Abstract
This review deals with the historical development of all N-F fluorinating agents developed so far. The unique properties of fluorine make fluorinated organic compounds attractive in many research areas and therefore fluorinating agents are important. N-F agents have proven useful by virtue of their easy handling. This reagent class includes many types of N-F compounds: perfluoro-N-fluoropiperidine, N-fluoro-2-pyridone, N-fluoro-N-alkylarenesulfonamides, N-fluoropyridinium salts and derivatives, N-fluoroquinuclidium salts, N-fluoro-trifluoromethanesulfonimide, N-fluoro-sultams, N-fluoro-benzothiazole dioxides, N-fluoro-lactams, N-fluoro-o-benzenedisulfonimide, N-fluoro-benzenesulfonimide, 1-alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts, N-fluoropyridinium-2-sulfonate derivatives, 1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane salts, N-fluorodinitroimidazole, N-fluoro-trichloro-1,3,5-triazinium salt, N-F ethano-Tröger’s base derivatives, N-fluoro-methanesulfonimide, N-fluoro-N-arylarenesulfonamides, bisN-F salts such as N,N’-difluorobipyridinium salts and N,N’-difluoro-1,4-diazoniabicyclo[2.2.2]octane salts, and their many derivatives and analogs, including chiral N-F reagents such as optically active N-fluoro-sultam derivatives, N-fluoro-alkaloid derivatives, DABCO-based N-F derivatives, and N-F binaphthyldisulfonimides. The synthesis and reactions of these reagents are described chronologically and the review also discusses the relative fluorination power of each reagent and their mechanisms chronicling developments from a historical perspective.
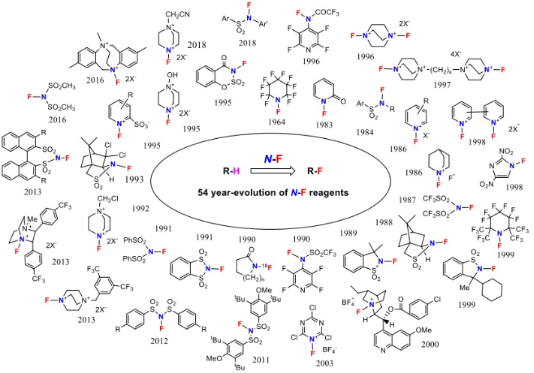
Graphical Abstract
Introduction
Fluorinated organic compounds occupy an important position in pharmaceuticals [1], agrochemicals [2], and materials [3]. Especially, in the first two areas, the presence of fluorine has attracted attention during the last decades. Nowadays, a considerable number of medicines [4,5] and agrochemicals [6] contain at least one fluorine atom in their structures. The fluorine atom has unique properties such as the highest electronegativity, extremely low polarization, strong C–F bonds, and the smallest size after a hydrogen atom [7]. Thus, introduction of fluorine into selective positions of a bioactive compound can produce remarkable changes in efficacy. Fluorine-scan/fluorine editing of a lead molecule is now a routine step in drug discovery [8]. Organofluorine compounds are very rare in nature [9] and therefore without natural compounds, chemical processes are required to generate building blocks. Molecular fluorine (F2) is a useful fluorinating reagent, however, unlike Cl2 and Br2, F2 is extremely reactive, toxic, and corrosive and its handling requires specialist skills and equipment. Therefore, easy-to-handle and selective fluorinating agents are essential for the wide-spread advancement of organofluorine chemistry to non-specialist chemists. Alternatives to F2, such as perchloryl fluoride (FClO3) [10] and the O-F reagents such as CF3OF [11], CF2(OF)2 [11], CsOSO2OF [12], CF3COOF [13], and CH3COOF [14] have been used as fluorinating agents for many years. However, these reagents have significant risks for safe handling. Although XeF2 [15] was considered as a safer alternative, it is expensive because of the scarcity of Xe in nature. The appearance of the safe and easy-to-handle N-F fluorinating agents described in this review have brought about a breakthrough in synthetic fluorine chemistry enabling an increasing number of researchers to engage in organofluorine chemistry. Their development has significantly contributed towards the current ‘golden age’ of fluorine chemistry. The N-F fluorinating agents now stand out as particularly useful electrophilic or radical fluorinating agents by virtue of their easy handling, efficiency, and selectivity. These non-hygroscopic nature and stability make them easier to handle than nucleophilic fluoride reagents. Potassium fluoride (KF) and naked fluoride anion salts are extremely sensitive to moisture, while HF seriously attacks human skin. The N-F fluorinating agents can be classified into two categories: these are neutral and cationic. This review covers the chronological advancement of these reagents regardless of their classification, as they advanced side by side.
Review
1. Historical progress of N-F fluorinating agents
1-1. Perfluoro-N-fluoropiperidine
The history of the N-F compounds acting as fluorine atom-transfer reagents can be traced back to 1964 when Banks and co-worker [16] first reported that perfluoro-N-fluoropiperidine (1-1) could fluorinate the sodium salt of 2-nitropropane to form 2-fluoro-2-nitropropane in a 40% yield (Scheme 1). The reaction with sodium diethyl malonate was also reported to produce the difluoromalonate, but in a very low yield of ca. 5%.
![[1860-5397-17-123-i1]](/bjoc/content/inline/1860-5397-17-123-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Fluorination with N-F amine 1-1.
Scheme 1: Fluorination with N-F amine 1-1.
N-F amine 1-1 is very volatile (bp 49.5 °C), and could only be prepared in 7.5% or 13% yield by electrochemical fluorination of pyridine or 2-fluoropyridine in anhydrous hydrogen fluoride [17,18] (Scheme 2). Not surprisingly, 1-1 did not become a popular reagent.
![[1860-5397-17-123-i2]](/bjoc/content/inline/1860-5397-17-123-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
In 1967, Banks et al. reported reactions of 1-1 with piperidine and triphenylphosphine, -arsine, and -stibine (Scheme 3, entries 1 and 2) [19]. The former reaction gave adduct 1-3 as a major product and the latter showed good fluorine-transfer to the hetero atoms. In 1972, German et al. reported the reaction with sodium phenoxide, which gave a small amount (5%) of fluorophenols 1-6 and a larger amount of adduct 1-7 (Scheme 3, entry 3) [20]. These data showed that the fluorinations with 1-1 were suppressed by the formation of perfluoro-3,4,5,6-tetrahydropyridine (1-5), which was reactive to the nucleophilic substrates existing in the reaction mixture.
![[1860-5397-17-123-i3]](/bjoc/content/inline/1860-5397-17-123-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In 1986, Banks and co-worker reported the preparation of polymeric analogues of perfluoro-N-fluoropiperidine (1-1) [21] and then in 1991, Banks et al. reported the improved yields of the reactions of 1-1 with sodium salts of 2-nitropropane, malonate esters, and a keto ester, and phenylmagnesium bromide [22]. However, the fluorinated products were still accompanied by considerable amounts of byproducts resulting from the reaction of the substrates with 1-5.
1-2. N-Fluoroperfluorosuccinimide and N-fluoroperfluoroglutarimide
In 1981, Yagupols’kii and co-worker reported the synthesis of N-fluoroperfluorosuccinimide (2-1) and N-fluoroperfluoroglutarimide (2-2) by reaction of precursor imides with XeF2 (Scheme 4) [23]. However, there were no reports on the fluorination capability of these N-F compounds. The purpose of this research was to establish if the presence of perfluoroacyl groups was sufficient to stabilize the Xe–N bond. Their experiment revealed that the intermediate F–Xe-N compounds were not detected, but N-fluoroimides 2-1 and 2-2 were formed. The stability of these N-F compounds was low, since they decomposed to H2NCO(CF2)nCOOH upon standing with air.
![[1860-5397-17-123-i4]](/bjoc/content/inline/1860-5397-17-123-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N-F perfluoroimides 2-1 and 2-2.
Scheme 4: Synthesis of N-F perfluoroimides 2-1 and 2-2.
1-3. 1-Fluoro-2-pyridone
In 1983, Purrington and co-worker synthesized 1-fluoro-2-pyridone (3-1) as a fluorination agent [24]. When 2-(trimethylsiloxy)pyridine was allowed to react with 5% F2 diluted with N2 in a freon solvent at −78 °C, 1-fluoro-2-pyridone was obtained in 63% yield (Scheme 5).
![[1860-5397-17-123-i5]](/bjoc/content/inline/1860-5397-17-123-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 1-fluoro-2-pyridone (3-1).
Scheme 5: Synthesis of 1-fluoro-2-pyridone (3-1).
The fluorination efficiency of 3-1 was higher than perfluoro-N-fluoropiperidine (1-1) and the yields of reaction with sodium diethyl malonates improved to between 9–39%; a difluorinated byproduct was obtained when the substituent R was H (Scheme 6). Further applications were investigated by the same authors in 1984 [25]. Grignard reagents and enamines could be fluorinated, but in very low yields and unfortunately, this reagent was not so stable.
![[1860-5397-17-123-i6]](/bjoc/content/inline/1860-5397-17-123-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Fluorination with 1-fluoro-2-pyridone (3-1).
Scheme 6: Fluorination with 1-fluoro-2-pyridone (3-1).
Notably, attempts to prepare N-fluorosuccinimide from succinimide, or one of its salts by reaction with fluorine (F2), trifluoromethyl hypofluorite, or perchloryl fluoride in a variety of solvents, and at temperatures ranging from −78 °C to room temperature, all but failed, as reported in the margin of the paper cited in [24].
1-4. N-Fluoro-N-alkylarenesulfonamides
In 1984, a series of stable N-fluoro-N-alkylsulfonamides 4-1a–g was reported by Barnette [26]. The treatment of N-alkylsulfonamides with very dilute F2 (1% or 5%) in N2 at −78 °C afforded the fluorinated products 4-1a–g. As detailed in Figure 1, various kinds of N-fluoro-N-alkylsulfonamides were synthesized by this method. However, the yields were low except for the case of compound 4-1d. In the cases of secondary and tertiary alkyl groups on the amine side, low yields were obtained and these were attributed to concomitant N–S-bond cleavage reactions with F2. This method, using the dilute F2, was inefficient for their production due to long reaction times.
