Abstract
In a previous study it was shown that the enantioselective α-fluorination of racemic α-chloroaldehydes with a chiral organocatalyst yielded the corresponding α-chloro-α-fluoroaldehydes with high enantioselectivity. It was also revealed that kinetic resolution of the starting aldehydes was involved in this asymmetric fluorination. This paper describes the determination of the absolute stereochemistry of a resulting α-chloro-α-fluoroaldehyde. Some information about the substrate scope and a possible reaction mechanism are also described which shed more light on the nature of this asymmetric fluorination reaction.

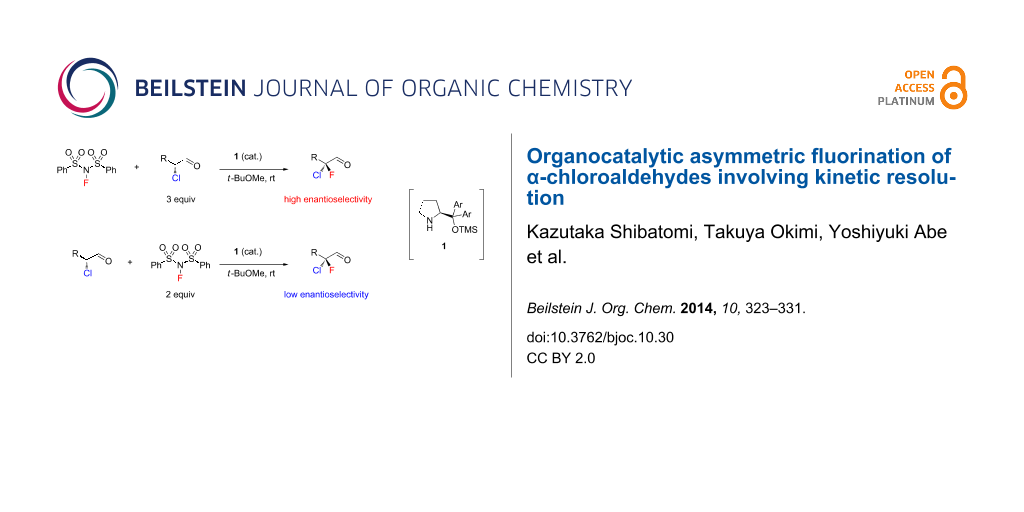
Graphical Abstract
Introduction
Fluorinated organic molecules are of considerable interest in pharmaceutical and agricultural chemistry owing to the unique properties of the fluorine atom [1,2]. These compounds, especially with one or more fluorinated stereogenic center(s), are fascinating building blocks for new drug candidates. Organocatalytic α-fluorination of aldehydes is known to be an efficient strategy for the enantioselective construction of fluorinated chiral carbon centers [3-6]; however, very few successful studies have been published on the fluorination of α-branched aldehydes [7]. During the course of our study on the enantioselective construction of such fluorinated stereogenic centers, we developed a method for the enantioselective synthesis of α-chloro-α-fluoroaldehydes via the organocatalytic α-fluorination of α-alkyl-α-chloroaldehydes, a type of α-branched aldehyde, mediated by the Jørgensen–Hayashi catalyst 1 [8]. The reaction yielded the desired α-chloro-α-fluoroaldehydes with high enantioselectivity when the starting aldehyde was used in excess over N-fluorobenzenesulfonimide (NFSI) in the reaction. However, when an excess NFSI with respect to the starting aldehyde was used, poor asymmetric induction was observed. In this paper, we describe the determination of the absolute stereochemistry of a resulting α-chloro-α-fluoroaldehyde using this methodology and discuss the possible reaction mechanism that involves kinetic resolution.
Results and Discussion
In our previous study [8], enantioselective fluorination of racemic 2-chloro-3-phenylpropanal (2a) was carried out with 3 equiv of NFSI in the presence of organocatalyst (S)-1 to yield the corresponding α-chloro-α-fluoroaldehyde 3a in good conversion. Isolation of the product and determination of enantiomeric purity were performed after reduction to primary alcohol 4a because 3a was unstable to silica gel chromatography. The reaction afforded 4a with high enantioselectivity along with the monochloro alcohol 5a, whose enantiomeric purity was determined to be 37% ee (Scheme 1) [8]. These results suggested that kinetic resolution of the starting aldehydes was involved in this asymmetric fluorination.
![[1860-5397-10-30-i1]](/bjoc/content/inline/1860-5397-10-30-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Organocatalytic enantioselective fluorination of α-chloroaldehyde 2a [8].
Scheme 1: Organocatalytic enantioselective fluorination of α-chloroaldehyde 2a [8].
To collect further information on the reaction mechanism, we sought to determine the absolute configuration of 4a. Recently, we reported the enantioselective synthesis of α-chloro-α-fluoro-β-keto esters via the sequential chlorination–fluorination of β-keto esters with the Cu(II) complex of SPYMOX [9], a spiro chiral oxazoline ligand developed by our research group [9-12]. In that study, we succeeded in determining the absolute stereochemistry of the α-chloro-α-fluoro-β-keto ester 6 by the X-ray crystallographic analysis of its derivative 7 (Scheme 2). Here, our aim was to transform chlorofluoro ester 6 to 4a in order to compare its optical rotation with that of 4a derived from 2a in the presence of catalyst (S)-1. As shown in Scheme 3, β-keto ester 6 was converted via the Barton–McCombie deoxygenation [13] into a simple ester 10, which was then reduced to the primary alcohol 4a by treatment with LiAlH4. Comparison of the optical rotations and retention times on chiral HPLC clearly showed that the asymmetric fluorination of 2a catalyzed by (S)-1 yielded 4a having the R configuration (Scheme 1).
