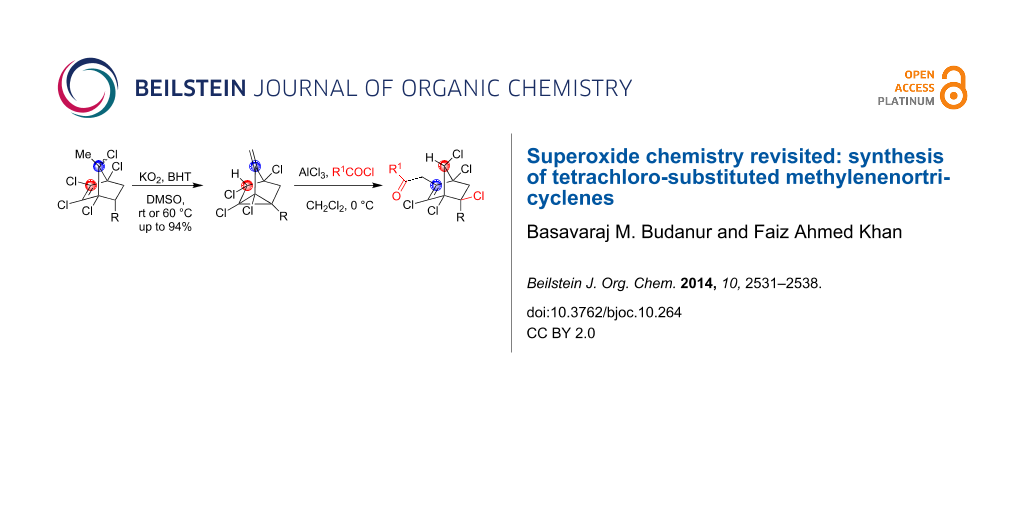
Abstract
An unexpected reactivity of the superoxide ion leading to the synthesis of tetrachloroaryl/vinyl-substituted nortricyclenes through its dual mode of action has been reported. KO2 was found to be superior and the only reagent to perform this kind of reaction over other conventional bases. Addition of the antioxidant BHT (2,6-di-tert-butyl-4-methylphenol) improved the yields of methylenenortricyclenes. A complete deuterium incorporation was observed in the superoxide-mediated reaction in DMSO-d6. Friedel–Crafts acylation reactions of 3-methylenenorticyclenes yielded 2-propanone-substituted pentachloronorbornenes.

Graphical Abstract
Introduction
The chemistry of the superoxide ion (O2−·) has been a subject of growing interest because of its presence in all aerobic organisms as a respiratory intermediate [1-6]. The study of the reactivity of the superoxide ion with organic substrates can facilitate understanding its role in metabolic processes [5]. Despite its biochemical importance, in organic synthesis, the superoxide ion has mainly been used for electron transfer reactions [7-9], oxidation [10-15], hydrolysis and substitution reactions [16-18]. Filippo and co-workers [19-22] have extensively studied the substitution reactions of alkyl halides and tosylates; oxidative cleavage reactions and hydrolysis of esters. Frimer and co-workers [23,24] have studied its reactivity with different functionalities. Jiang and co-workers [25] used KO2 as an alternative oxidation reagent in a Winterfeldt reaction instead of O2/KOt-Bu and many others reported reactions of superoxide which acts as oxidant, reductant, oxygen nucleophile, or Brønsted base. However, the full potential of the superoxide ion as a reagent in organic synthesis is still underexplored.
A very few and scattered reports appeared in the literature for the conversion of bicyclic norbornene to substituted nortricyclenes (Scheme 1). In 1966 Kempter and co-workers [26] have synthesized nortricyclenes from norcamphor using o-amino acetophenone hydrochloride. Ladenberger and co-worker [27] observed the rearrangement of a lithiated-tetrachloronorbornyl intermediate to a tricyclene derivative in a study about stability of vinyl carbanions. Li and co-workers [28,29] have synthesized aryl/benzyl substituted nortricyclenes using palladium and zinc powder from norbornadiene. The bromination of norbornene [30-32] is another route to the nortricyclenes. Here, in this study we report an efficient method for the synthesis of aryl- and vinyl-substituted tetrachloromethylenenortricyclenes from the Diels–Alder (DA) adducts of pentachloro-5-methylcyclopentadiene 1 and styrenes, by concurrent dehydrohalogenation and rearrangement reactions, starting from the commercially available inexpensive reagent potassium superoxide.
![[1860-5397-10-264-i1]](/bjoc/content/inline/1860-5397-10-264-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of Nortricyclenes from Norbornenes.
Scheme 1: Synthesis of Nortricyclenes from Norbornenes.
