021/ol070340q |
| 2. | Filippini, M.-H.; Rodriguez, J. Chem. Rev. 1999, 99, 27–76. doi:10.1021/cr970029u |
| 37. | Young, G. L.; Smith, S. A.; Taylor, R. J. K. Tetrahedron Lett. 2004, 45, 3797–3801. doi:10.1016/j.tetlet.2004.03.083 |
| 18. | Zhang, J.; Coqueron, P.-Y.; Vors, J.-P.; Ciufolini, M. A. Org. Lett. 2010, 12, 3942–3945. doi:10.1021/ol101704r |
| 19. | Zhang, J.; Ciufolini, M. A. Org. Lett. 2011, 13, 390–393. doi:10.1021/ol102678j |
| 20. | Zhang, J.; Ciufolini, M. A. Org. Lett. 2009, 11, 2389–2392. doi:10.1021/ol900455m |
| 21. | Cardwell, K. S.; Hermitage, S. A.; Sjolin, A. Tetrahedron Lett. 2000, 41, 4239–4242. doi:10.1016/S0040-4039(00)00573-6 |
| 22. | Swaminathan, S.; Singh, A. K.; Li, W.-S.; Venit, J. J.; Natalie, K. J., Jr.; Simpson, J. H.; Weaver, R. E.; Silverberg, L. J. Tetrahedron Lett. 1998, 39, 4769–4772. doi:10.1016/S0040-4039(98)00904-6 |
| 23. | Šagud, I.; Faraguna, F.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2011, 76, 2904–2908. doi:10.1021/jo1025942 |
| 24. | Webb, M. R.; Addie, M. S.; Crawforth, C. M.; Dale, J. W.; Franci, X.; Pizzonero, M.; Donald, C.; Taylor, R. J. K. Tetrahedron 2008, 64, 4778–4791. doi:10.1016/j.tet.2008.01.116 |
| 25. | Hoffman, T. J.; Rigby, J. H.; Arseniyadis, S.; Cossy, J. J. Org. Chem. 2008, 73, 2400–2403. doi:10.1021/jo702305g |
| 33. | Šindler-Kulyk, M.; Stiplošek, Z.; Vojnović, D.; Metelko, B.; Marinić, Ž. Heterocycles 1991, 32, 2357–2363. doi:10.3987/COM-91-5846 |
| 14. | Palmer, D. C. Oxazoles: Synthesis, Reactions, and Spectroscopy. In The Chemistry of heterocyclic compounds, Part A; Taylor, E. C.; Wipf, P., Eds.; John Wiley and Sons Inc.: New Jersey, 2003; pp 94–118. |
| 15. | Yeh, V. S. C. Tetrahedron 2004, 60, 11995–12042. doi:10.1016/j.tet.2004.10.001 |
| 16. | Jin, Z. Nat. Prod. Rep. 2011, 28, 1143–1191. doi:10.1039/c0np00074d |
| 17. | Jin, Z. Nat. Prod. Rep. 2013, 30, 869–915. doi:10.1039/c3np70006b |
| 34. | Reeves, J. T.; Song, J. J.; Tan, Z.; Lee, H.; Yee, N. K.; Senanayake, C. H. Org. Lett. 2007, 9, 1875–1878. doi:10.1021/ol070340q |
| 35. | Mulder, R. J.; Shafer, C. M.; Molinski, T. F. J. Org. Chem. 1999, 64, 4995–4998. doi:10.1021/jo9906328 |
| 36. | Addie, M. S.; Taylor, R. J. K. J. Chem. Soc., Perkin Trans. 1 2000, 527–531. doi:10.1039/a908987j |
| 11. | Butković, K.; Basarić, N.; Lovreković, K.; Marinić, Ž.; Višnjevac, A.; Kojić-Prodić, B.; Šindler-Kulyk, M. Tetrahedron Lett. 2004, 45, 9057–9060. doi:10.1016/j.tetlet.2004.10.036 |
| 12. | Butković, K.; Vuk, D.; Marinić, Ž.; Penić, J.; Šindler-Kulyk, M. Tetrahedron 2010, 66, 9356–9362. doi:10.1016/j.tet.2010.10.013 |