![[1860-5397-10-30-i2]](/bjoc/content/inline/1860-5397-10-30-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Determination of absolute configuration of α-chloro-α-fluoro-β-keto ester 6 by X-ray analysis [9].
Scheme 2: Determination of absolute configuration of α-chloro-α-fluoro-β-keto ester 6 by X-ray analysis [9].
![[1860-5397-10-30-i3]](/bjoc/content/inline/1860-5397-10-30-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Transformation of α-chloro-α-fluoro-β-keto ester 6 to chlorofluoro alcohol 4a.
Scheme 3: Transformation of α-chloro-α-fluoro-β-keto ester 6 to chlorofluoro alcohol 4a.
An investigation of the substrate scope of the organocatalytic fluorination of α-chloroaldehydes was performed as shown in Table 1. The reaction of 2a with 3 equiv of NFSI yielded 4a in 87% ee along with monochloro alcohol 5a in 37% ee (Table 1, entry 2) as described above. On the other hand, the reaction with 2 equiv of NFSI against to 2a showed poor enantioselectivity (31% ee, Table 1, entry 1). We also examined the reaction with 2 equiv of 2a based on NFSI. The reaction yielded 4a in 75% ee (lower ee than that in Table 1, entry 2), and the enantiomeric purity of the recovered 5a was increased to 52% ee (Table 1, entry 3). Similar trends were observed in the fluorination with some other substrates 2b–2g (Table 1, entries 4–14). These results strongly suggested that the high asymmetric induction in this fluorination requires not only control of enantiofacial selection during electrophilic fluorination of the enamine intermediates, but also a high level of kinetic resolution of the starting aldehydes.
Table 1: Enantioselective fluorination of α-chloroaldehydes.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-30-i7.svg?max-width=637&scale=1.0)
|
||||||
| entry | R | 2:NFSI | t (h) | % yield of 4b | % ee of 4c | % ee of 5c,d |
|---|---|---|---|---|---|---|
| 1e | Bn (2a) | 1:2 | 11 | 78 | 31 (R) | – |
| 2e,f | Bn | 3:1 | 6 | 98 | 87 (R) | 37 (S) |
| 3 | Bn | 2:1 | 6 | 96 | 75 (R) | 52 (S) |
| 4 | n-Hex (2b) | 1:2 | 11 | 82 | 31 | – |
| 5e,f | n-Hex | 3:1 | 10 | 97 | 80 | 35 (S) |
| 6 | n-Hex | 2:1 | 19 | 92 | 68 | 49 (S) |
| 7 | –(CH2)3OCH2OCH3 (2c) | 1:2 | 19 | 83 | 23 | – |
| 8f | –(CH2)3OCH2OCH3 | 3:1 | 10 | 90 | 78 | 33 (S) |
| 9f | –(CH2)3CO2Et (2d) | 3:1 | 4 | 90 | 80 | 20 |
| 10g | c-Hex (2e) | 1:2 | 48 | 88 | 42 | – |
| 11g | c-Hex | 3:1 | 24 | 92 | 96 | 15 |
| 12 | Ph (2f) | 1:2 | 12 | 61 | 72 | – |
| 13e,f | Ph | 3:1 | 10 | 82 | 90 | 5 |
| 14e,g | t-Bu (2g) | 3:1 | 30 | 87 | 99 | 29 |
aReactions were carried out in t-BuOMe with 15 mol % of (S)-1 unless otherwise noted. bIsolated yield based on 2 or NFSI. cDetermined by chiral HPLC or GC analysis. dMonochloro alcohol 5 was recovered in nearly quantitative yield. eSimilar result was reported in Ref. [8]. f10 mol % of (S)-1 was used. gReaction was carried out with 30 mol % of (S)-1 at 30 °C.
From these results, we proposed a reaction mechanism for the fluorination of α-chloroaldehydes, as shown in Scheme 4. Catalyst (S)-1 reacts with (R)-2a to form iminium intermediate I, which undergoes deprotonation from the side opposite to the bulky substituent X (X = CAr2OTMS) of the pyrrolidine ring to afford enamine intermediate (Z)-11 (path A). Then, NFSI attacks (Z)-11 from the side opposite to X to yield (R)-3a. Although deprotonation may also occur from the same side as X to give (E)-11 (path B), the reaction through path B is considered to be very slow because the steric repulsion between the counter anion (OH–) and X would prevent deprotonation. Further, the resulting (E)-11 would be a thermodynamically unfavorable product because of steric repulsion between the methylene group on the pyrrolidine ring and the benzyl substituent on 2a. Alternatively, (S)-2a reacts with (S)-1 to form iminium intermediate II, which also undergoes deprotonation to form (E)- or (Z)-11. In these cases, deprotonation from the side opposite to X (path C) is considered to be slow because the resulting (E)-11 is a thermodynamically unfavorable form, as described above, and deprotonation from the same side as X (path D) is also slow because of steric repulsion between the counter anion (OH–) and X. Thus, it is difficult to control the geometry of enamine intermediate 11 when starting from (S)-2a, and hence, the enantioselectivity of the fluorination is significantly decreased because the fluorination occurs from the side opposite to X, regardless of the geometry of 11. For these reasons, high enantioselectivity was observed when 2a was employed in excess in the reaction, whereas an excess of NFSI led to poor asymmetric induction. In the former reaction, the major enantiomer of the recovered 5a was the S-form (Table 1, entries 2 and 3). This result also supports the proposed mechanism.