Results and Discussion
We have successfully utilized polyhalogenated bicyclic compounds for the synthesis of natural and aesthetically pleasing unnatural products. During our endeavour, we were selectively removing the halogens, utilizing them as transformation tools [33-36]. Our continued interest in this direction led us to explore the chemistry of alkylpentachlorocyclopentadienes as they have not been studied systematically even though discovered more than half a century ago. Alkylpentachlorocyclopentadienes can be easily prepared from hexachlorocyclopentadiene by treatment with the corresponding phosphite esters by a known protocol [37]. 5-Alkylpentachlorocyclopentadienes were rarely used as dienes in DA reactions; this may be because of their inverse-electron demand thus restricting them to DA reactions with electron-rich dienophiles. During our studies we noticed that pentachloro-5-methylcyclopentadiene (1) underwent a smooth DA reaction with styrenes (Table 1).
Table 1: Preparation of pentachloro-7-methyl substituted norbornenesa,b.
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-264-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | R | Product(s)a | Time(h) | Yield(%)c |
|---|---|---|---|---|
| 1 | Ph | 3a/4a | 6 | 83 |
| 2 | 4-OMeC6H4 | 3b/4b | 8 | 82 |
| 3 | 3,4,5-(OMe)3C6H2 | 3c/4c | 14 | 82 |
| 4 | 2,5-(OMe)2C6H3 | 3d/4d | 12 | 87 |
| 5d | 4-NO2C6H4 | 3e | 8 | 75 |
| 6 | 4-biphenyl | 3f/4f | 15 | 94 |
| 7 | 1-naphthyl | 3g/4g | 16 | 92 |
| 8 | 2-MeC6H4 | 3h/4h | 15 | 86 |
| 9e | 4-iPrC6H4 | 3i/4i | 23 | 89 |
| 10d | 4-Py | 3j | 15 | 77 |
| 11 | 2-BrC6H4 | 3k/4k | 30 | 77 |
| 12d | 2,4-(Cl)2C6H3 | 3l | 20 | 70 |
| 13 | -COMe | 3m/4m | 10 | 95 |
| 14d | CN | 3n | 17 | 92 |
| 15 | -(CH2)6- | 3o/4o | 15 | 81 |
| 16 | α-Me-vinyl | 3pf | – | 83 |
aUnless otherwise mentioned anti:syn ratio was ~4:1 and was calculated based on 1H NMR of the crude product; bdienophiles were prepared according to the known procedures [38-40]; cisolated yields; donly anti (3) isomers were isolated in these cases; eanti:syn ratio was 93:7; f3p was prepared by Wittig reaction on 3m.
We then prepared the series of DA adducts 3/4 from pentachloro-5-methylcyclopentadiene 1 and corresponding dienophiles 2 [38-40] by heating at 120–130 °C in a sealed tube. Styrene derivatives containing groups like methoxy, alkyl, naphthyl, and biphenyl (Table 1, entries 1–4, 6–9 ), methyl vinyl ketone (Table 1, entry 13) and cyclooctene (Table 1, entry 15) have furnished DA adducts in excellent yields and good diastereoselectivity ~4:1 (anti:syn) in 6–15 h. Some dienophiles such as p-nitrostyrene (Table 1, entry 5), 4-vinylpyridine (Table 1, entry 10), chlorinated styrene (Table 1, entry 12) and acrylonitrile (Table 1, entry 14) gave corresponding adducts in good yield and single anti-diastereomer only. The stereostructures of the DA adducts 3 and 4 were deduced from the literature established [41] 1H NMR pattern of the downfield shift (δ = ~1.8) for the syn methyl protons in the pentachloro DA adducts 4 and upfield shift (δ = ~1.6) for the anti-methyl protons in the pentachloro DA adducts 3.
Having adduct 3a in hand, we turned our attention to a dehydrochlorination to create a double bond at the C-7 position. Unfortunately the use of conventional bases like KOt-Bu, DBU, 2,6-lutidine, NaOH and NaOMe did not yield the desired product even at elevated temperatures. Then we looked for a hydrodechlorination at the C-7 position and found from the literature that zinc/AcOH [42,43] can be used for the reduction of chlorine at the C-7 position of a vinylacetate DA adduct of hexachlorocyclopentadiene, but only 6% of the bis-reduced product was obtained. We have slightly modified these conditions using an excess of zinc on substrate 3a, but failed to get the reduced product.
A literature search also revealed that potassium superoxide has been used for selective monodehalogenation and elimination reactions in polyhalogenated compounds [44,45]. Treatment of adduct 3a with potassium superoxide at 60 °C under argon atmosphere gave a colourless compound. Initially, we expected it to be the elimination product because of the two olefinic peaks in the 1H NMR spectrum but the presence of a singlet at δ 4.2 and only two olefinic peaks in the 13C NMR are not in accordance with our expectations. The single crystal X-ray analysis of the product (Figure 1) sorted out all the ambiguity associated with structure. To our pleasant surprise the structure of the product was methylenenortricyclene 5a which seems to have been formed from a concurrent elimination as well as a rearrangement reaction.