| 13. | Butković, K.; Marinić, Ž.; Molčanov, K.; Kojić-Prodić, B.; Šindler-Kulyk, M. Beilstein J. Org. Chem. 2011, 7, 1663–1670. doi:10.3762/bjoc.7.196 |
| 9. | Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2006, 71, 9382–9392. doi:10.1021/jo061435t |
| 10. | Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. J. Org. Chem. 2003, 68, 7524–7527. doi:10.1021/jo0346454 |
| 26. | Griesbeck, A. G.; Fiege, M.; Lex, J. J. Chem. Commun. 2000, 589–590. doi:10.1039/b000578i |
| 27. | Weuthen, M.; Scharf, H.-D.; Runsink, J. Chem. Ber. 1987, 120, 1023–1026. doi:10.1002/cber.19871200623 |
| 28. | Griesbeck, A. G.; Bondock, S. Can. J. Chem. 2003, 81, 555–559. doi:10.1139/v03-029 |
| 29. | Bondock, S.; Griesback, A. G. Monatsh. Chem. 2006, 137, 765–777. doi:10.1007/s00706-006-0474-4 |
| 30. | Griesbeck, A. G.; Franke, M.; Neudörfl, J.; Kotaka, H. Beilstein J. Org. Chem. 2011, 7, 127–134. doi:10.3762/bjoc.7.18 |
| 31. | Zhang, Y.; Wang, L.; Zhang, M.; Fun, H.-K.; Xu, J.-H. Org. Lett. 2004, 26, 4893–4895. doi:10.1021/ol048028t |
| 32. | Wang, L.; Huang, Y.-C.; Liu, Y.; Fun, H.-K.; Zhang, Y.; Xu, J.-H. J. Org. Chem. 2010, 75, 7757–7768. doi:10.1021/jo101764f |
| 6. | Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062 |
| 40. | Dale, W. J.; Starr, L.; Strobel, C. W. J. Org. Chem. 1961, 26, 2225–2227. doi:10.1021/jo01351a016 |
| 41. | Škorić, I.; Basarić, N.; Marinić, Ž.; Šindler-Kulyk, M. Heterocycles 2001, 55, 1889–1896. doi:10.3987/COM-01-9314 |
| 48. | Murakami, M.; Itahashi, T.; Ito, Y. J. Am. Chem. Soc. 2002, 124, 13976–13977. doi:10.1021/ja021062n |
| 49. | Hoffmann, N. Photochem. Photobiol. Sci. 2012, 11, 1613–1641. doi:10.1039/c2pp25074h |
| 43. | Lansbury, P. T.; Nienhouse, E. J. J. Am. Chem. Soc. 1966, 88, 4290–4291. doi:10.1021/ja00970a052 |
| 44. | Lansbury, P. T.; Nienhouse, E. J.; Scharf, D. J.; Hilfiker, F. R. J. Am. Chem. Soc. 1970, 92, 5649–5657. doi:10.1021/ja00722a018 |
| 45. | Wilt, J. W.; Rasmussen, R. R. J. Org. Chem. 1975, 40, 1031–1036. doi:10.1021/jo00896a007 |
| 46. | Kitahonoki, K.; Takano, Y.; Matsura, A.; Kotera, K. Tetrahedron 1969, 25, 335–353. doi:10.1016/S0040-4020(01)82628-4 |
| 47. | Balci, M.; Harmandar, M. Tetrahedron Lett. 1984, 25, 237–240. doi:10.1016/S0040-4039(00)99849-6 |
| 42. | Šindler-Kulyk, M.; Laarhoven, W. H. Recl. Trav. Chim. Pays-Bas 1979, 98, 187–191. doi:10.1002/recl.19790980410 |
| 6. | Vidaković, D.; Škorić, I.; Horvat, M.; Marinić, Ž.; Šindler-Kulyk, M. Tetrahedron 2008, 64, 3928–3934. doi:10.1016/j.tet.2008.02.062 |
© 2014 Šagud et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)