![[1860-5397-10-30-i4]](/bjoc/content/inline/1860-5397-10-30-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
To test the proposed reaction mechanism, we carried out the fluorination of enantioenriched 2a (61% ee, R favored) with 2 equiv of NFSI in the presence of each enantiomer of catalyst 1. As expected from the mechanism, good enantioselectivity was observed when (S)-1 was employed in the reaction, whereas the reaction proceeded more slowly to yield 4a with poor enantioselectivity in the presence of (R)-1 (Scheme 5).
![[1860-5397-10-30-i5]](/bjoc/content/inline/1860-5397-10-30-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Fluorination of the enantiomers of 2a.
Scheme 5: Fluorination of the enantiomers of 2a.
Finally, we were curious to know whether a similar kinetic resolution would be observed in the fluorination of α,α-dialkylaldehydes. We examined the fluorination of racemic α,α-dialkylaldehyde 12 in the presence of catalyst 1 (Scheme 6). The reaction with 3 equiv of rac-12 based on NFSI afforded the corresponding product 13 in higher enantioselectivity than that obtained in the reaction with 2 equiv of NFSI, along with 27% ee of 14; however the enantiomeric excess of 13 was not sufficiently high (47% ee). These results suggested that the reaction proceeded by a similar mechanism as shown in Scheme 4.
![[1860-5397-10-30-i6]](/bjoc/content/inline/1860-5397-10-30-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Enantioselective fluorination of α-branched aldehyde 12.
Scheme 6: Enantioselective fluorination of α-branched aldehyde 12.
Conclusion
In conclusion, we succeeded in the highly enantioselective fluorination of α-chloroaldehydes to afford α-chloro-α-fluoroaldehydes mediated by chiral organocatalyst 1. It was revealed that kinetic resolution of the racemic α-chloroaldehydes occurred during this fluorination reaction, which played an important role in the asymmetric induction.
Experimental
Experiments involving moisture- and/or air-sensitive compounds were performed in oven-dried flasks under an atmosphere of dry argon. All reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) using pre-coated silica gel plates with F254 indicator. Visualization was accomplished with UV light (254 nm), or phosphomolybdic acid, potassium permanganate, or anisaldehyde staining. Column chromatography was performed over silica gel (40–100 μm). 1H, 13C, and 19F NMR spectra were acquired on a JEOL JNM-ECX500 spectrometer. Chemical shift values (δ) are reported in ppm (1H: δ 0.00 for tetramethylsilane; 19F: δ 0.00 for trichlorofluoromethane; 13C: δ 77.0 for residual chloroform). IR spectra were measured on a JASCO FT/IR-230 spectrometer. Elemental analysis was performed with a Yanaco CHN CORDER MT-6. High-performance liquid chromatography (HPLC) analyses were performed with a JASCO PU-1586 with a UV-1575 UV–vis detector using a chiral column. GC analysis was performed with a Shimadzu model 2014 instrument. Optical rotations were measured on a JASCO P-1030 polarimeter.
α-Chloro aldehydes 2 were prepared with N-chlorosuccinimide in the presence of organocatalyst according to the procedure reported by Jørgensen [14] and were distilled before use. Racemic forms were synthesized with DL-proline catalyst, and optically active 2a was synthesized with L-prolinamide catayst, whose enantiopurity was slightly decreased during the distillation.
We confirmed that the optical purity of fluorinated products 4 did not change even after chromatographic purification using achiral silica gel and subsequent solvent evaporation. Therefore, we concluded that the enantiomers did not undergo self-disproportionation during the purification process [15-19].
Transformation of 6 to (R)-4a
Compound 8 was synthesized from 6 (94% de) according to the procedure reported in [9]. A flame-dried flask under argon was charged with 8 (anti/syn = 10:1, 0.35 mmol) and 1,2-dichloroethane (2 mL). 1,1′-Thiocarbonyldiimidazole (0.6 mmol) was added to this solution, and the mixture was stirred for 16 h at ambient temperature. The mixture was quenched by adding saturated aqueous NaHCO3 and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude mixture was purified by silica gel column chromatography (1:1 hexane/Et2O) to give 9 in 98% yield (anti/syn = 10:1).
9: 1H NMR (400 MHz, CDCl3) δ 8.33 (s, 1H), 7.83–7.72 (m, 6H), 7.06 (s, 1H), 6.74 (d, J = 21.6 Hz, 1H), 4.87 (td, J = 10.8, 4.4 Hz, 1H), 2.01–1.94 (m, 1H), 1.74–1.56 (m, 4H), 1.55–1.36 (m, 2H), 1.18–0.96 (m, 2H), 0.92 (d, J = 6.4 Hz, 3H), 0.74 (d, J = 7.2 Hz, 3H), 0.52 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 180.6, 163.6 (d, J = 29.4 Hz), 137.1, 131.4, 130.3, 129.9, 129.8, 128.8, 128.6, 117.8, 103.3 (d, J = 262.8 Hz), 84.5 (d, J = 19.8 Hz), 78.8, 46.8, 40.1, 33.9, 31.5, 26.2, 22.8, 22.0, 20.7, 15.2; 19F NMR (376 MHz, CDCl3) δ −132.1 (d, J = 21.4 Hz); FTIR (neat) υmax: 2955, 1762, 1464, 1395, 1288, 1212, 1102, 992, 952, 742, 475 cm−1; anal calcd (%) for C23H28ClFN2O3S: C, 59.15; H, 6.04; N, 6.00; found: C, 59.18; H, 5.96; N, 6.40.