![[1860-5397-10-264-1]](/bjoc/content/figures/1860-5397-10-264-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystal structure of 5a with 30% thermal ellipsoids.
Figure 1: X-ray crystal structure of 5a with 30% thermal ellipsoids.
In superoxide chemistry many aprotic solvents have been used. We prefer DMSO because the solubility of potassium superoxide is better in DMSO compared to other solvents [16]. As the phase-transfer catalyst can enhance the extent of solubility of the KO2 [16], we tried to use 18-crown-6 and to change the temperature to effect the rate of the reaction. We did not notice any significant difference in the rate under these conditions.
To study the scope and limitations and to probe the electronic and steric effects of this reaction, substituted styrene adducts were subjected to the reaction with KO2 (3 equiv) in DMSO and the results are summarized in Scheme 2. The substrates endowed with electron donating groups, for example anti-adduct 3b gave methylenenortricyclene 5b, whereas its syn-isomer 4b, after consumption of all the starting material in 3 h furnished eliminated product 6a, along with 5b. Further treatment of the inseparable mixture of 5b/6a with KO2 afforded exclusively nortricyclene 5b in 80% overall yield from 4b. The 3,4,5-trimethoxy-substituted DA adducts 3c/4c gave title product 5c at rt. The o-methoxy-substituted adducts 3d/4d gave nortricyclene 5d in good yield (78%). The substrate bearing an NO2 group on the phenyl ring 3e (Table 1, entry 5) was completely unreactive even with excess of KO2 at up to 100 °C. The formation of a nitrobenzyl radical anion [9,22] by one-electron transfer might be the reason for this.
![[1860-5397-10-264-i2]](/bjoc/content/inline/1860-5397-10-264-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: KO2-mediated synthesis of tetrachloro-substituted 3-methylenenortricyclenes. Reaction conditions: All the substrates are a mixture of syn and anti with KO2 (3 equiv)/DMSO at 60 °C, isolated yields are given. aReaction performed at rt, bsyn-isomers 4b and 4h gave mixtures of 5 and 6. cReaction performed at 100 °C.
Scheme 2: KO2-mediated synthesis of tetrachloro-substituted 3-methylenenortricyclenes. Reaction conditions: A...
Biphenyl-substituted adducts 3f/4f yielded nortricyclene derivative 5f smoothly at rt. But naphthyl-substituted adducts 3g/4g furnished the title product 5g at 60 °C, probably due to steric effects (Scheme 2). The other substrates having alkyl substituents on the phenyl ring 3h, 3i/4i afforded the corresponding nortricyclenes in good yields, whereas syn-isomer 4h yielded an inseparable mixture of 5h and 6b. We have examined the influence of heteroarene substitution towards this reaction using adduct 3j which afforded nortricyclene 5i in 60% and rearranged product 7 in 16% yield at rt after 30 min. Adducts 3k/4k containing bromine at ortho position, a relatively bulky substituent close to the reactive site, took a little longer time to afford the nortricyclene 5k. The 2,4-dichloro-substituted adduct 3l gave a good yield of the desired product 5m (78%) (Scheme 2).
In order to explore this unexpected reaction with respect to substrates containing other functional groups which are known to stabilize the anion were subjected with KO2 (Table 1); the acetyl- and cyano-substituted adducts 3m/4m and 3n (Table 1, entries 13, 14) did not yield any product and resulted in substantial loss of starting material. The olefin-substituted compound 3p (Table 1, entry 16) underwent a smooth reaction to afford nortricyclene 5m (Scheme 2). The cyclooctane-fused adducts 3o/4o (Table 1, entry 15) afforded exclusively the elimination product 6c in excellent yield.
As the pKa of the conjugate acid of the superoxide ion, the hydroperoxyl radical is 4.88 [46] in water which is similar to that of acetic acid; it implies that the superoxide ion is a weak base. Although basicity of the anion is higher in nonprotic solvents than in protic solvents [18], KOAc can be a surrogate to KO2. Treating adduct 3a with KOAc in DMSO did not yield any product which suggested that there is a unique and exceptional degree of reactivity of the superoxide ion species which triggered this kind of reaction.
To elucidate the reaction mechanism, we have performed a deuterium labelling experiment by treating 3c with KO2 in DMSO-d6 at rt. Absence of the peak at δ 4.26 (singlet) in 1H NMR suggest the incorporation of deuterium and a triplet at δ 71.0, arising from C–D coupling, instead of sharp peak at δ 71.3 in 13C NMR further confirmed the deuteration at the chlorine-attached carbon atom in 5n (Scheme 3). Further, in order to understand the mechanistic aspects of the reaction we have treated 3f/4f under standard conditions by using the additive radical quencher and antioxidant BHT (5 mol %) afforded the product 5f in improved yield of 94% (Scheme 3). Similarly, substrates 3c/4c and 3i/4i afforded 5c and 5i in 91% and 89% yield, respectively. This observation provides added evidence that the mechanism involved might be ionic.