![[1860-5397-17-123-1]](/bjoc/content/figures/1860-5397-17-123-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthesis of N-F sulfonamides 4-1a–g.
Figure 1: Synthesis of N-F sulfonamides 4-1a–g.
N-Fluoro-N-alkyl-p-toluenesulfonamides 4-1b,c,f proved to be efficient fluorinating agents in the fluorination of carbanions (Scheme 7). The yields of reactions with sodium malonates and Grignard reagents were largely improved to up to 81% and 50%, respectively. The carbanions of aromatics, ketones, nitroalkanes, amides, etc. could also be reasonably well fluorinated and this study showed great progress. However, although these fluorinating agents were stable and easy-to-handle, their fluorinating power was low. They could fluorinate only reactive carbanions, but not aromatics, olefins, vinyl acetates, trimethylsilyl or alkyl enol ethers, and so on.
![[1860-5397-17-123-i7]](/bjoc/content/inline/1860-5397-17-123-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Fluorination with N-F reagent 4-1b,c,f.
Scheme 7: Fluorination with N-F reagent 4-1b,c,f.
Soon after (1986), Schwartz and co-worker reported the stereospecific synthesis of alkenyl fluorides with N-fluoro-N-tert-butylbenzenesulfonamide (4-1h), a compound which is soluble at low temperature [27]. Alkenyllithium reagents, generated in situ, reacted at −120 °C with 4-1h in THF/Et2O/pentane to give the desired alkenyl fluorides 4-2 in good yields (Scheme 8). However, reactions that were run above −120 °C, or in pure ether or THF gave higher yields of the protonated products 4-3.
![[1860-5397-17-123-i8]](/bjoc/content/inline/1860-5397-17-123-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Fluorination of alkenyllithiums with N-F 4-1h.
Scheme 8: Fluorination of alkenyllithiums with N-F 4-1h.
1-5. N-Fluoropyridinium salts and their derivatives
In 1986, Umemoto et al. reported N-fluoropyridinium triflate and its derivatives 5-4 as new stable cationic fluorinating agents. These possessed either electron-donating or -withdrawing substituents on the pyridinium nuclei, and were the first reactive, easy-to-handle fluorinating agents with wide application [28,29]. The work continued with additional disclosures until 1991 [30-34]. Before that, reactive fluorinating reagents were difficult to handle because of toxicity, a tendency to explode, instability, and/or hygroscopicity, while easy-to-handle reagents had limited application because of their low reactivity.
Umemoto et al. found that the hygroscopic pyridine·F2 complex 5-2a decomposing vigorously at temperatures above −2 °C [35], which was formed by the fluorination of pyridine (10% F2/N2) at low temperature in a freon solvent, could undergo straightforward counteranion replacement with a non-nucleophilic anion. Therefore, exchange with salts such as sodium triflate in acetonitrile generated non-hygroscopic N-fluoropyridinium triflate salts as highly thermally stable reagents as illustrated in Scheme 9 [28]. Moreover, they found that the fluorination power (reactivity) of these N-fluoropyridinium salts could be tuned by the substituents on the pyridinium nuclei.
![[1860-5397-17-123-i9]](/bjoc/content/inline/1860-5397-17-123-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of N-fluoropyridinium triflate (5-4a).
Scheme 9: Synthesis of N-fluoropyridinium triflate (5-4a).
The transformation of the unstable pyridine·F2 complex to stable N-fluoropyridinium salts could be conducted by direct fluorination (10% F2/N2) of the pyridine in acetonitrile and in the presence of a suitable salt at low temperature (method B in Scheme 10). This one-step process was successfully applied to many other pyridine derivatives (method B in Figure 2).
![[1860-5397-17-123-i10]](/bjoc/content/inline/1860-5397-17-123-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthetic methods for N-F-pyridinium salts.
Scheme 10: Synthetic methods for N-F-pyridinium salts.
![[1860-5397-17-123-2]](/bjoc/content/figures/1860-5397-17-123-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthesis of various N-fluoropyridinium salts. Note: athis yield was the one by the improved method reported in [37]; bthis improved method was reported in [38].
Figure 2: Synthesis of various N-fluoropyridinium salts. Note: athis yield was the one by the improved method...
It was finally shown that the stable N-fluoropyridinium salts 5-4 could be synthesized in four different ways using 10% F2/N2 [28,31,33] (Scheme 10): method A (stepwise method) involved the fluorination of a pyridine derivative with F2/N2, to form a pyridine·F2 complex 5-2, followed by treatment with a non-nucleophilic anion salt, acid, or silyl derivative. Method B (one-step method) involved the fluorination of a pyridine derivative with F2/N2 in the presence of a non-nucleophilic anion salt. Method C involved mixing a pyridine derivative with an acid or its silyl derivative, forming a salt 5-5, before fluorination. Method D involved mixing a pyridine derivative with a Lewis acid, forming complex 5-6, and then the fluorination with F2/N2 was carried out.
In total, sixty-two stable N-fluoropyridinium salts possessing different non-nucleophilic counteranions and electron-withdrawing or -donating groups were efficiently synthesized [33]. Figure 2 shows 31 examples and their methods of preparation. Umemoto and co-worker also reported the synthesis of a polymer version, poly(vinyl-N-fluoropyridinium salts) of these reagents [36].
The reactivities of many N-fluoropyridinium salts were examined [32] and mainly five kinds of N-fluoropyridinium salts, shown in Scheme 11, emerged as useful fluorinating agents due to their availability. The fluorination power greatly changed depending on the electron density at the nitrogen, an aspect controlled by the electronic nature of the substituents. The fluorinating power increased in the order of 2,4,6-triMe 5-4j < unsubstituted 4a < 3,5-diCl 4t < 2,6-diCl 4r < pentachloro 4v, in good agreement with the decreasing order of the pKa values of the pyridines. For example, in order to fluorinate phenol, triMe 5-4j needed heating at 100 °C in a haloalkane solvent for 24 h, whereas pentachloro 5-4v required only room temperature within 0.1 h for a successful reaction. The salt 5-4v was so powerful that it fluorinated an equimolar amount of benzene in dichloromethane in 2 h at 40 °C. In general, triflate salts were more effective than BF4 salts because of the higher solubility of the triflate salts in a haloalkane solvent.
![[1860-5397-17-123-i11]](/bjoc/content/inline/1860-5397-17-123-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Fluorination power order of N-fluoropyridinium salts.
Scheme 11: Fluorination power order of N-fluoropyridinium salts.
As outlined in Scheme 12, fluorinations of many kinds of substrates with these N-fluoropyridinium salts were performed. It was shown that less reactive substrates can be fluorinated well with the more powerful reagents, and reactive substrates can be fluorinated with less powerful reagents, a match process which minimizes side reactions. Thus, these N-fluoropyridinium salts made possible the fluorination of a diversity of nucleophilic organic compounds with different reactivities ranging across aromatics, carbanions (Grignard reagents, enolate anions), active methylene compounds, olefins, silyl enol ethers, vinyl acetates, sulfides and so on, under mild conditions with high selectivity and yields [29-32]. All these reactions could be carried out routinely using standard glassware in normal laboratory environments and without any specialist training.
![[1860-5397-17-123-i12]](/bjoc/content/inline/1860-5397-17-123-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Fluorinations with N-F salts 5-4.
Scheme 12: Fluorinations with N-F salts 5-4.
Some interesting observations are noted: N-Fluoro-2,6-bis(methoxymethyl)pyridinium triflate (5-4l) fluorinated the trimethylsilyl ether of γ-butyrolactone and 1-cyclohexenyl acetate in much higher yields than the N-fluoro-2,4,6-trimethyl salt 5-4j [32]. Thus, as seen in Scheme 13, 5-4l converted the Corey lactone 5-7 via its silyl ether 5-8 to the fluorinated lactone 5-9 in a very satisfactory overall yield [32]. N-Fluoro-2,6-bis(CH2OAc)pyridinium triflate 5-4n [32], N-fluoro-2,6-bis(COOMe)pyridinium triflate 5-4y [30,32], and N-fluoro-2-cyano- and -2,6-dicyanopyridinium tetrafluoroborates [39] were useful reagents, too. These non-chlorinated reagents avoid chlorinated byproducts, an occurrence that was observed when other powerful fluorinating agents such as N-fluoro-2,3,4,5,6-pentachloropyridinium salts were used [40].
![[1860-5397-17-123-i13]](/bjoc/content/inline/1860-5397-17-123-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Fluorination of Corey lactone 5-7 with N-F-bis(methoxymethyl) salt 5-4l.
Scheme 13: Fluorination of Corey lactone 5-7 with N-F-bis(methoxymethyl) salt 5-4l.
In 1991, N-fluoropyridinium pyridine heptafluorodiborate (NFPy), C5H5NF(C5H5N)B2F7, was introduced as a fluorinating agent [41]. NFPy was prepared by the reaction of fluorine with pyridine·BF3 complex and its fluorination ability is shown in Scheme 14. However, it was subsequently reported that the correct structure was N-fluoropyridinium pyridinium tetrafluoroborate trifluorohydroxyborate, C5H5NF(C5H5NH)BF4(BF3OH), which was a 1:1 mixture of the N-fluoropyridinium salt and N-hydropyridinium salt (anion parts; BF4 and BF3OH), based on an X-ray diffraction study of a commercial sample [42].
![[1860-5397-17-123-i14]](/bjoc/content/inline/1860-5397-17-123-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-6. N-Fluoroquinuclidinium fluoride
In 1986 as the N-F pyridinium reagents were emerging, Banks et al. disclosed the quaternary ammonium N-F reagent, N-fluoroquinuclidinium fluoride (6-1) [43]. They subsequently followed with more detailed results in 1988 [44]. Quinuclidine was fluorinated by neat fluorine in trichlorofluoromethane at −72 °C, affording the product 6-1 in 86% yield (Scheme 15). Fluorination examples with 6-1 are shown in Scheme 16.
![[1860-5397-17-123-i15]](/bjoc/content/inline/1860-5397-17-123-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of the N-F reagent, N-fluoroquinuclidinium fluoride (6-1).