A flame-dried flask under argon was charged with 9 (0.22 mmol) and benzene (3.6 mL). Tributyltin hydride (0.45 mmol) was added to this solution, and the mixture was stirred for 30 min at room temperature. The solvent was removed under reduced pressure and the crude mixture was purified by silica gel column chromatography (5:1 hexane/CH2Cl2) to give 10 in 42% yield.
10: 1H NMR (400 MHz, CDCl3) δ 7.35–7.25 (m, 5H), 4.74 (td, J = 10.8, 4.4 Hz, 1H), 3.65 (d, J = 6.8 Hz, 1H), 3.59 (d, J = 3.2 Hz, 1H), 1.85–1.76 (m, 2H), 1.72–1.63 (m, 2H), 1.50–1.41 (m, 2H), 1.37–1.19 (m, 1H), 1.09–0.98 (m, 1H), 0.95–0.91 (m, 1H), 0.89 (d, J = 2.4 Hz, 3H), 0.87 (dd, J = 3.2 Hz, 3H), 0.74 (dd, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.3 (d, J = 27.4 Hz), 132.3, 130.7, 130.6, 128.5, 127.9, 106.1 (d, J = 257.9 Hz), 77.9, 46.8, 40.0, 34.0, 31.4, 26.1, 23.3, 22.0, 20.8, 16.1; 19F NMR (376 MHz, CDCl3) δ −116.6 (dd, J = 22.9, 19.6 Hz); FTIR (neat) υmax: 2954, 1754, 1458, 1282, 1216, 1145, 1043, 952, 704, 624, 471 cm−1; anal calcd (%) for C19H26ClFO2: C, 66.95; H, 7.69; found: C, 67.02; H, 7.96.
A flame-dried flask under argon was charged with 10 (0.07 mmol) and Et2O (0.2 mL). LiAlH4 (0.11 mmol) was added to this solution at −78 °C, and the mixture was stirred for 1 h at room temperature. The reaction mixture was quenched with saturated aqueous NH4Cl and the mixture was extracted with Et2O. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude mixture was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give (R)-4a in 74% yield, with an enantiomeric purity of 94% ee.
4a: [α]D = −2.8 (c 1.5, CHCl3). HPLC (99:1 hexane/2-propanol; 1 mL/min; using a CHIRALPAK IC column (0.46 cm Ø × 25 cm)): 11.4 min (major) and 11.9 min (minor). These analytical data were identical to those of 4a synthesized from 2a with (S)-1.
General procedure for the asymmetric fluorination of α-chloroaldehydes 2
To a solution of α-chloroaldehyde 2 (1.5 mmol) in t-BuOMe (2 mL) was added catalyst 1 (0.05 mmol) and NFSI (0.5 mmol). The reaction mixture was stirred at room temperature for the time given in Table 1 and then poured into MeOH/CH2Cl2 (1:4, 5 mL) at 0 °C. To this solution, NaBH4 (5 mmol) was added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched with saturated aqueous NH4Cl, and the mixture was extracted with Et2O. The organic layer was dried over Na2SO4, concentrated, and chromatographed on silica gel to give 4, along with monochloro alcohol 5.
The results of all spectroscopic analyses of compounds 4a, 4b, 4f, 4g, and 5a–5f were identical to those described in our previous report [8] and in references [20,21]. Absolute configuration of 5a–5c was confirmed by comparing their optical rotation to that reported in the above-mentioned literature [20].
(R)-2-Chloro-2-fluoro-3-phenylpropan-1-ol (4a, 87% ee): 1H NMR (500 MHz, CDCl3) δ 7.35–7.30 (m, 5H), 3.88–3.71 (m, 2H), 3.46 (dd, J = 32.3, 15.0 Hz, 2H), 2.15 (br, 1H); 13C NMR (125 MHz, CDCl3) δ 133.3 (d, J = 3.8 Hz), 130.7, 128.4, 127.6, 114.8 (d, J = 247 Hz), 67.2 (d, J = 26.4 Hz), 44.6 (d, J = 21.4 Hz); 19F NMR (470 MHz, CDCl3) δ −114.2 (m); [α]D = −2.7 (c 1.5, CHCl3). The enantiopurity was determined by HPLC (99:1 hexane/2-propanol; 1 mL/min; using a CHIRALPAK IC column (0.46 cm Ø × 25 cm)): 11.4 min (major) and 11.9 min (minor).
2-Chloro-2-fluorooctan-1-ol (4b, 80% ee): 1H NMR (500 MHz, CDCl3) δ 3.91–3.78 (m, 2H), 2.14–2.05 (m, 3H), 1.59–1.54 (m, 2H), 1.37–1.29 (m, 6H), 0.90 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 116.1 (d, J = 245 Hz), 68.3 (d, J = 26.4 Hz), 38.5 (d, J = 21.3 Hz), 31.5, 28.9, 23.3 (d, J = 3.8 Hz), 22.5, 14.0; 19F NMR (470 MHz, CDCl3) δ −113.9 (br); [α]D = +2.0 (c 0.5, CHCl3). The enantiopurity was determined by GC (100–150 °C, 3 °C/min; using a Chiral DEX B-DM column): 12.4 min (major) and 13.3 min (minor).