![[1860-5397-10-264-i3]](/bjoc/content/inline/1860-5397-10-264-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on known chemistry in the literature, initially we have assumed two different possible pathways for the formation of product 6: The nucleophilic substitution of the superoxide ion followed by elimination (path A) or the direct dehydrohalogenation (path B) [47] (Scheme 4). We ruled out path A based on our observation that even after adding BHT, the reaction underwent smoothly. The second step involved the deprotonation of a benzylic proton by the superoxide ion [48]. The resulting benzylic anion I undergoes an unexpected rearrangement by adding onto the double bond possessing the vicinal chlorines [8] and the resulting carbanion is protonated by the solvent DMSO to afford the nortricyclene 5.
![[1860-5397-10-264-i4]](/bjoc/content/inline/1860-5397-10-264-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Plausible mechanism of the KO2-mediated reaction.
Scheme 4: Plausible mechanism of the KO2-mediated reaction.
In the next part of our study, we have investigated the acylation reactions as it was reported that acylation reactions of parent nortricyclenes [49] proceed through initial formation of chloroacetylated norbornane which was rapidly dehydrohalogenated upon storage at rt to give 1-acetylnortricyclene. We have applied the standard acylation reaction conditions (AlCl3, R1COCl, 1:1) to 3-methylenenortricyclenes to observe acylation on the double bond and not on the cyclopropane ring to give 2-propanone-substituted pentachloronorbornene derivatives 8 (Table 2). The structure of 8a was unambiguously proved by single crystal X-ray analysis (Figure 2).
Table 2: Acylation reaction of 3-methylenenortricyclenes.
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-264-i7.svg?max-width=637&scale=1.0)
|
||
| R | R1 | product/yield(%)a |
|---|---|---|
| Ph, 5a | Me | 8a/76 |
| Ph, 5a | Ph | 8b/61 |
| BiPh, 5f | Me | 8c/68b |
aIsolated yields; bacetylation also occurred at the 4'-positon of the biphenyl ring.
![[1860-5397-10-264-2]](/bjoc/content/figures/1860-5397-10-264-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of 8a with 30% thermal ellipsoids.
Figure 2: X-ray crystal structure of 8a with 30% thermal ellipsoids.
A plausible mechanism involving the initial nucleophilic attack of the exocyclic olefin on the acylium ion with concomitant cyclopropane ring opening leading to benzylic cation II followed by its combination with chloride ion leading to 8 is proposed (Scheme 5).
![[1860-5397-10-264-i5]](/bjoc/content/inline/1860-5397-10-264-i5.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism of the acylation reaction of 3-methylenenortricyclenes.
Scheme 5: Plausible mechanism of the acylation reaction of 3-methylenenortricyclenes.
Conclusion
In summary, we have demonstrated the preparation of a variety of Diels–Alder adducts pentachloro-7-methyl-5-aryl/vinyl-substituted norbornenes and utilized them in the synthesis of tetrachloro aryl/vinyl-substituted methylenenortricyclenes efficiently with good to excellent yields using inexpensive and commercially available potassium superoxide. The reaction mechanism of the KO2-mediated reaction was derived through a controlled experiment with BHT which improved the yields of nortricyclenes and a deuterium labelling experiment using DMSO-d6. Further, acylation reactions of the synthesized methylenenortricyclens afforded the 2-propanone-substituted pentachloronorbornenes in good yields. The studies towards further synthetic applications of KO2 are currently underway in our lab.
Supporting Information
Experimental procedures and analytical data, including copies of 1H and 13C NMR spectra for all new compounds. Crystallographic data for structures 5a and 8a have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC 1021351 and 1021303. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.ukor via: http://www.ccdc.cam.ac.uk).
| Supporting Information File 1: Experimental part and NMR spectra. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Knowles, P. F.; Gibson, J. F.; Pick, F. M.; Bray, R. C. Biochem. J. 1969, 111, 53.
Return to citation in text: [1] -
Fridovich, I. Acc. Chem. Res. 1972, 5, 321. doi:10.1021/ar50058a001
Return to citation in text: [1] -
Nanni, E. J., Jr.; Stallings, M. D.; Sawyer, D. T. J. Am. Chem. Soc. 1980, 102, 4481. doi:10.1021/ja00533a029
Return to citation in text: [1] -
Sawyer, D. T.; Valentine, J. S. Acc. Chem. Res. 1981, 14, 393. doi:10.1021/ar00072a005
Return to citation in text: [1] -
Lee-Ruff, E. Chem. Soc. Rev. 1977, 6, 195. doi:10.1039/CS9770600195
Return to citation in text: [1] [2] -
Gibian, M. J.; Sawyer, D. T.; Ungermann, T.; Tangpoonpholvivat, R.; Morrison, M. M. J. Am. Chem. Soc. 1979, 101, 640. doi:10.1021/ja00497a026
Return to citation in text: [1] -
Peters, J. W.; Foote, C. S. J. Am. Chem. Soc. 1976, 98, 873. doi:10.1021/ja00419a058
Return to citation in text: [1] -
Lee-Ruff, E.; Lever, A. B. P.; Rigaudy, J. Can. J. Chem. 1976, 54, 1837.