Scheme 15: Synthesis of the N-F reagent, N-fluoroquinuclidinium fluoride (6-1).
![[1860-5397-17-123-i16]](/bjoc/content/inline/1860-5397-17-123-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Fluorinations achieved with N-F fluoride 6-1.
Scheme 16: Fluorinations achieved with N-F fluoride 6-1.
This reagent proved to be very hygroscopic and deteriorated in air. In terms of fluorination yields, N-fluoroquinuclidinium fluoride (6-1) was however superior to Purrington’s 1-fluoro-2-pyridone (3-1) [24,25], but inferior to Barnette’s N-fluoro-N-alkylarenesulfonamides 4-1 [26] and Umemoto’s N-fluoropyridinium salts 5-4 [29,32].
1-7. N-Fluoroperfluoroalkanesulfonimides
In 1987, DesMarteau et al. reported the synthesis of a series of N-fluoroperfluoroalkanesulfonimides 7-1. These were prepared by reacting N,N-bis(perfluoroalkanesulfonyl)amides with 100% F2 at −196 °C to 22 °C [45] (Scheme 17). All of these N-fluoroperfluoroalkanesulfonimides were stable over time at 22 °C, if stored in a fluoropolymer container. However, 7-1d decomposed at the melting point of 60 °C.
![[1860-5397-17-123-i17]](/bjoc/content/inline/1860-5397-17-123-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of N-F imides 7-1a–g.
Scheme 17: Synthesis of N-F imides 7-1a–g.
It was shown that N-fluorotrifluoromethanesulfonimide 7-1a (mp −69.8 °C) had a much higher reactivity than the sulfonamide reagents such as Barnette’s N-fluoro-N-alkylarenesulfonamides, since the electronic density on the nitrogen was greatly decreased by two strong electron-withdrawing CF3SO2 groups. Reagent 7-1a reacted slowly with benzene and toluene under neat conditions, whereas activated aromatics such as phenol, cresol, and naphthalene were fluorinated in chloroform at 22 °C (Scheme 18). The N-F imide reagent 7-1a fluorinated the sodium salt of diethyl 1-methylmalonate at −10 °C to give the corresponding fluoro product in high yield (96%).
![[1860-5397-17-123-i18]](/bjoc/content/inline/1860-5397-17-123-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Fluorination with (CF3SO2)2NF, 7-1a.
Scheme 18: Fluorination with (CF3SO2)2NF, 7-1a.
Later (1991 and 1992), the same laboratory reported fluorination reactions of functionalized carbonyl compounds, 1,3-dicarbonyl derivatives, olefins, and steroids with 7-1a [46-49] (Scheme 19).
![[1860-5397-17-123-i19]](/bjoc/content/inline/1860-5397-17-123-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Fluorination reactions of various substrates with 7-1a.
Scheme 19: Fluorination reactions of various substrates with 7-1a.
1-8. N-Fluoroquinuclidinium triflate
In 1988, Banks and co-worker developed the stable and nonhygroscopic N-fluoroquinuclidinium triflate (8-1) [50], which was an alternative to N-fluoroquinuclidinium fluoride (6-1) (Scheme 20). The triflate 8-1 was prepared in high yield by the counteranion replacement reaction developed by Umemoto and co-worker [28]. The fluorinating power of triflate 8-1 was the same as that of the fluoride 6-1, but its nonhygroscopic nature made it a useful fluorinating agent in terms of handling and storage. Details on the synthesis and reactivities of triflate 8-1 and other salts having CF3COO−, C3F7COO−, and BF4− counterions were reported in 1991 [51].
![[1860-5397-17-123-i20]](/bjoc/content/inline/1860-5397-17-123-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of N-F triflate 8-1.
Scheme 20: Synthesis of N-F triflate 8-1.
1-9. Optically active N-fluorosultams
In 1988, the first optically active N-F fluorinating agents, chiral N-fluorosultams, were synthesized by Lang and co-worker [52]. A camphor-derived imine was reduced or methylated, followed by direct fluorination (10% F2/N2) to give optically active N-F reagents 9-1 and 9-2 in 75% and 80% yield, respectively (Scheme 21). These N-fluorosultams were stable below 100 °C.
![[1860-5397-17-123-i21]](/bjoc/content/inline/1860-5397-17-123-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of chiral N-fluoro sultams 9-1 and 9-2.
Scheme 21: Synthesis of chiral N-fluoro sultams 9-1 and 9-2.
The enantioselectivities of products were examined after the fluorination of different metal enolates. In the best case a 63% yield and 70% enantiomeric excess (ee) was obtained. Even though other products gave less satisfactory outcomes, the potential of the N-F fluorinating agents for an enantioselective fluorination was demonstrated (Scheme 22).
![[1860-5397-17-123-i22]](/bjoc/content/inline/1860-5397-17-123-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Fluorination with chiral N-fluoro sultams 9-1 and 9-2.
Scheme 22: Fluorination with chiral N-fluoro sultams 9-1 and 9-2.
1-10. N-Fluoro-3,3-dimethylbenzothiazole dioxide
In 1989, Lang and co-worker developed the saccharin-derived N-fluorosultam N-fluoro-3,3-dimethyl-2,3-dihydro-1,2-benzothiazole-1,1-dioxide (10-2) from the known precursor 10-1 (Scheme 23) [53]. A direct fluorination using 10% F2/N2 at −40 °C gave N-F reagent 10-2 in 74% yield. Another method, via the N-trimethylsilylation, was also reported but in low efficiency. This N-F reagent is a colorless, thermally stable (<200 °C) solid with a mp of 114–116 °C.
![[1860-5397-17-123-i23]](/bjoc/content/inline/1860-5397-17-123-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of saccharin-derived N-fluorosultam 10-2.
Scheme 23: Synthesis of saccharin-derived N-fluorosultam 10-2.
The reagent 10-2 having no α-proton to the N-F site proved to be a good choice for fluorinating enolate anions (Scheme 24). The side reaction, involving HF elimination, and which was a problem in reactions with the Barnette’s reagents 4-1 having the α-proton(s) except for 4-1b [26], was avoided here. The HF elimination is a decomposition process that is observed with N-F reagents that have an α-proton and occurs under strong base conditions.
![[1860-5397-17-123-i24]](/bjoc/content/inline/1860-5397-17-123-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Fluorination with N-fluorosultam 10-2.
Scheme 24: Fluorination with N-fluorosultam 10-2.
1-11. Perfluoro[N-fluoro-N-(4-pyridyl)methanesulfonamide]
In 1990, Banks and co-worker reported perfluoro[N-fluoro-N-(4-pyridyl)methanesulfonamide] (11-2) [54]. Starting from pentafluoropyridine, the precursor 11-1 was prepared as illustrated in Scheme 25. Treatment of 11-1 with neat F2 in acetonitrile at −10 °C under reduced pressure gave N-fluoro-sulfonamide 11-2 in 89% yield. This product was however a 9:1 mixture of the N-F reagent 11-2 and the protonated compound of 11-1.
![[1860-5397-17-123-i25]](/bjoc/content/inline/1860-5397-17-123-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Synthesis of N-F reagent 11-2.
Scheme 25: Synthesis of N-F reagent 11-2.
The fluorination of benzene and anisole under excess substrate conditions gave fluorobenzene and fluoroanisoles in 88% and 98% yield, respectively. The reaction with sodium diethyl phenylmalonate gave the fluorinated product in 93% yield (Scheme 26).
![[1860-5397-17-123-i26]](/bjoc/content/inline/1860-5397-17-123-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Fluorination with N-F reagent 11-2.
Scheme 26: Fluorination with N-F reagent 11-2.
1-12. N-Fluorolactams
A new class of N-F fluorinating agents, N-fluorolactams 12-1, was synthesized by Sathyamurthy et al. in 1990 [55]. These compounds had already been prepared by Grakauskas and co-worker in 1970 [56], but in low yields and the N-fluorolactams were not recognized as fluorinating agents at that time. For positron emission tomography (PET), Sathyamurthy et al. allowed these lactams to react with 0.05% 18F2/Ne in a freon and obtained the N-[18F]fluorolactams 12-1 in good yields (Scheme 27, entry 1). The 18F-transfer ability was demonstrated by fluorination reactions with various Grignard reagents in up to 51% yield (Scheme 27, entry 2). In the event the β-elimination of HF proved to be an obstacle for the fluorination of strong bases such as phenyllithium.
![[1860-5397-17-123-i27]](/bjoc/content/inline/1860-5397-17-123-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis and reaction of N-fluorolactams 12-1.
Scheme 27: Synthesis and reaction of N-fluorolactams 12-1.
1-13. N-Fluoro-o-benzenedisulfonimide
In 1991, Davis and co-worker reported N-fluoro-o-benzenedisulfonimide (NFOBS, 13-2) as a fluorination reagent. NFOBS was prepared by the direct fluorination of its corresponding sulfonimide 13-1 with 10% F2/N2 in the presence of NaF and in high yield (Scheme 28) [57].
![[1860-5397-17-123-i28]](/bjoc/content/inline/1860-5397-17-123-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
NFOBS is a stable and crystalline solid of mp 139–140 °C (dec) with good fluorinating power. Its usefulness was demonstrated through fluorination reactions with different types of carbanions (Scheme 29). For example, reactions with enolates gave α-fluorinated products in 65–100% yields. Azaenolates could be fluorinated in moderate yields. A fluorination reaction with phenylmagnesium bromide provided fluorobenzene in 80% yield, an outcome which was better than that of N-fluoropyridinium triflate 5-4j (Umemoto’s reagent, 58%) and N-fluorosulfonamide 4-1b (Barnette’s reagent, 50%). NFOBS was able to fluorinate 1,3-dimethoxybenzene under neat conditions, while attempts to fluorinate toluene and acetophenone failed. The details of these reactions and applications were described in a full paper later published in 1995 [58].
![[1860-5397-17-123-i29]](/bjoc/content/inline/1860-5397-17-123-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-14. N-Fluorobenzenesulfonimide (NFSI)
In 1991, N-fluorobenzenesulfonimide (NFSI, 14-2) was synthesized by Differding and co-worker [59]. It was prepared from benzenesulfonimide 14-1 in good yield by its reaction with 10% F2/N2 in acetonitrile at −40 °C (Scheme 30). NFSI is a stable and non-hygroscopic crystalline solid with a mp of 114–116 °C.