2-Chloro-2-fluoro-5-(methoxymethoxy)pentan-1-ol (4c, 78% ee): 1H NMR (500 MHz, CDCl3) δ 4.63 (s, 2H), 3.99–3.79 (m, 2H), 3.66–3.55 (m, 2H), 3.37 (s, 3H), 2.40–2.12 (m, 3H), 2.01–1.82 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 115.8 (d, J = 245 Hz), 96.4, 68.4 (d, J = 26.4 Hz), 66.8, 55.3, 35.4 (d, J = 21.6 Hz), 23.9 (d, J = 4.8 Hz); 19F NMR (470 MHz, CDCl3) δ −114.3 (m); [α]D22 = +4.6 (c 0.16, CHCl3); anal calcd (%) for C7H14ClFO3: C, 41.91; H, 7.03; Cl, 17.67; F, 9.47; O, 23.92; found: C, 44.91; H, 7.51. The enantiopurity was determined after conversion into the corresponding 2-naphthoate 15c.
A flame-dried flask under argon was charged with 4c (0.10 mmol) and CH2Cl2 (1.0 mL). Triethylamine (0.20 mmol), 2-naphthoyl chloride (0.15 mmol), and 4-dimethylaminopyridine (0.01 mmol) were added to this solution, and the mixture was stirred for 2 h at 0 °C. The mixture was diluted by saturated aqueous NaHCO3, and extracted with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude mixture was purified by silica gel column chromatography (hexane/ethyl acetate 5:1) to give the desired 2-naphthoate 15c in 82% yield.
2-Chloro-2-fluoro-5-(methoxymethoxy)pentyl 2-naphthoate (15c, 78% ee): 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.08 (d, J = 10.4 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.93–7.86 (m, 2H), 7.67–7.53 (m, 2H), 4.83–4.66 (m, 2H), 4.61 (s, 2H), 3.62 (t, J = 5.80 Hz, 2 H), 3.34 (s, 3H), 2.49–2.19 (m, 2H), 2.11–1.88 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 165.6, 135.8, 132.4, 131.6, 129.4, 128.6, 128.4, 127.8, 126.8, 126.3, 125.2, 112.8 (d, J = 247 Hz), 96.4, 68.1 (d, J = 26.8 Hz), 66.6, 55.2, 36.2 (d, J = 22.0 Hz), 23.9 (d, J = 3.83 Hz); 19F NMR (470 MHz, CDCl3) δ −111.7 (m); [α]D22 = +7.5 (c 0.36, CHCl3); anal calcd (%) for C18H20ClFO4: C, 60.93; H, 5.68; Cl, 9.99; F, 5.35; O, 18.04; found: C, 60.95; H, 5.85. The enantiopurity was determined by HPLC (50:1 hexane/2-propanol; 0.5 mL/min; using a CHIRALPAK ID column (0.46 cm Ø × 25 cm)): 25.1 min (major) and 30.5 min (minor).
Ethyl 5-chloro-5-fluoro-6-hydroxyhexanoate (4d, 80% ee): 1H NMR (500 MHz, CDCl3) δ 4.14 (q, J = 7.3 Hz, 2H), 3.94–3.80 (m, 2H), 2.58 (s, 1H), 2.44–2.34 (m, 2H), 2.28–2.07 (m, 2H), 1.95–1.86 (m, 2H), 1.26 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.1, 115.4 (d, J = 246.5 Hz), 68.1 (d, J = 26.5 Hz), 60.6, 37.4 (d, J = 22.8 Hz), 33.3, 18.9 (d, J = 4.8 Hz), 14.2; 19F NMR (470 MHz, CDCl3) δ −114.0 (m); [α]D13 = −1.48 (c 1.1, CHCl3); anal calcd (%) for C8H14ClFO3: C, 45.19; H, 6.64; found: C, 44.65; H, 6.67. The enantiopurity was determined after conversion into the corresponding 2-naphthoate 15d by a procedure similar to that employed for the synthesis of 15c. The crude mixture was purified by silica gel column chromatography (hexane/EtOAc = 10:1) to give 81% yield of 15d.
2-Chloro-6-ethoxy-2-fluoro-6-oxohexyl 2-naphthoate (15d, 80% ee): 1H NMR (500 MHz, CDCl3) δ 8.65 (s, 1H), 8.07 (d, J = 8.8 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H), 7.92–7.89 (m, 2H), 7.64–7.55 (m, 2H), 4.79–4.66 (m, 2H), 4.13 (q, J = 7.0 Hz, 2H), 2.48–2.38 (m, 2H), 2.37–2.17 (m, 2H), 2.09–1.95 (m, 2H), 1.24 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 172.7, 165.6, 135.7, 132.4, 131.6, 129.5, 128.6, 128.4, 127.8, 126.8, 126.2, 125.1, 112.5 (d, J = 247.6 Hz), 67.9 (d, J = 27.6 Hz), 60.5, 38.4 (d, J = 22.8 Hz), 33.4, 18.9 (d, J = 4.8 Hz), 14.2; 19F NMR (470 MHz, CDCl3) δ −111.8 (m); [α]D21 = +7.1 (c 0.31, CHCl3); anal calcd (%) for C19H20ClFO4: C, 62.21; H, 5.50; found: C, 62.92; H, 6.07. The enantiopurity was determined by HPLC (50:1 hexane/2-propanol; 1.0 mL/min; using a CHIRALPAK IB-3 column (0.46 cm Ø × 25 cm)): 19.5 min (minor) and 24.9 min (major).