Return to citation in text: [1] [2] -
Poupko, R.; Rosenthal, I. J. Phys. Chem. 1973, 77, 1722. doi:10.1021/j100632a027
Return to citation in text: [1] [2] -
Moro-oka, Y.; Foote, C. S. J. Am. Chem. Soc. 1976, 98, 1510. doi:10.1021/ja00422a038
Return to citation in text: [1] -
Neckers, D. C.; Hauck, G. J. Org. Chem. 1983, 48, 4691. doi:10.1021/jo00172a046
Return to citation in text: [1] -
Frimer, A. A.; Farkash-Solomon, T.; Aljadeff, G. J. Org. Chem. 1986, 51, 2093. doi:10.1021/jo00361a030
Return to citation in text: [1] -
Frimer, A. A.; Gilinsky-Sharon, P.; Aljadeff, G.; Gottlieb, H. E.; Hameiri-Buch, J.; Marks, V.; Philosof, R.; Rosental, Z. J. Org. Chem. 1989, 54, 4853. doi:10.1021/jo00281a030
Return to citation in text: [1] -
Frimer, A. A.; Gilinsky-Sharon, P. J. Org. Chem. 1995, 60, 2796. doi:10.1021/jo00114a031
Return to citation in text: [1] -
Chen, Y.-J.; Huang, Y.-P. Tetrahedron Lett. 2000, 41, 5233. doi:10.1016/S0040-4039(00)00828-5
Return to citation in text: [1] -
Corey, E. J.; Nicolaou, K. C.; Shibasaki, M.; Machida, Y.; Shiner, C. S. Tetrahedron Lett. 1975, 16, 3183. doi:10.1016/S0040-4039(00)91450-3
Return to citation in text: [1] [2] [3] -
Fillippo, J. S., Jr.; Chern, C.-I.; Valentine, J. S. J. Org. Chem. 1975, 40, 1678. doi:10.1021/jo00899a048
Return to citation in text: [1] -
Chern, C.-I.; DiCosimo, R.; De Jesus, R.; Filippo, J. S., Jr. J. Am. Chem. Soc. 1978, 100, 7317. doi:10.1021/ja00491a032
Return to citation in text: [1] [2] -
Filippo, J. S., Jr.; Romano, L. J.; Chern, C.-I.; Valentine, J. S. J. Org. Chem. 1976, 41, 586. doi:10.1021/jo00865a050
Return to citation in text: [1] -
Filippo, J. S., Jr.; Chern, C.-I.; Valentine, J. S. J. Org. Chem. 1976, 41, 1077. doi:10.1021/jo00868a037
Return to citation in text: [1] -
Rosenthal, I.; Frimer, A. Tetrahedron Lett. 1976, 17, 2805. doi:10.1016/S0040-4039(00)93844-9
Return to citation in text: [1] -
Kornblum, N.; Singaram, S. J. Org. Chem. 1979, 44, 4727. doi:10.1021/jo00393a063
Return to citation in text: [1] [2] -
Frimer, A. A.; Rosenthal, I.; Hoz, S. Tetrahedron Lett. 1977, 18, 4631. doi:10.1016/S0040-4039(01)83587-5
Return to citation in text: [1] -
Rosenthal, I.; Frimer, A. Tetrahedron Lett. 1975, 16, 3731. doi:10.1016/S0040-4039(00)91321-2
Return to citation in text: [1] -
Jiang, W.; Zhang, X.; Sui, Z. Org. Lett. 2003, 5, 43. doi:10.1021/ol0271279
Return to citation in text: [1] -
Kempter, G.; Mühlstädt, M.; Heilmann, D.-C. D. Angew. Chem., Int. Ed. Engl. 1966, 5, 248. doi:10.1002/anie.196602481
Return to citation in text: [1] -
Schlosser, M.; Ladenberger, V. Angew. Chem., Int. Ed. Engl. 1966, 5, 519. doi:10.1002/anie.196605191
Return to citation in text: [1] -
Li, C.-S.; Cheng, C.-H.; Cheng, S.-S.; Shaw, J.-S. J. Chem. Soc., Chem. Commun. 1990, 1774. doi:10.1039/C39900001774