![[1860-5397-17-123-i30]](/bjoc/content/inline/1860-5397-17-123-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
NFSI was shown to fluorinate a variety of nucleophiles. As seen in Scheme 31, trimethylsilyl enol ethers, enolate anions of ketones and esters, and aryl- and vinyllithiums were fluorinated with NFSI in moderate to high yields. Although aromatics such as anisole, toluene, and acetanilide could also be fluorinated by NFSI, these reactions required neat conditions and high temperatures, indicating that NFSI was not so powerful.
![[1860-5397-17-123-i31]](/bjoc/content/inline/1860-5397-17-123-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-15. N-Fluorosaccharin and N-fluorophthalimide
In 1991, Gakh et al. reported the synthesis of N-fluorosaccharin (15-1) and N-fluorophthalimide (15-2) in their studies on the reactivity of cesium fluoroxysulfate (Cs+−OSO2OF) [60]. Sodium salts of saccharin and phthalimide reacted with cesium fluoroxysulfate in acetonitrile at 0–5 °C to give 15-1 and 15-2 in 69% and 48% yields, respectively (Scheme 32). However, they did not report any fluorination reactions with 15-1 and 15-2. A transfer reaction failed to generate N-fluorosuccinimide from the reaction of the sodium salt of succinimide with cesium fluoroxysulfate.
![[1860-5397-17-123-i32]](/bjoc/content/inline/1860-5397-17-123-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Synthesis of N-fluorosaccharin (15-1) and N-fluorophthalimide (15-2).
Scheme 32: Synthesis of N-fluorosaccharin (15-1) and N-fluorophthalimide (15-2).
1-16. 1-Alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts
In 1992, Banks et al. reported a new series of N-fluoro diquaternary ammonium salts, 1-alkyl-4-fluoro-1,4-diazoniabicylco[2.2.2]octane salts 16-3a–d (Scheme 33) [42]. The salts 16-3 were synthesized in high yields after alkylation of 1,4-diazabicyclo[2.2.2]octane (16-1), followed by the Umemoto’s fluorination/counteranion replacement reaction of the resulting monoquaternary ammonium salts 16-2 with 10% F2/N2 in acetonitrile at −40 to −20 °C. The resultant salts 16-3 proved to be stable, non-hygroscopic, crystalline solids with high fluorinating power. The salt 16-3a was chosen as the commercial reagent, SelectfluorTM, from a cost-effectiveness viewpoint.
![[1860-5397-17-123-i33]](/bjoc/content/inline/1860-5397-17-123-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
The fluorinating power of reagents 16-3 strengthened as the electronegativity of the R group increased in the order of CH3 < CH2Cl < CH2CF3. The reagents 16-3 were able to fluorinate enol and conjugated enol acetates of steroids, sodium malonates, enamines, Grignard reagents, and aromatic compounds under mild conditions. In each case Selectfluor gave the corresponding fluorinated products in good to high yields (Scheme 34).
![[1860-5397-17-123-i34]](/bjoc/content/inline/1860-5397-17-123-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Fluorination with N-F salts 16-3.
Scheme 34: Fluorination with N-F salts 16-3.
As can be seen in Figure 3 and Figure 4, two years later (1994), Banks et al. reported mono- and difluorinations of various 1,3-dicarbonyl compounds using Selectfluor (16-3a) [61].
![[1860-5397-17-123-3]](/bjoc/content/figures/1860-5397-17-123-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Monofluorination with Selectfluor (16-3a).
Figure 3: Monofluorination with Selectfluor (16-3a).
![[1860-5397-17-123-4]](/bjoc/content/figures/1860-5397-17-123-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Difluorination with Selectfluor (16-3a).
Figure 4: Difluorination with Selectfluor (16-3a).
In 1995, the same group reported that Selectfluor reacted with quinuclidine to form N-fluoroquinuclidinium tetrafluoroborate in quantitative yield [62] (Scheme 35). They described this as a “transfer fluorination” since there was an intermolecular transfer of the fluorine atom of Selectfluor to the nitrogen of quinuclidine. In 1996, full details were published on the reactivities of all 16-3 reagents and the syntheses of 16-3 and intermediates 16-2 including additionally C2H5 and C8H17 as R group and PF6− and FSO3− as anion X− [63,64].
![[1860-5397-17-123-i35]](/bjoc/content/inline/1860-5397-17-123-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Transfer fluorination of Selectfluor (16-3a).
Scheme 35: Transfer fluorination of Selectfluor (16-3a).
The generous provision of free samples of Selectfluor after its commercialization resulted in many publications concerning its applications from a diversity of research groups and in a short time period (1992–1995): These included fluorinations of alkylbenzenes (entry 1, Scheme 36) [65], Grignard reagents (entry 2) [65], electron-rich alkenes (entry 3) [65,66], alkyl sulfides (entry 4) [65,67], 1,3-dicarbonyl compounds [65,68], phosphonate esters (entry 5) [65], steroidal silyl enol ethers and enol acetates (entry 6) [65], pyrimidine bases and nucleosides (entry 7) [67,69], phenylalkynes (entry 8) [70], anthraquinones (entry 9) [71], vinylstannanes (entry 10) [72], trialkylstannylindoles [73], and cyclopentadienylthallium (entry 11, Scheme 36) [74].
![[1860-5397-17-123-i36]](/bjoc/content/inline/1860-5397-17-123-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Fluorination of substrates with Selectfluor (16-3a).
Scheme 36: Fluorination of substrates with Selectfluor (16-3a).
1-17. Optically active N-fluoro-2,10-(3,3-dichlorocamphorsultam)
In 1993, Davis et al. reported the synthesis of the second optically active N-fluoro-2,10-(3,3-dichlorocamphorsultam) 17-2 [75] (Scheme 37).
![[1860-5397-17-123-i37]](/bjoc/content/inline/1860-5397-17-123-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Synthesis of chiral N-fluoro-sultam 17-2.
Scheme 37: Synthesis of chiral N-fluoro-sultam 17-2.
The maximum enantioselectivity of enolates of β-ketoesters with (−)-9-1 or (+)-9-2, first prepared by Lang in 1988 (see section 1-9), was 70% ee. The asymmetric fluorination with (+)- or (−)-17-2 afforded up to 75% ee as indicated in Scheme 38. The dichloro reagent 17-2 gave higher yields than the non-chloro reagent 9-1 because of the greater reactivity of 17-2.
![[1860-5397-17-123-i38]](/bjoc/content/inline/1860-5397-17-123-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Asymmetric fluorination with chiral 17-2.
Scheme 38: Asymmetric fluorination with chiral 17-2.
1-18. Zwitterionic N-fluoropyridinium salts
In 1995, Umemoto and co-worker disclosed a zwitterionic N-fluoropyridinium salt system 18-2 which had a broad fluorinating power and high selectivity [76]. A series of N-fluoropyridinium-2-sulfonates with electron-withdrawing or -donating substituents were synthesized in high yields by fluorination of the corresponding pyridinium-2-sulfonates 18-1 with 10% F2/N2 in acetonitrile or a mixture of acetonitrile/water at −40 to −10 °C (Figure 5). In a few cases, a catalytic amount of triethylamine was used to improve the yields. The starting pyridinium-2-sulfonates possessing an electron-withdrawing group(s) were prepared in high yields from the reaction of the corresponding 2-chloropyridines with sodium sulfite. All of these N-F reagents are easy-to-handle and stable crystalline solids.
![[1860-5397-17-123-5]](/bjoc/content/figures/1860-5397-17-123-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Synthesis of Zwitterionic N-fluoropyridinium salts 18-2a–h.
Figure 5: Synthesis of Zwitterionic N-fluoropyridinium salts 18-2a–h.
The nature of the lipophilic alkyl or trifluoromethyl substituents had a significant effect on the reactivity. Previously, N-fluoropyridinium-2-sulfonate and its 6-chloro derivative had been synthesized and shown to have excellent selectivity in fluorination reactions, but these reagents exhibited a low reactivity due to their low solubility in organic solvents [32].
Their fluorinating power increased in the order of 18-2a < 2b ≈ 2c ≈ 2d ≈ 2e < 2f < 2g < 2h, consistent with the order of the pKa values of the pyridines (Scheme 39). The least powerful 18-2a was suitable for the fluorination of reactive carbanions and easily oxidizable sulfides, whereas the most powerful 18-2h was suitable for less-reactive substrates such as olefins, aromatics, and neutral active methylene compounds. N-Fluoro-6-(trifluoromethyl)pyridinium-2-sulfonate (18-2f’) was prepared later [77].
![[1860-5397-17-123-i39]](/bjoc/content/inline/1860-5397-17-123-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Fluorinating power order of zwitterionic N-fluoropyridinium salts.
Scheme 39: Fluorinating power order of zwitterionic N-fluoropyridinium salts.
As shown in Scheme 40, the 18-2 pyridinium series proved to be effective fluorinating agents for a wide range of substrates. Moreover, an extremely high ortho-selectivity was observed in the fluorination of phenol. This could be attributed to a hydrogen-bonding interaction between the sulfonate anion and the phenol hydroxy group in the transition state. This assumption could also explain the high o-selectivity obtained in the fluorinations of naphthol, phenylurethane, and trimethylsilyl ether of phenol, and the exclusive 6-selectivity observed in the fluorination of conjugated enol triisopropylsilyl ethers of steroids. An additional advantage of the 18-2 reagents is that, after the fluorinations, the resulting pyridine-sulfonic acids are easily removed in an aqueous work-up.
![[1860-5397-17-123-i40]](/bjoc/content/inline/1860-5397-17-123-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Fluorination with zwitterionic 18-2.
Scheme 40: Fluorination with zwitterionic 18-2.
As illustrated in Scheme 41, triflic acid was shown to promote the fluorination of anisole with 18-2h. While the reaction time was ≈30 h without the additive (entry 1, Scheme 41), 1 equiv of TfOH shortened this to less than 1 h (entry 2). Therefore, it would appear that this additive acted as a catalyst. With 0.1 equiv of TfOH, the reaction time was 5.5 h (entry 3) which can be attributed to the strong electron-withdrawing effect of the 2-SO3H substituent formed by the protonation of the sulfonate anion in 18-2h.