2-Chloro-2-cyclohexyl-2-fluoroethan-1-ol (4e, 96% ee): 1H NMR (500 MHz, CDCl3) δ 4.02–3.83 (m, 2H), 2.19–2.08 (m, 1H), 1.98–1.92 (m, 1H), 1.89–1.78 (m, 3H), 1.74–1.66 (m, 1H), 1.39–1.11 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 119.0 (d, J = 247 Hz), 66.8 (d, J = 26.4 Hz), 44.5 (d, J = 20.4 Hz), 27.3 (d, J = 6.0 Hz), 26.1 (d, J = 3.6 Hz), 25.9, 25.7, 25.6; 19F NMR (470 MHz, CDCl3) δ −117.8 (m); [α]D22 = −6.2 (c 0.64, CHCl3); anal calcd (%) for C8H14ClFO: C, 53.19; H, 7.81; Cl, 19.62; F, 10.52; O, 8.86; found: C, 52.52; H, 7.88. The enantiopurity was determined after conversion into the corresponding 2-naphthoate 15e by a procedure similar to that employed for the synthesis of 15c. The crude mixture was purified by silica gel column chromatography (hexane/ethyl acetate = 20:1) to give 81% yield of 15e.
2-Chloro-2-cyclohexyl-2-fluoroethyl 2-naphthoate (15e, 96% ee): 1H NMR (500 MHz, CDCl3) δ 8.65 (s, 1H), 8.08 (d, J = 10.32 Hz, 1H), 7.99 (d, J = 8.41 Hz, 1H), 7.94–7.87 (m, 2H), 7.66–7.54 (m, 2 H), 4.75 (br d, J = 17.5 Hz, 1H), 4.75 (br d, J = 19.0 Hz, 1H), 2.25–2.16 (m, 1H), 2.09–2.00 (m, 1H), 1.96–1.80 (m, 3H), 1.76–1.66 (m, 1H), 1.49–1.35 (m, 1H), 1.36–1.15 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 165.6, 135.7, 132.4, 131.6, 129.5, 128.6, 128.4, 127.8, 126.8, 126.5, 125.2, 116.0 (d, J = 248.2 Hz), 66.7 (d, J = 25.9 Hz), 45.4 (d, J = 20.1 Hz), 27.4 (d, J = 5.8 Hz), 26.1 (d, J = 2.8 Hz), 25.8, 25.7, 25.6; 19F NMR (470 MHz, CDCl3) δ −114.3 (m); [α]D24 = −13.7 (0.36, CHCl3); anal calcd (%) for C19H20ClFO2: C, 68.16; H, 6.02; Cl, 10.59; F, 5.67; O, 9.56; found: C, 68.03; H, 5.98. The enantiopurity was determined by HPLC (200:1 hexane/2-propanol; 0.5 mL/min; using a CHIRALCEL OJ-H column (0.46 cm Ø × 25 cm)): 22.5 min (major) and 25.4 min (minor).
2-Chloro-2-fluoro-2-phenylethanol (4f, 90% ee): 1H NMR (500 MHz, CDCl3) δ 7.59–7.53 (m, 2H), 7.46–7.40 (m, 3H), 4.15–4.04 (m, 2H), 2.15 (t, J = 7.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 137.7 (d, J = 22.6 Hz), 129.8, 128.6, 125.3 (d, J = 7.5 Hz), 112.9 (d, J = 247 Hz), 70.2 (d, J = 26.4 Hz); 19F NMR (470 MHz, CDCl3) δ −118.2 (t, J = 18.8 Hz); [α]D = −76.5 (c 0.6, CHCl3). The enantiopurity was determined by HPLC (99:1 hexane/2-propanol; 1 mL/min; using a CHIRALPAK IC column (0.46 cm Ø × 25 cm)): 19.1 min (major) and 21.1 min (minor).
2-Chloro-2-fluoro-3,3-dimethylbutan-1-ol (4g) and 2-Chloro-3,3-dimethylbutan-1-ol (5g): 4g and 5g were inseparable by column chromatography. Therefore, isolation and determination of their enantiopurity were performed after the conversion into the corresponding 2-naphthoates 15g and 16g by a procedure similar to that employed for the synthesis of 15c. The crude mixture was purified by silica gel column chromatography (hexane/CH2Cl2 = 3:1) to give 87% yield of 15g, along with 80% yield of 16g.
2-Chloro-2-fluoro-3,3-dimethylbutyl 2-naphthoate (15g, 99% ee): 1H NMR (400 MHz, CDCl3) δ 8.68 (s, 1H), 8.12 (d, J = 8.9 Hz, 1H), 7.98 (d, J = 8.2 Hz, 1H), 7.92–7.88 (m, 2H), 7.63–7.54 (m, 2H), 4.89–4.78 (m, 2H), 1.26 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 166.0, 135.7, 132.4, 131.6, 129.5, 128.5, 128.3, 127.8, 126.7, 126.6, 125.2, 119.1 (d, J = 251.9 Hz), 66.0 (d, J = 25.2 Hz), 40.8 (d, J = 20.4 Hz), 25.5 (d, J = 3.6 Hz); 19F NMR (470 MHz, CDCl3) δ −120.3 (m); [α]D20 = −22.5 (c 1.4, CHCl3); anal calcd (%) for C17H18ClFO2: C, 66.13; H, 5.88; found: C, 65.88; H, 6.10. The enantiopurity was determined by HPLC (200:1 hexane/2-propanol; 1.0 mL/min; using a CHIRALPAK IB-3 column (0.46 cm Ø × 25 cm)): 9.7 min (minor) and 14.0 min (major).