Return to citation in text: [1] -
Li, C.-S.; Jou, D.-C.; Cheng, C.-H. Organometallics 1993, 12, 3945. doi:10.1021/om00034a029
Return to citation in text: [1] -
Roberts, J. D.; Trumbull, E. R., Jr.; Bennett, W.; Armstrong, R. J. Am. Chem. Soc. 1950, 72, 3116. doi:10.1021/ja01163a086
Return to citation in text: [1] -
Ver Nooy, C. D.; Rondestvedt, C. S., Jr. J. Am. Chem. Soc. 1955, 77, 3583. doi:10.1021/ja01618a048
Return to citation in text: [1] -
Schmerling, L.; Luvisi, J. P.; Welch, R. W. J. Am. Chem. Soc. 1956, 78, 2819. doi:10.1021/ja01593a047
Return to citation in text: [1] -
Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558. doi:10.1021/ja001956c
Return to citation in text: [1] -
Khan, F. A.; Dash, J. J. Org. Chem. 2003, 68, 4556. doi:10.1021/jo034023i
Return to citation in text: [1] -
Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424. doi:10.1021/ja017371f
Return to citation in text: [1] -
Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, C. J. Org. Chem. 2002, 67, 3783. doi:10.1021/jo025521e
Return to citation in text: [1] -
Mark, V.; Wann, R. E.; Godt, H. C., Jr. Org. Synth. 1963, 43, 90. doi:10.15227/orgsyn.043.0090
Return to citation in text: [1] -
Cho, S. J.; Jensen, N. H.; Kurome, T.; Kadari, S.; Manzano, M. L.; Malberg, J. E.; Caldarone, B.; Roth, B. L.; Kozikowski, A. P. J. Med. Chem. 2009, 52, 1885. doi:10.1021/jm801354e
Return to citation in text: [1] [2] -
Khan, Z. A.; Iwaoka, M.; Wirth, T. Tetrahedron 2010, 66, 6639. doi:10.1016/j.tet.2010.03.062
Return to citation in text: [1] [2] -
Okuma, K.; Sakai, O.; Shioji, K. Bull. Chem. Soc. Jpn. 2003, 76, 1675. doi:10.1246/bcsj.76.1675
Return to citation in text: [1] [2] -
Wellman, M. A.; Burry, L. C.; Letourneau, J. E.; Bridson, J. N.; Miller, D. O.; Burnell, D. J. J. Org. Chem. 1997, 62, 939. doi:10.1021/jo961710j
Return to citation in text: [1] -
Williamson, K. L.; Li Hsu, Y.-F.; Young, E. I. Tetrahedron 1968, 24, 6007. doi:10.1016/S0040-4020(01)90984-6
Return to citation in text: [1] -
Williamson, K. L.; Li Hsu, Y.-F.; Lacko, R.; He Youn, C. J. Am. Chem. Soc. 1969, 91, 6129. doi:10.1021/ja01050a035
Return to citation in text: [1] -
Dureja, P.; Walia, S.; Mukerjee, S. K. J. Agric. Food Chem. 1984, 32, 1217. doi:10.1021/jf00126a003
Return to citation in text: [1] -
Dureja, P.; Caslda, J. E.; Ruzo, L. O. Tetrahedron Lett. 1982, 23, 5003. doi:10.1016/S0040-4039(00)85557-4
Return to citation in text: [1] -
Behar, D.; Czapski, G.; Rabani, J.; Dorfman, L. M.; Schwarz, H. A. J. Phys. Chem. 1970, 74, 3209. doi:10.1021/j100711a009
Return to citation in text: [1] -
Afanas’ev, I. B. Superoxide Ion: Chemistry and Biological Implications; CRC Press: Boca Raton, FL, 1989; Vol. 1.
And references cited therein.