![[1860-5397-17-123-i41]](/bjoc/content/inline/1860-5397-17-123-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Activation of salt 18-2h with TfOH.
Scheme 41: Activation of salt 18-2h with TfOH.
1-19. 1-Fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane salts (NFTh)
In 1995, Poss and Shia disclosed the synthesis and fluorination reactivity of 1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (19-2, NFTh/AccufluorTM) in a patent [78]. According to the patent, NFTh was prepared by fluorination of 1,4-diazabicyclo[2.2.2]octane N-oxide (19-1) in acetonitrile or water with 10% F2/N2 in the presence of HBF4, NaBF4, BF3 etherate, and/or BF3 at −35 °C or 8–9 °C. Scheme 42 shows the preparation of NFTh, a white solid that decomposes at 125 °C.
![[1860-5397-17-123-i42]](/bjoc/content/inline/1860-5397-17-123-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
The fluorination scope of NFTh is shown in Scheme 43 [79,80]. NFTh reacted with aromatics, enols, ketones, activated olefins, and substrates with active methylene groups to produce the corresponding fluorinated products in good to high yields. NFTh is a powerful fluorinating agent comparable to Selectfluor. Since NFTh has an acidic proton, its effectiveness is curtailed in the case of anionic substrates.
![[1860-5397-17-123-i43]](/bjoc/content/inline/1860-5397-17-123-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-20. N-Fluorobenzo-oxathiazone dioxide
In 1995, Cabrera and co-worker claimed 3-fluorobenzo-1,2,3-oxathiazin-4-one 2,2-dioxide (20-2) to be a useful fluorinating agent; 20-2 is a stable crystalline compound that was prepared in 83% yield by fluorination of its precursor 20-1 with 5% F2/N2 in acetonitrile and in the presence of NaF at −40 °C [81] (Scheme 44). They also tried to prepare N-fluorosaccharin (20-3) from saccharin with the F2/N2 system, but failed. Reagent 20-3 was synthesized using cesium fluoroxysulfate in 1991 (see section 1-15).
![[1860-5397-17-123-i44]](/bjoc/content/inline/1860-5397-17-123-i44.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 44: Synthesis of 3-fluorobenzo-1,2,3-oxathiazin-4-one 2,2-dioxide (20-2).
Scheme 44: Synthesis of 3-fluorobenzo-1,2,3-oxathiazin-4-one 2,2-dioxide (20-2).
Reagent 20-2 proved useful for the fluorination of both neutral and anionic nucleophiles under mild conditions. Scheme 45 illustrates some pertinent examples. Phenyl Grignard reagent, active methylene compounds, and a conjugated enol acetate of a steroid were all fluorinated in moderate to high yields. The fluorination of anisole required high temperature and neat conditions suggesting that the fluorination power of 20-2 is not so high.
![[1860-5397-17-123-i45]](/bjoc/content/inline/1860-5397-17-123-i45.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-21. Perfluoro[N-fluoro-N-(4-pyridyl)acetamide]
In 1996, the Banks group reported perfluoro[N-fluoro-N-(4-pyridyl)acetamide] (21-3) as a carboxamide analogue of perfluoro[N-fluoro-N-(4-pyridyl)methanesulfonamide] (11-2, see section 1-11) [82]. Its precursor, 21-2, was prepared from pentafluoropyridine by either one of two methods (Scheme 46). Precursor 21-2 was treated with neat F2 at 10–20 mmHg pressure in acetonitrile at −35 °C to produce the N-F carboxamide 21-3 in 75% yield but the resulting product was a 79:18 mixture of the desired N-F product 21-3 and the protonated compound 21-1.
![[1860-5397-17-123-i46]](/bjoc/content/inline/1860-5397-17-123-i46.svg?scale=2.0&max-width=1024&background=FFFFFF)
As a reagent N-F carboxamide 21-2 fluorinated electron-rich substrates such as sodium diethyl (phenyl)malonate, 1-morpholinocyclohexene, phenol, and anisole (Scheme 47). The fluorination power of the carboxamide 21-2 was less than that of its N-F sulfonamide analog 11-2.
![[1860-5397-17-123-i47]](/bjoc/content/inline/1860-5397-17-123-i47.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 47: Fluorination with N-F amide 21-2.
Scheme 47: Fluorination with N-F amide 21-2.
1-22. N,N’-Difluoro-1,4-diazoniabicyclo[2.2.2]octane salts
In 1996, Umemoto and co-worker reported the successful synthesis of various N,N’-difluoro-1,4-diazoniabicyclo[2.2.2]octane salts 22-1a–f in a pure form and in good to excellent yields (Scheme 48) [83]. All the salts were fully identified by elemental analysis and spectral analysis. Previously, in 1992, Banks and co-workers described how attempts to synthesize 22-1 proved to be unsatisfactory [42] and in 1995, they reported their synthesis characterized only by 19F NMR and described that the salts were moisture-sensitive [84].
![[1860-5397-17-123-i48]](/bjoc/content/inline/1860-5397-17-123-i48.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 48: Synthesis of N,N’-difluorodiazoniabicyclo[2.2.2]octane salts 22-1.
Scheme 48: Synthesis of N,N’-difluorodiazoniabicyclo[2.2.2]octane salts 22-1.
The synthesis is sensitive to solvents and the amount of Brønsted acids (HX). N,N’-Difluoro-1,4-diazoniabicyclo[2.2.2]octane bistriflate (22-1a), bis(hydrogensulfate) 22-1b, hydrogensulfate bishydrogenfluoride fluoride 22-1c, bishexafluoroantimonate 22-1e, and bishexafluorophosphate 22-1f were prepared in a pure form by fluorination of 1,4-diazabicyclo[2.2.2]octane (DABCO) with 10% F2/N2 in hexafluoroisopropanol in the presence of two molar equivalents or fewer of HX (one-step method, Scheme 48). The bisSbF6 salt 22-1e and bisPF6 22-1f could also be prepared in a pure form by using acetonitrile as the solvent. However, bisBF4 salt 22-1d could not be satisfactorily synthesized by this one-step protocol because an inseparable mixture of 22-1d and the DABCO·2HBF4 salt formed.
BisBF4 22-1d was considered to be commercially valuable because of its low preparative cost. Umemoto and co-worker found a clean counteranion replacement reaction of the intermediate (F−)x(HF)y(HSO4−)z salts (x + z = 2) by using BF3 etherate and thus solved the problem with a simple two-step one-pot method; (step 1) fluorination of a mixture of DABCO and 1.5 molar equivalents of H2SO4 with 10% F2/N2 in acetonitrile at −20 °C, and (step 2) treatment of the resulting reaction mixture with 2.1 molar equivalents of BF3 etherate at −20 °C to room temperature, gave pure 22-1d as a white precipitate (Scheme 49). By applying this telescoped method, pure 22-1d was easily obtained in 88% yield only after simple filtration of the reaction mixture and no further purification was needed. Pure 22-1d is non-hygroscopic and forms a stable crystalline solid with a high decomposition point of ca. 170 °C.
![[1860-5397-17-123-i49]](/bjoc/content/inline/1860-5397-17-123-i49.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 49: One-pot synthesis of N,N’-difluoro-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate salt (22-1d).
Scheme 49: One-pot synthesis of N,N’-difluoro-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate salt (22-1d...
The bisOTf salt 22-1a, bisBF4 22-1d, bisSbF6 22-1e, and bisPF6 22-1f are easy-to-handle because they are non-hygroscopic and stable crystals. As shown in Figure 6, 22-1a,d,e mediated a quantitative conversion of anisole to isomers of fluoroanisole at room temperature after 15 min, whereas Selectfluor (16-3a) only produced fluoroanisole with a 19% conversion. This demonstrated that 22-1 is a much more powerful reagent than Selectfluor, a power that is attributed to the high electronegativity of the fluorine atom compared to the ClCH2 group.
![[1860-5397-17-123-6]](/bjoc/content/figures/1860-5397-17-123-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fluorination of anisole with 22-1a, d, e.
Figure 6: Fluorination of anisole with 22-1a, d, e.
By using the bistetrafluoroborate salt 22-1d, many substrates such as aromatics, active methylene compounds and their salts, olefins, and enol acetates were efficiently fluorinated (Scheme 50). It was shown also that only one of the two N–F groups of 22-1 is used for C-fluorination, while the other N–F plays an activating role.
![[1860-5397-17-123-i50]](/bjoc/content/inline/1860-5397-17-123-i50.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 50: Fluorination with N,N’-diF bisBF4 22-1d.
Scheme 50: Fluorination with N,N’-diF bisBF4 22-1d.
1-23. Bis-N-fluoro reagents derived from precursors containing two heterocycles
In 1997, the Banks group reported bis-N-fluoro reagents 23-1–3 and related salts 23-4,5 by the fluorination of precursors containing two heterocycles (Scheme 51) [85]. Reagents 23-1–3 and -5 were obtained in good yields and 23-4 was also obtained, but as an impure product. The reagents 23-1 and 23-2 are bis-Selectfluor-type reagents and 23-3 is a bis-N-fluoropyridinium reagent. The fluorinating ability of reagents 23-2, -4, and -5 were examined as shown in Scheme 52.
![[1860-5397-17-123-i51]](/bjoc/content/inline/1860-5397-17-123-i51.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 51: Synthesis of bis-N-F reagents 23-1–5.
Scheme 51: Synthesis of bis-N-F reagents 23-1–5.
![[1860-5397-17-123-i52]](/bjoc/content/inline/1860-5397-17-123-i52.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-24. N,N’-Difluorobipyridinium salts
In 1998, the Umemoto group reported a new series of N,N’-difluorobipyridinium salts 24-2, N,N’-difluoro-2,2’-, -2,4’-, -3,3’-, and -4,4’-bipyridinium salts. These were synthesized by the direct fluorination of bipyridines 24-1 with 10–20% F2/N2 in the presence of a Lewis acid, a Brønsted acid, or its metal salt in acetonitrile or as a 50:1 mixture of acetonitrile/formic acid at −40 to 0 °C. The yields were good to excellent [86] (Figure 7). The trimer 24-3 and polymer homologues 24-4 were also prepared. The N,N’-difluorobipyridinium salts are stable and generally furnished non-hygroscopic free flowing materials, however, this was less the case for those derivatives with electron-withdrawing substituents.