2-Chloro-3,3-dimethylbutyl 2-naphthoate (16g, 29% ee): 1H NMR (500 MHz, CDCl3) δ 8.65 (s, 1H), 8.09 (d, J = 8.6 Hz, 1H), 7.98 (d, J = 7.6 Hz, 1H), 7.91–7.89 (m, 2H), 7.62–7.55 (m, 2H), 4.81 (dd, J = 3.1, 11.9 Hz, 1H), 4.45 (dd, J = 8.8, 11.9 Hz, 1H), 4.11 (dd, J = 3.1, 8.8 Hz, 1H), 1.16 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 166.5, 135.6, 132.4, 131.3, 129.4, 128.3, 128.2, 127.7, 127.0, 126.7, 125.2, 70.1, 66.4, 35.2, 27.0; [α]D20 = +16.2 (c 1.3, CHCl3); anal. calcd (%) for C17H19ClO2: C, 70.22; H, 6.59; found: C, 69.92; H, 6.88. The enantiopurity was determined by HPLC (200:1 hexane/2-propanol; 1.0 mL/min; using a CHIRALPAK AS-H column (0.46 cm Ø × 25 cm)): 7.2 min (major) and 8.3 min (minor).
2-Fluoro-3-(4-isopropylphenyl)-2-methylpropan-1-ol (13, 47% ee) [7]: 1H NMR (500 MHz, CDCl3) δ 7.16 (s, 4H), 3.61–3.56 (m, 2H), 2.96 (br d, J = 16.5 Hz, 1H), 2.96 (br d, J = 20.5 Hz, 1H), 2.91–2.85 (m, 1H), 1.82 (br s, 1H), 1.27 (d, J = 21.8 Hz, 3H), 1.24 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 147.3, 133.2 (d, J = 4.8 Hz), 130.3, 126.3, 97.4 (d, J = 170 Hz), 67.5 (d, J = 22.8 Hz), 41.9 (d, J = 22.8 Hz), 33.7, 24.0, 20.9 (d, J = 22.8); 19F NMR (470 MHz, CDCl3) δ −154.7 (m); [α]D25 = −7.0 (c 0.60, CHCl3); The enantiopurity was determined by HPLC (99:1 hexane/2-propanol; 1 mL/min; using a CHIRALCEL OJ column (0.46 cm Ø × 25 cm)): 17.4 min (major) and 21.8 min (minor).
3-(4-Isopropylphenyl)-2-methylpropan-1-ol (14, 27% ee) [22]: 1H NMR (500 MHz, CDCl3) δ 7.15 (d, J = 8.0 Hz, 2H), 7.10 ( d, J = 8.0 Hz, 2H), 3.54 (dd, 5.0, 5.7 Hz, 1H), 3.47 (dd, J = 4.6, 6.1 Hz, 1H), 2.88 (m, 1H), 2.71 (dd, J = 6.5, 6.9 Hz, 1H), 2.41 (dd, J = 5.3, 8.1 Hz, 1H), 1.93 (m, 1H), 1.24 (d, J = 6.9 Hz, 6H), 0.92 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.4, 137.8, 129.0, 126.3, 67.8, 39.3, 37.8, 33.7, 24.1, 16.6; [α]D25 = −2.3 (c 0.15, CHCl3); The enantiopurity was determined by HPLC (99:1 hexane/2-propanol; 1 mL/min; using a CHIRALPAK IC-3 column (0.46 cm Ø × 25 cm)): 17.6 min (minor) and 19.9 min (major).
References
-
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons: Chichester, 2009. doi:10.1002/9781444312096
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delphon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, 2008. doi:10.1002/9780470281895
Return to citation in text: [1] -
Marigo, M.; Fielenbach, D.; Braunton, A.; Kjærsgaard, A.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2005, 44, 3703. doi:10.1002/anie.200500395
Return to citation in text: [1] -
Beeson, T. D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 8826. doi:10.1021/ja051805f
Return to citation in text: [1] -
Steiner, D. D.; Mase, N.; Barbas, C. F., III. Angew. Chem., Int. Ed. 2005, 44, 3706. doi:10.1002/anie.200500571
Return to citation in text: [1] -
Enders, D.; Hüttl, M. R. M. Synlett 2005, 991. doi:10.1055/s-2005-864813
Return to citation in text: [1] -
Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K. A. Chem.–Eur. J. 2006, 12, 6039. doi:10.1002/chem.200600495
Return to citation in text: [1] [2] -
Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944. doi:10.1021/ol201007e
Return to citation in text: [1] [2] [3] [4] -
Shibatomi, K.; Muto, T.; Smikawa, Y.; Narayama, A.; Iwasa, S. Synlett 2009, 241. doi:10.1055/s-0028-1087675
Return to citation in text: [1] -
Shibatomi, K.; Soga, Y.; Narayama, A.; Fujisawa, I.; Iwasa, S. J. Am. Chem. Soc. 2012, 134, 9836. doi:10.1021/ja304806j
Return to citation in text: [1] -
Narayama, A.; Shibatomi, K.; Soga, Y.; Muto, T.; Iwasa, S. Synlett 2013, 24, 375. doi:10.1055/s-0032-1318027
Return to citation in text: [1] -
Barton, D. H. R.; McCombie, S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574. doi:10.1039/p19750001574
Return to citation in text: [1] -
Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jørgensen, K. A. J. Am. Chem. Soc. 2004, 126, 4790. doi:10.1021/ja049231m
Return to citation in text: [1] -
Soloshonok, V. A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A. E. Chem. Soc. Rev. 2012, 41, 4180. doi:10.1039/c2cs35006h
Return to citation in text: [1] -
Han, J.; Nelson, D. J.; Sorochinsky, A. E.; Soloshonok, V. A. Curr. Org. Synth. 2011, 8, 310. doi:10.2174/157017911794697303
Return to citation in text: [1] -
Ueki, H.; Yasumoto, M.; Soloshonok, V. A. Tetrahedron: Asymmetry 2010, 21, 1396. doi:10.1016/j.tetasy.2010.04.040
Return to citation in text: [1] -
Soloshonok, V. A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J. S.; Singleton, D. A. J. Am. Chem. Soc. 2007, 129, 12112. doi:10.1021/ja065603a