Return to citation in text: [1] -
Tezuka, M.; Ohkatsu, Y.; Osa, T. Bull. Chem. Soc. Jpn. 1975, 48, 1471. doi:10.1246/bcsj.48.1471
Return to citation in text: [1] -
Hart, H.; Martin, R. A. J. Org. Chem. 1959, 24, 1267. doi:10.1021/jo01091a027
Return to citation in text: [1]
| 49. | Hart, H.; Martin, R. A. J. Org. Chem. 1959, 24, 1267. doi:10.1021/jo01091a027 |
| 1. | Knowles, P. F.; Gibson, J. F.; Pick, F. M.; Bray, R. C. Biochem. J. 1969, 111, 53. |
| 2. | Fridovich, I. Acc. Chem. Res. 1972, 5, 321. doi:10.1021/ar50058a001 |
| 3. | Nanni, E. J., Jr.; Stallings, M. D.; Sawyer, D. T. J. Am. Chem. Soc. 1980, 102, 4481. doi:10.1021/ja00533a029 |
| 4. | Sawyer, D. T.; Valentine, J. S. Acc. Chem. Res. 1981, 14, 393. doi:10.1021/ar00072a005 |
| 5. | Lee-Ruff, E. Chem. Soc. Rev. 1977, 6, 195. doi:10.1039/CS9770600195 |
| 6. | Gibian, M. J.; Sawyer, D. T.; Ungermann, T.; Tangpoonpholvivat, R.; Morrison, M. M. J. Am. Chem. Soc. 1979, 101, 640. doi:10.1021/ja00497a026 |
| 16. | Corey, E. J.; Nicolaou, K. C.; Shibasaki, M.; Machida, Y.; Shiner, C. S. Tetrahedron Lett. 1975, 16, 3183. doi:10.1016/S0040-4039(00)91450-3 |
| 17. | Fillippo, J. S., Jr.; Chern, C.-I.; Valentine, J. S. J. Org. Chem. 1975, 40, 1678. doi:10.1021/jo00899a048 |
| 18. | Chern, C.-I.; DiCosimo, R.; De Jesus, R.; Filippo, J. S., Jr. J. Am. Chem. Soc. 1978, 100, 7317. doi:10.1021/ja00491a032 |
| 38. | Cho, S. J.; Jensen, N. H.; Kurome, T.; Kadari, S.; Manzano, M. L.; Malberg, J. E.; Caldarone, B.; Roth, B. L.; Kozikowski, A. P. J. Med. Chem. 2009, 52, 1885. doi:10.1021/jm801354e |
| 39. | Khan, Z. A.; Iwaoka, M.; Wirth, T. Tetrahedron 2010, 66, 6639. doi:10.1016/j.tet.2010.03.062 |
| 40. | Okuma, K.; Sakai, O.; Shioji, K. Bull. Chem. Soc. Jpn. 2003, 76, 1675. doi:10.1246/bcsj.76.1675 |
| 10. | Moro-oka, Y.; Foote, C. S. J. Am. Chem. Soc. 1976, 98, 1510. doi:10.1021/ja00422a038 |
| 11. | Neckers, D. C.; Hauck, G. J. Org. Chem. 1983, 48, 4691. doi:10.1021/jo00172a046 |
| 12. | Frimer, A. A.; Farkash-Solomon, T.; Aljadeff, G. J. Org. Chem. 1986, 51, 2093. doi:10.1021/jo00361a030 |
| 13. | Frimer, A. A.; Gilinsky-Sharon, P.; Aljadeff, G.; Gottlieb, H. E.; Hameiri-Buch, J.; Marks, V.; Philosof, R.; Rosental, Z. J. Org. Chem. 1989, 54, 4853. doi:10.1021/jo00281a030 |
| 14. | Frimer, A. A.; Gilinsky-Sharon, P. J. Org. Chem. 1995, 60, 2796. doi:10.1021/jo00114a031 |
| 15. | Chen, Y.-J.; Huang, Y.-P. Tetrahedron Lett. 2000, 41, 5233. doi:10.1016/S0040-4039(00)00828-5 |
| 38. | Cho, S. J.; Jensen, N. H.; Kurome, T.; Kadari, S.; Manzano, M. L.; Malberg, J. E.; Caldarone, B.; Roth, B. L.; Kozikowski, A. P. J. Med. Chem. 2009, 52, 1885. doi:10.1021/jm801354e |
| 39. | Khan, Z. A.; Iwaoka, M.; Wirth, T. Tetrahedron 2010, 66, 6639. doi:10.1016/j.tet.2010.03.062 |
| 40. | Okuma, K.; Sakai, O.; Shioji, K. Bull. Chem. Soc. Jpn. 2003, 76, 1675. doi:10.1246/bcsj.76.1675 |
| 7. | Peters, J. W.; Foote, C. S. J. Am. Chem. Soc. 1976, 98, 873. doi:10.1021/ja00419a058 |
| 8. | Lee-Ruff, E.; Lever, A. B. P.; Rigaudy, J. Can. J. Chem. 1976, 54, 1837. |
| 9. | Poupko, R.; Rosenthal, I. J. Phys. Chem. 1973, 77, 1722. doi:10.1021/j100632a027 |
| 33. | Khan, F. A.; Prabhudas, B.; Dash, J.; Sahu, N. J. Am. Chem. Soc. 2000, 122, 9558. doi:10.1021/ja001956c |
| 34. | Khan, F. A.; Dash, J. J. Org. Chem. 2003, 68, 4556. doi:10.1021/jo034023i |
| 35. | Khan, F. A.; Dash, J. J. Am. Chem. Soc. 2002, 124, 2424. doi:10.1021/ja017371f |