![[1860-5397-17-123-7]](/bjoc/content/figures/1860-5397-17-123-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Synthesis of N,N’-difluorobipyridinium salts 24-2.
Figure 7: Synthesis of N,N’-difluorobipyridinium salts 24-2.
Controlled fluorination reactions with these reagents (Figure 8) clearly showed a power order of 2,2’- (24-2a) >> 2,4’- (24-2c) > 3,3’- (24-2f) ≈ 4,4’- (24-2g) >> N-fluoropyridinium triflate (Scheme 53), which is again in good agreement with the pKa values of the bipyridines. The 2,2’-isomers had the highest power (reactivity) among the dipyridinium isomers, and they were much more reactive than the monomeric N-fluoropyridinium triflate (5-4a).
![[1860-5397-17-123-8]](/bjoc/content/figures/1860-5397-17-123-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Controlled fluorination of N,N’-diF 24-2.
Figure 8: Controlled fluorination of N,N’-diF 24-2.
![[1860-5397-17-123-i53]](/bjoc/content/inline/1860-5397-17-123-i53.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 53: Fluorinating power of N,N’-diF salts 24-2 and N-F salt 5-4a.
Scheme 53: Fluorinating power of N,N’-diF salts 24-2 and N-F salt 5-4a.
In contrast to the N,N’-difluoro-1,4-diazoniabicyclo[2.2.2]octane salts 22-1, both of the N-F moieties in 24-2 were effective in fluorinations and they reacted sequentially. N,N’-Difluoro-2,2’-bipyridinium bis(tetrafluoroborate) (24-2b, X = BF4, SynFluorTM, MEC-31) was chosen as a representative of this class to assess fluorination capability because of its low production cost and high fluorine content per molecule. As a result, SynFluorTM (24-2b) proved to be a practical reagent for the fluorination of many substrates affording the products in good to high yields (Scheme 54 and Scheme 55). SynFluor can be considered a powerful fluorinating agent with a high fluorine content, F: 103 g/1 kg (comparison; 54 g/1 kg for Selectfluor).
![[1860-5397-17-123-i54]](/bjoc/content/inline/1860-5397-17-123-i54.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 54: Fluorination reactions with SynfluorTM (24-2b).
Scheme 54: Fluorination reactions with SynfluorTM (24-2b).
![[1860-5397-17-123-i55]](/bjoc/content/inline/1860-5397-17-123-i55.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 55: Additional fluorination reactions with SynfluorTM (24-2b).
Scheme 55: Additional fluorination reactions with SynfluorTM (24-2b).
1-25. N-Fluoro-2,4-dinitroimidazole
In 1998, Laali et al. reported the fluorination of polycyclic aromatic hydrocarbons (PAHs) with N-fluoro-2,4-dinitroimidazole (25-1) [87] (Scheme 56). This reagent is a white solid that was prepared by Forohar et al. through fluorination of 2,4-dinitroimidazole (5% F2 in N2 at −40 °C) [88]. Although additional information on 25-1 was not available, it was noticed [87] that the polynitro compound 25-1 is potentially dangerous towards detonation.
![[1860-5397-17-123-i56]](/bjoc/content/inline/1860-5397-17-123-i56.svg?scale=2.0&max-width=1024&background=FFFFFF)
Laali et al. attempted to fluorinate some polycyclic aromatics with Selectfluor, but intractable mixtures were obtained. Thus, they examined the utility of 25-1 in the fluorination of polycyclic aromatics. Although a detailed optimization was conducted, the fluorination of over 20 polycyclic aromatics was poor giving products in very low yields, varying from 27% to 3% depending on the substrate. The fluorinations were accompanied by tar formation and dimerization. Scheme 57 illustrates some representative examples.
![[1860-5397-17-123-i57]](/bjoc/content/inline/1860-5397-17-123-i57.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 57: Fluorination of polycyclic aromatics with 25-1.
Scheme 57: Fluorination of polycyclic aromatics with 25-1.
1-26. Perfluoro-(N-fluoro-2,2,6,6-tetramethylpiperidine) and its 2,6-dimethyl analogue
In 1999, the Banks group reported the syntheses of perfluoro-(N-fluoro-2,2,6,6-tetramethylpiperidine) (26-1) and its 2,6-dimethyl analogue 26-2, reagents which do not have α-fluorine atoms adjacent to the N-F site [89] (Scheme 58). These reagents overcame the drawback of perfluoro-N-fluoropiperidine (1-1) which does contain α-fluorine atoms (see section 1-1).
![[1860-5397-17-123-i58]](/bjoc/content/inline/1860-5397-17-123-i58.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 58: Synthesis of 26-1 and dimethyl analog 26-2.
Scheme 58: Synthesis of 26-1 and dimethyl analog 26-2.
The fluorinating ability of reagents 26-1 and -2 was tested in fluorinations of the sodium salt of a keto ester (Scheme 59). Reagents 26-1 and -2 were superior to 1-1 and 26-3 where the α-fluorine atoms can be eliminated after fluorine-transfer from the N-F site.
![[1860-5397-17-123-i59]](/bjoc/content/inline/1860-5397-17-123-i59.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 59: Fluorination with reagents 26-1, 26-2, 1-1, and 26-3.
Scheme 59: Fluorination with reagents 26-1, 26-2, 1-1, and 26-3.
1-27. N-Fluoro-3-ethyl-3-methyldioxobenzothiazinone and (R)- and (S)-N-fluoro-3-cyclohexyl-3-methylbenzoisothiazole dioxide
In 1999, Takeuchi et al. reported N-fluoro-3-ethyl-3-methyl-1,1-dioxo-2,3-dihydro-1H-1λ6-benzo[e]1,2-thiazin-4-one (27-2) as an efficient reagent for the fluorination of carbanions [90]. The precursor 27-1, which was prepared in a three step protocol from saccharin, was fluorinated with FClO3 to give 27-2 in good yield (Scheme 60). Direct fluorination of 27-1 with 10% F2/N2 had failed because of decomposition. Various ketone enolates were successfully fluorinated with reagent 27-2 in good to high yields [90].
![[1860-5397-17-123-i60]](/bjoc/content/inline/1860-5397-17-123-i60.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 60: Synthesis of N-F reagent 27-2.
Scheme 60: Synthesis of N-F reagent 27-2.
In addition, Takeuchi et al. reported that optically active N-fluorosultams, (R)- and (S)-N-fluoro-3-cyclohexyl-3-methyl-2,3-dihydrobenzo[1,2-d]isothiazole 1,1-dioxides 27-6 (Scheme 61) were efficient reagents for the asymmetric fluorination of enolates [91].
![[1860-5397-17-123-i61]](/bjoc/content/inline/1860-5397-17-123-i61.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 61: Synthesis of chiral N-F reagents 27-6.
Scheme 61: Synthesis of chiral N-F reagents 27-6.
To showcase his methodology, they first synthesized acyclic optically active N-fluoro sulfonamides 27-7–9 (Scheme 62) and attempted the enantioselective fluorination of some enolates in 1997 [92]. However, the best result was an enantiomeric excess of 48% with a chemical yield of 53% after the enolate anion of 2-benzyl-α-tetralone was treated with 27-8.
![[1860-5397-17-123-i62]](/bjoc/content/inline/1860-5397-17-123-i62.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 62: Synthesis of chiral N-F 27-7–9.
Scheme 62: Synthesis of chiral N-F 27-7–9.
As summarized in Scheme 61, Takeuchi et al. prepared the enantiomers of cyclic N-F sulfonamide 27-6 by the approach used by Lang for N-fluorosultams [53]. A cyclohexyl ring was introduced nucleophilically into imine 27-3 and the resulting 27-4 was treated with (−)-menthoxyacetyl chloride, followed by separation of the diastereomers. The chiral auxiliary was then removed with LiOH and the resulting sultams 27-5 as single enantiomers were fluorinated with 15% F2/He in the presence of KF to produce optically pure N-fluorosultam reagents (R)- and (S)-27-6.
Enantioselective fluorinations of typical enolates were then performed (Scheme 63). The (R)-27-6 reagent gave up to 79% yield and 88% enantiomeric excess in the case of 2-benzyl-α-tetralone.
![[1860-5397-17-123-i63]](/bjoc/content/inline/1860-5397-17-123-i63.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 63: Asymmetric fluorination with 27-6.
Scheme 63: Asymmetric fluorination with 27-6.
1-28. (R)- and (S)-N-fluoro-3-tert-butyl-7-nitro-benzothiazine 1,1-dioxides and spiro-type analogues
In 2000, the Takeuchi group reported their second series of chiral N-fluorosultam reagents, (R)- and (S)-N-fluoro-3-tert-butyl-7-nitro-3,4-dihydro-2H-benzo[e][1,2]thiazine 1,1-dioxides 28-3 [93] (Scheme 64). Racemic 28-1 reacted with (+)-10-camphorsulfonyl chloride to give a mixture of the diastereomers of 28-2, which was accompanied by 45% of unreacted 28-1. Interestingly, pure (R)-isomer 28-1 was obtained in 20% yield from the unreacted 28-1. The diastereomer separation of 28-2 was achieved by column chromatography to separate the (R)- and (S)-isomers of 28-2 in 21 and 31% yields, respectively. Deprotection followed by fluorination with FClO3 gave (R)- and (S)-28-3 in good yields. X-ray crystallography was used to determine the structure and confirm the absolute stereochemistry of (S)-28-3.
![[1860-5397-17-123-i64]](/bjoc/content/inline/1860-5397-17-123-i64.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 64: Synthesis of chiral N-F reagents 28-3.
Scheme 64: Synthesis of chiral N-F reagents 28-3.
The optically active reagents (R)- and (S)-28-3 were effective in the enantioselective fluorination of cyclic ketones, as is illustrated with representative in Scheme 65.