Return to citation in text: [1] -
Soloshonok, V. A. Angew. Chem., Int. Ed. 2006, 45, 766. doi:10.1002/anie.200503373
Return to citation in text: [1] -
Amatore, M.; Beeson, T. D.; Brown, S. P.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2009, 48, 5121. doi:10.1002/anie.200901855
Return to citation in text: [1] [2] -
Lutje Spelberg, J. H.; van Hylckama Vlieg, J. E. T.; Bosma, T.; Kellogg, R. M.; Janssen, D. B. Tetrahedron: Asymmetry 1999, 10, 2863. doi:10.1016/S0957-4166(99)00308-0
Return to citation in text: [1] -
Rosini, G.; Paolucci, C.; Boschi, F.; Marotta, E.; Righi, P.; Tozzi, F. Green Chem. 2010, 12, 1747. doi:10.1039/c0gc00013b
Return to citation in text: [1]
| 7. | Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K. A. Chem.–Eur. J. 2006, 12, 6039. doi:10.1002/chem.200600495 |
| 20. | Amatore, M.; Beeson, T. D.; Brown, S. P.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2009, 48, 5121. doi:10.1002/anie.200901855 |
| 21. | Lutje Spelberg, J. H.; van Hylckama Vlieg, J. E. T.; Bosma, T.; Kellogg, R. M.; Janssen, D. B. Tetrahedron: Asymmetry 1999, 10, 2863. doi:10.1016/S0957-4166(99)00308-0 |
| 20. | Amatore, M.; Beeson, T. D.; Brown, S. P.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2009, 48, 5121. doi:10.1002/anie.200901855 |
| 1. | Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons: Chichester, 2009. doi:10.1002/9781444312096 |
| 2. | Bégué, J.-P.; Bonnet-Delphon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, 2008. doi:10.1002/9780470281895 |
| 8. | Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682 |
| 9. | Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944. doi:10.1021/ol201007e |
| 8. | Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682 |
| 8. | Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682 |
| 7. | Brandes, S.; Niess, B.; Bella, M.; Prieto, A.; Overgaard, J.; Jørgensen, K. A. Chem.–Eur. J. 2006, 12, 6039. doi:10.1002/chem.200600495 |
| 14. | Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jørgensen, K. A. J. Am. Chem. Soc. 2004, 126, 4790. doi:10.1021/ja049231m |
| 3. | Marigo, M.; Fielenbach, D.; Braunton, A.; Kjærsgaard, A.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2005, 44, 3703. doi:10.1002/anie.200500395 |
| 4. | Beeson, T. D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2005, 127, 8826. doi:10.1021/ja051805f |
| 5. | Steiner, D. D.; Mase, N.; Barbas, C. F., III. Angew. Chem., Int. Ed. 2005, 44, 3706. doi:10.1002/anie.200500571 |
| 6. | Enders, D.; Hüttl, M. R. M. Synlett 2005, 991. doi:10.1055/s-2005-864813 |
| 15. | Soloshonok, V. A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A. E. Chem. Soc. Rev. 2012, 41, 4180. doi:10.1039/c2cs35006h |
| 16. | Han, J.; Nelson, D. J.; Sorochinsky, A. E.; Soloshonok, V. A. Curr. Org. Synth. 2011, 8, 310. doi:10.2174/157017911794697303 |
| 17. | Ueki, H.; Yasumoto, M.; Soloshonok, V. A. Tetrahedron: Asymmetry 2010, 21, 1396. doi:10.1016/j.tetasy.2010.04.040 |
| 18. | Soloshonok, V. A.; Ueki, H.; Yasumoto, M.; Mekala, S.; Hirschi, J. S.; Singleton, D. A. J. Am. Chem. Soc. 2007, 129, 12112. doi:10.1021/ja065603a |
| 19. | Soloshonok, V. A. Angew. Chem., Int. Ed. 2006, 45, 766. doi:10.1002/anie.200503373 |
| 9. | Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944. doi:10.1021/ol201007e |
| 10. | Shibatomi, K.; Muto, T.; Smikawa, Y.; Narayama, A.; Iwasa, S. Synlett 2009, 241. doi:10.1055/s-0028-1087675 |
| 11. | Shibatomi, K.; Soga, Y.; Narayama, A.; Fujisawa, I.; Iwasa, S. J. Am. Chem. Soc. 2012, 134, 9836. doi:10.1021/ja304806j |
| 12. | Narayama, A.; Shibatomi, K.; Soga, Y.; Muto, T.; Iwasa, S. Synlett 2013, 24, 375. doi:10.1055/s-0032-1318027 |
| 9. | Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944. doi:10.1021/ol201007e |
| 9. | Shibatomi, K.; Narayama, A.; Soga, Y.; Muto, T.; Iwasa, S. Org. Lett. 2011, 13, 2944. doi:10.1021/ol201007e |
| 8. | Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682 |
| 8. | Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682 |
| 22. | Rosini, G.; Paolucci, C.; Boschi, F.; Marotta, E.; Righi, P.; Tozzi, F. Green Chem. 2010, 12, 1747. doi:10.1039/c0gc00013b |
| 8. | Shibatomi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2008, 47, 5796. doi:10.1002/anie.200801682 |
| 13. | Barton, D. H. R.; McCombie, S. W. J. Chem. Soc., Perkin Trans. 1 1975, 1574. doi:10.1039/p19750001574 |
© 2014 Shibatomi et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)