| 36. | Khan, F. A.; Dash, J.; Sahu, N.; Sudheer, C. J. Org. Chem. 2002, 67, 3783. doi:10.1021/jo025521e |
| 37. | Mark, V.; Wann, R. E.; Godt, H. C., Jr. Org. Synth. 1963, 43, 90. doi:10.15227/orgsyn.043.0090 |
| 26. | Kempter, G.; Mühlstädt, M.; Heilmann, D.-C. D. Angew. Chem., Int. Ed. Engl. 1966, 5, 248. doi:10.1002/anie.196602481 |
| 28. | Li, C.-S.; Cheng, C.-H.; Cheng, S.-S.; Shaw, J.-S. J. Chem. Soc., Chem. Commun. 1990, 1774. doi:10.1039/C39900001774 |
| 29. | Li, C.-S.; Jou, D.-C.; Cheng, C.-H. Organometallics 1993, 12, 3945. doi:10.1021/om00034a029 |
| 30. | Roberts, J. D.; Trumbull, E. R., Jr.; Bennett, W.; Armstrong, R. J. Am. Chem. Soc. 1950, 72, 3116. doi:10.1021/ja01163a086 |
| 31. | Ver Nooy, C. D.; Rondestvedt, C. S., Jr. J. Am. Chem. Soc. 1955, 77, 3583. doi:10.1021/ja01618a048 |
| 32. | Schmerling, L.; Luvisi, J. P.; Welch, R. W. J. Am. Chem. Soc. 1956, 78, 2819. doi:10.1021/ja01593a047 |
| 23. | Frimer, A. A.; Rosenthal, I.; Hoz, S. Tetrahedron Lett. 1977, 18, 4631. doi:10.1016/S0040-4039(01)83587-5 |
| 24. | Rosenthal, I.; Frimer, A. Tetrahedron Lett. 1975, 16, 3731. doi:10.1016/S0040-4039(00)91321-2 |
| 19. | Filippo, J. S., Jr.; Romano, L. J.; Chern, C.-I.; Valentine, J. S. J. Org. Chem. 1976, 41, 586. doi:10.1021/jo00865a050 |
| 20. | Filippo, J. S., Jr.; Chern, C.-I.; Valentine, J. S. J. Org. Chem. 1976, 41, 1077. doi:10.1021/jo00868a037 |
| 21. | Rosenthal, I.; Frimer, A. Tetrahedron Lett. 1976, 17, 2805. doi:10.1016/S0040-4039(00)93844-9 |
| 22. | Kornblum, N.; Singaram, S. J. Org. Chem. 1979, 44, 4727. doi:10.1021/jo00393a063 |
| 27. | Schlosser, M.; Ladenberger, V. Angew. Chem., Int. Ed. Engl. 1966, 5, 519. doi:10.1002/anie.196605191 |
| 44. | Dureja, P.; Walia, S.; Mukerjee, S. K. J. Agric. Food Chem. 1984, 32, 1217. doi:10.1021/jf00126a003 |
| 45. | Dureja, P.; Caslda, J. E.; Ruzo, L. O. Tetrahedron Lett. 1982, 23, 5003. doi:10.1016/S0040-4039(00)85557-4 |
| 41. | Wellman, M. A.; Burry, L. C.; Letourneau, J. E.; Bridson, J. N.; Miller, D. O.; Burnell, D. J. J. Org. Chem. 1997, 62, 939. doi:10.1021/jo961710j |
| 42. | Williamson, K. L.; Li Hsu, Y.-F.; Young, E. I. Tetrahedron 1968, 24, 6007. doi:10.1016/S0040-4020(01)90984-6 |
| 43. | Williamson, K. L.; Li Hsu, Y.-F.; Lacko, R.; He Youn, C. J. Am. Chem. Soc. 1969, 91, 6129. doi:10.1021/ja01050a035 |
| 48. | Tezuka, M.; Ohkatsu, Y.; Osa, T. Bull. Chem. Soc. Jpn. 1975, 48, 1471. doi:10.1246/bcsj.48.1471 |
| 18. | Chern, C.-I.; DiCosimo, R.; De Jesus, R.; Filippo, J. S., Jr. J. Am. Chem. Soc. 1978, 100, 7317. doi:10.1021/ja00491a032 |
| 47. |
Afanas’ev, I. B. Superoxide Ion: Chemistry and Biological Implications; CRC Press: Boca Raton, FL, 1989; Vol. 1.
And references cited therein. |
| 9. | Poupko, R.; Rosenthal, I. J. Phys. Chem. 1973, 77, 1722. doi:10.1021/j100632a027 |
| 22. | Kornblum, N.; Singaram, S. J. Org. Chem. 1979, 44, 4727. doi:10.1021/jo00393a063 |
| 46. | Behar, D.; Czapski, G.; Rabani, J.; Dorfman, L. M.; Schwarz, H. A. J. Phys. Chem. 1970, 74, 3209. doi:10.1021/j100711a009 |
| 16. | Corey, E. J.; Nicolaou, K. C.; Shibasaki, M.; Machida, Y.; Shiner, C. S. Tetrahedron Lett. 1975, 16, 3183. doi:10.1016/S0040-4039(00)91450-3 |
| 16. | Corey, E. J.; Nicolaou, K. C.; Shibasaki, M.; Machida, Y.; Shiner, C. S. Tetrahedron Lett. 1975, 16, 3183. doi:10.1016/S0040-4039(00)91450-3 |
© 2014 Budanur and Khan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)