![[1860-5397-17-123-i65]](/bjoc/content/inline/1860-5397-17-123-i65.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 65: Asymmetric fluorination with 28-3.
Scheme 65: Asymmetric fluorination with 28-3.
In addition, the Takeuchi group reported a spiro-type analogue, (2'S,3R,5'R)-2-fluoro-2'-methylethyl-5'-methyl-2H,4H-spiro[benzo[e][1,2]thiazine-3,1'-cyclohexane]-1,1-dione (28-7a) [94], as their third chiral reagent in this sultam series. As can be seen in Scheme 66, 28-4 was treated with BuLi followed by reaction with optically active menthone to give 28-5, which was converted to enantiomers of spiro sultams 28-6a and 28-6b in high yield. The spiro sultams 28-6a and -6b were separated and each was fluorinated with FClO3 to give (2'S,3R,5'R)-isomer 28-7a and (2'S,3S,5'R)-isomer 28-7b in 81% and 44% yield, respectively.
![[1860-5397-17-123-i66]](/bjoc/content/inline/1860-5397-17-123-i66.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 66: Synthesis of chiral N-F reagents 28-7.
Scheme 66: Synthesis of chiral N-F reagents 28-7.
Figure 9 summarizes the outcomes of asymmetric fluorination reactions of enolates of aryl ketones using 28-7a and -7b. In the event isomer 28-7a yielded much better ees than 28-7b. Although 28-7a gave a maximum 70% ee (entry 6, Figure 9), it was less than that obtained (74% ee) by N-fluorosultam (R)-27-6 with the same substrate (see the previous section 1-27).
![[1860-5397-17-123-9]](/bjoc/content/figures/1860-5397-17-123-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Asymmetric fluorination with 28-7.
Figure 9: Asymmetric fluorination with 28-7.
1-29. N-Fluorinated cinchona alkaloid derivatives by combination with Selectfluor
In 2000, Shibata and Takeuchi reported a far more practical enantioselective fluorination method. They discovered that the fluorination of carbanions with Selectfluor occurred in a highly enantioselective manner when carried out in the presence of cinchona alkaloid derivatives [95]. This method consisted of two simple steps. Firstly, the cinchona alkaloid is reacted with Selectfluor in acetonitrile at room temperature for 1 h, and then this is followed by addition of the substrate. The resulting mixture is stirred at a suitable temperature. They proposed that Selectfluor transfers fluorine to the alkaloid to give a chiral N-F alkaloid species, in a manner that followed the fluorine transfer reported by Banks when quinuclidine was N-fluorinated with Selectfluor [62] (Scheme 67). Subsequently, in 2001, Shibata et al. presented full details of their studies including the definitive identification of N-fluorinated cinchona alkaloids by X-ray crystallography analysis and further applications [96]. This method proved to be far more practical than the enantiomeric sulfonamide-type N-F reagents developed until then.
![[1860-5397-17-123-i67]](/bjoc/content/inline/1860-5397-17-123-i67.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 67: In situ formation of N-fluorinated cinchona alkaloids with SelectfluorTM.
Scheme 67: In situ formation of N-fluorinated cinchona alkaloids with SelectfluorTM.
Many cinchona alkaloid derivatives were tested for their ability to fluorinate substrates such as cyclic and acyclic ketones and esters. Scheme 67 shows two typical examples of N-F cinchona alkaloid species, 29-2 and 29-4, formed from dihydroquinine 4-chlorobenzoate (DHQB, 29-1) and dihydroquinidine acetate (DHQDA, 29-3) with Selectfluor. As a result, high chemical yields and high enantiomeric excesses were obtained for many substrates as summarized in Scheme 68. The DHQB/Selectfluor combination was effective for the fluorination of silyl enol ethers of indanones and tetralones, forming the fluorinated products in up to 91% ee. The DHQDA/Selectfluor combination was effective also for acyclic esters, with outcomes up to 87% ee, and for cyclic keto esters, up to 80% ee. For oxindoles, the (DHQD)2PYR/Selectfluor combination was effective too, generating the products with up to 82% ee.
![[1860-5397-17-123-i68]](/bjoc/content/inline/1860-5397-17-123-i68.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 68: Asymmetric fluorination with N-F alkaloids formed in situ.
Scheme 68: Asymmetric fluorination with N-F alkaloids formed in situ.
1-30. Enantiopure N-fluorocinchona alkaloid salts
Only six days after the report by Shibata and Takeuchi appeared (Oct. 2000), Cahard et al. reported the synthesis and application of similar enantiopure N-fluoro salts of cinchona alkaloids through the reaction of the alkaloids with Selectfluor [97]. Following the precedent from Banks’s fluorine-transfer reaction from Selectfluor to the N-site of quinuclidine [62], Cahard et al. isolated four N-fluorocinchona alkaloid salts, F-CD-BF4 30-1, F-CN-BF4 30-2, F-QN-BF4 30-3, and F-QD-BF4 30-4 in good yields (Scheme 69). Soon after in 2001, they reported the X-ray structural analysis of F-CD-BF4 30-1 [98].
![[1860-5397-17-123-i69]](/bjoc/content/inline/1860-5397-17-123-i69.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 69: Synthesis of N-fluorocinchona alkaloids with Selectfluor.
Scheme 69: Synthesis of N-fluorocinchona alkaloids with Selectfluor.
The enantioselective fluorination of the sodium enolate of 2-methyl-1-tetralone was examined using these N-F alkaloid salts (Scheme 70). F-CD-BF4 30-1 gave the highest result with a 56% ee. F-CD-BF4 30-1 is a nonhygroscopic, free-flowing solid with a high decomposition point (189 °C, dec).
![[1860-5397-17-123-i70]](/bjoc/content/inline/1860-5397-17-123-i70.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 70: Asymmetric fluorination with 30-1–4.
Scheme 70: Asymmetric fluorination with 30-1–4.
Enantioselective fluorinations of the sodium enolates of other ketones and β-keto esters with F-CD-BF4 30-1 were also investigated. The trimethylsilyl enol ether of 2-methyl-1-tetralone was examined, too (up to 61%ee, 93% yield). These experiments gave the fluorinated products in excellent chemical yields but with lower enantiomeric excesses than the Shibata and Takeuchi’s protocol.
1-31. N-Fluoro-p-chlorobenzoylquinine salt: another N-F reagent of cinchona alkaloid
In 2003, Cahard et al. reported the transfer fluorination of a cinchona alkaloid, p-chlorobenzoylquinine (pClBzQN, 31-1) using not only SelectfluorTM (F-TEDA-BF4) but also other powerful N-F fluorinating agents such as 1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate (NFTh, 19-2), N-fluorobenzenesulfonimide (NFSI), and N-fluoro-2,6-dichloropyridinium tetrafluoroborate (5-4s) (Scheme 71) [99]. These reactions produced F-pClBzQN-X 31-2 quantitatively at 20 °C within 30 min in acetonitrile or in an ionic liquid such as 1-hexyl-3-methylimidazolinium hexafluorophosphate [hmim][PF6]. However, the less powerful reagents, N-fluoroquinuclidinium tetrafluoroborate, N-fluoro-N-methyl-p-toluenesulfonamide (4-1a), N-fluoropyridinium triflate (5-4a), N-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (5-4k), and N,N’-difluoro-2,2’-bipyridinium bistetrafluoroborate (24-2b) failed. Among the five effective fluorine-transfer reagents, the 2,6-dichloropyridinium salt 5-4s was more cost effective because it has a higher fluorine content (5-4s, 3.94 mmol/g) than the others (Selectfluor, 2.82 mmol/g; F-TEDA-OTf, 2.09 mmol/g; NFTh, 3.11 mmol/g; NFSI, 3.17 mmol/g).
![[1860-5397-17-123-i71]](/bjoc/content/inline/1860-5397-17-123-i71.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 71: Transfer fluorination from various N-F reagents.
Scheme 71: Transfer fluorination from various N-F reagents.
The enantioselective fluorination of F-pClBzQN-X 31-2 was examined on trimethylsilyl enol ethers of methyl- and benzylindanones 31-3 and -4 (Figure 10). High chemical yields and ees (88–97% and 81–85% ee) were obtained in the reactions of benzyl derivative 31-4 with F-pClBzQN-X [X = BF4 or N(SO2Ph)2] prepared in situ from the N-F reagents and pClBzQN (entries 4–8 in Figure 10).
![[1860-5397-17-123-10]](/bjoc/content/figures/1860-5397-17-123-10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Asymmetric fluorination of silyl enol ethers.
Figure 10: Asymmetric fluorination of silyl enol ethers.
1-32. N-Fluoro-2,4,6-trichloro-1,3,5-triazinium tetrafluoroborate
In 2003, Banks et al. reported the synthesis of N-fluoro-2,4,6-trichloro-1,3,5-triazinium tetrafluoroborate (32-2) and its fluorination of aromatic compounds [100]. Although the syntheses of N-fluoro-2,4,6-trifluoro-1,3,5-triazinium hexafluoroarsenate and N-fluoro-2,4,6-trichloro-1,3,5-triazinium hexafluoroarsenate had been reported a decade earlier (1993) [101], their fluorination ability had not been disclosed.
Salt 32-2 was prepared in high yield in a small-scale batch reactor (Scheme 72). Accordingly, 100% F2 (1 equiv) and BF3 (1 equiv) gas were condensed into a stainless steel autoclave cooled at −196 °C, which contained cyanuric chloride (32-1, 1 equiv) in CFCl3. This was followed by gradual warming to room temperature and the reaction mixture was stored for 5 days. The resultant salt 32-2 is a moisture-sensitive white solid that decomposes at 153–155 °C.
![[1860-5397-17-123-i72]](/bjoc/content/inline/1860-5397-17-123-i72.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 72: Synthesis of N-fluoro salt 32-2.
Scheme 72: Synthesis of N-fluoro salt 32-2.
Reagent 32-2 reacted with deactivated benzenes such as chlorobenzene and nitrobenzene at ambient temperature (Scheme 73). This outcome indicated that 32-2 was a more powerful fluorinating reagent than the N-fluoropentachloropyridinium salts 5-4v,w. However, reagent 32-2 is not easy-to-handle because of its moisture-sensitivity.
![[1860-5397-17-123-i73]](/bjoc/content/inline/1860-5397-17-123-i73.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 73: Reactivity of N-fluorotriazinium salt 32-2.
Scheme 73: Reactivity of N-fluorotriazinium salt 32-2.
1-33. Bulky N-fluorobenzenesulfonimides (NFBSI)
In 2011, Shibata et al. reported a bulky NFSI analog, N-fluoro-(3,5-di-tert-butyl-4-methoxy)benzenesulfonimide (NFBSI, 33-3) [102]. This stable and crystalline reagent was synthesized in 57% yield by fluorination of bis(3,5-di-tert-butyl-4-methoxybenzenesulfonyl)amide (33-2) with 10% F2/N2 in the presence of NaF in acetonitrile at −40 °C (Scheme 74). X-ray crystal structure analysis revealed that the fluorine atom of NFBSI was surrounded by four bulky tert-butyl substituents.
![[1860-5397-17-123-i74]](/bjoc/content/inline/1860-5397-17-123-i74.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 74: Synthesis of bulky N-fluorobenzenesulfonimide NFBSI 33-3.
Scheme 74: Synthesis of bulky N-fluorobenzenesulfonimide NFBSI 33-3.
This feature improved the enantioselectivity in cinchona alkaloid-catalyzed enantioselective fluorinations. The enantiomeric excesses of the products obtained with NFBSI 33-3 increased by 18% compared to that of (PhSO2)2NF (NFSI, 14-2), while the chemical yields decreased (Scheme 75). Since the active fluorination agent in these reactions is considered to be the in situ generated N-fluorocinchona alkaloid salt, after fluorine transfer from NFBSI or NFSI to generate a counter anion, the bulky in situ generated counter anion may help enhance the % ee of the products.
![[1860-5397-17-123-i75]](/bjoc/content/inline/1860-5397-17-123-i75.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-34. p-Substituted N-fluorobenzenesulfonimides
In 2012, Yang et al. reported an efficient process for the preparation of p-substituted N-fluorobenzenesulfonimides 34-3 [103]. The traditional process involved the direct fluorination of benzensulfonimides in acetonitrile in the presence of a large excess of NaF. They prepared the sodium salts 34-2 (precipitates) with 2% aq NaOH solution and treated the air-dried precipitates with 10% F2/N2 in acetonitrile (Scheme 76). This process considerably improved the yields of the N-fluorobenzenesulfonimides 34-3.
![[1860-5397-17-123-i76]](/bjoc/content/inline/1860-5397-17-123-i76.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 76: Synthesis of p-substituted N-fluorobenzenesulfonimides 34-3.
Scheme 76: Synthesis of p-substituted N-fluorobenzenesulfonimides 34-3.
In 2013, the same group reported the enantioselective fluorination of oxindoles 34-5 with 34-3 in the presence of a catalytic amount of bis-cinchona alkaloid (DHQD)2PHAL 34-4 (Figure 11) [104]. In these reactions, the actual enantioselective fluorinating agents should be the N-F cinchona alkaloid salts formed in situ after the transfer from 34-3. Electron-donating groups (R) afforded higher enantioselectivities compared to R = H (NFSI, 14-2), while chemical yields were decreased. For example, while NFSI afforded 56% ee and 66% chemical yield (entry 1, Figure 11), R = OMe (34-3a) and tert-butyl (34-3b) gave 94% ee and 96% ee (entries 2 and 3 in Figure 11), but with only 49% and 17% chemical yields, respectively. Electron-withdrawing groups, R = CF3 34-3c and OCF3 34-3d, failed to furnish the fluorinated product under the same conditions (entries 8 and 9, Figure 11).
![[1860-5397-17-123-11]](/bjoc/content/figures/1860-5397-17-123-11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 11: Asymmetric fluorination with 34-3 and a chiral catalyst 34-4.
Figure 11: Asymmetric fluorination with 34-3 and a chiral catalyst 34-4.
A year later (2014), the Yang group published the synthesis of other substituted NFSI derivatives and examined the enantioselective fluorination of 2-oxindoles catalyzed by chiral palladium complexes [105].
1-35. Two Selectfluor analogues, 1-fluoro-4-[3’,4’-bis(trifluoromethyl)phenylmethyl]- and -(pentafluorophenyl)methyl-1,4-diazoniabicyclo[2.2.2]octane salts
In March 2013, Toste et al. reported the enantioselective 1,4-fluoroamination of conjugated dienes using Selectfluor in an anionic, phase-transfer catalysis in a nonpolar solvent [106]. Diene 35-1 was treated with Selectfluor and (R)-TCYP 35-3 as a catalyst to give 35-2 in 91% chemical yield and 96% ee (Scheme 77). In this case, the actual fluorinating species was considered to be the salt containing 1-chloromethy-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane as the cation and (R)-TCYP as the anion, which could be formed in the non-polar organic layer.
![[1860-5397-17-123-i77]](/bjoc/content/inline/1860-5397-17-123-i77.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 77: 1,4-Fluoroamination with Selecfluor and a chiral catalyst.
Scheme 77: 1,4-Fluoroamination with Selecfluor and a chiral catalyst.
However, since the reaction with the less reactive diene 35-6a occurred in very low yield (<10%) (Figure 12, entry 1), they synthesized two new Selectfluor analogues, 1-fluoro-4-[3’,4’-bis(trifluoromethyl)phenylmethyl]- and -(pentafluorophenyl)methyl-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborates 35-5a and 35-5b, by treating the precursor 35-4 with XeF2 in the presence of sodium tetrafluoroborate [106] (Scheme 78).
![[1860-5397-17-123-12]](/bjoc/content/figures/1860-5397-17-123-12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 12: Asymmetric fluoroamination with 35-5a, b.
Figure 12: Asymmetric fluoroamination with 35-5a, b.
![[1860-5397-17-123-i78]](/bjoc/content/inline/1860-5397-17-123-i78.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 78: Synthesis of Selectfluor analogs 35-5a, b.
Scheme 78: Synthesis of Selectfluor analogs 35-5a, b.
Reagent 35-5a showed much improved chemical yields (65–75%) and enantioselectivities (73–89% ee) in fluorocyclization reactions with diene 35-6 (Figure 12, entries 2, 4, and 5) than reagent 35-5b. Although there may be an electron-deficiency effect, this could also be attributed to the strong lipophilic effect of the bis(CF3)phenyl group in 35-5a to the organic layer (PhCF3).
1-36. Chiral dicationic DABCO-based N-F reagents
In June 2013, Gouverneur et al. reported the synthesis of chiral dicationic DABCO-based N-F reagents 36-5 that made possible the asymmetric electrophilic fluorocyclization of monoolefins with carbon nucleophiles [107]. This can be contrasted with Toste’s method, described in section 1-35 above, for asymmetric fluorocyclization of dienes with nitrogen nucleophiles using Selectfluor and an optically active phase-transfer catalyst, a reaction which did not work for monoolefins with carbon nucleophiles.
As shown in Scheme 79, the chiral DABCO core 36-3 was prepared from an enantiopure vicinal diamine 36-1 using the reported method. The precursor 36-4 was fluorinated with either 10% F2/N2 or N-fluoropentachloropyridinium triflate (5-1v) to produce optically active products 36-5a, -5b, and -5c in high yields. This fluorination which uses the shelf-stable, easy-to-handle N-fluoropentachloropyridinium triflate (5-1v) was advantageous as it allowed the in situ formation of the chiral reagents 36-5.
![[1860-5397-17-123-i79]](/bjoc/content/inline/1860-5397-17-123-i79.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 79: Synthesis of chiral dicationic DABCO-based N-F reagents 36-5.
Scheme 79: Synthesis of chiral dicationic DABCO-based N-F reagents 36-5.
The solubility, reactivity, and enantioselectivity of these types of reagents could be tuned by varying the substituents on the aryl rings and the p-trifluoromethyl-derivative 36-5b proved to be the most efficient for this type of reaction (Scheme 80). Up to 99% chemical yield and an average of 71% ee were obtained for a series of indene derivatives. However, the enantioselectivity was low (19% ee) for the case of a dihydronaphthalene derivative (the bottom in Scheme 80). Racemates of these fluoro products were prepared in high yields with N-fluoro-2,6-dichloropyridinium triflate (5-1r).
![[1860-5397-17-123-i80]](/bjoc/content/inline/1860-5397-17-123-i80.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 80: Asymmetric fluorocyclization with chiral 36-5b.
Scheme 80: Asymmetric fluorocyclization with chiral 36-5b.
1-37. Chiral N-fluorobinaphthyldisulfonimides
In June 2013, the same month as the report described above from Gouverneur’s lab, Shibata, Ma, and Cahard disclosed a chiral N-fluorobinaphthyldisulfonimide 37-2a and its bis[3,5-bis(trifluoromethyl)phenyl] derivative 37-2b as enantioselective fluorination agents, compounds which were prepared in 67% and 27% yields, respectively, by fluorination of the NH precursors 37-1 with 0.2% F2/N2 (Scheme 81) [108]. In both cases, side-products were formed but not characterized. The precursors 37-1 had been synthesized by List [109] and Giernoth [110] and previously used as organocatalysts.
![[1860-5397-17-123-i81]](/bjoc/content/inline/1860-5397-17-123-i81.svg?scale=2.0&max-width=1024&background=FFFFFF)
The fluorination ability of 37-2a and -2b was evaluated with β-keto ester derivatives (Scheme 82). Moderate yields and good enantioselectivities of the fluorinated products were obtained. Reagent 37-2b clearly gave a better selectivity than the unsubstituted 37-2a (86% ee vs 17% ee).
![[1860-5397-17-123-i82]](/bjoc/content/inline/1860-5397-17-123-i82.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 82: Asymmetric fluorination with chiral 37-2a,b.
Scheme 82: Asymmetric fluorination with chiral 37-2a,b.
For an enantioselective synthesis of the potent Maxi-K potassium channel opener MaxPost 37-4 (R = H), the N-Boc-protected substrate 37-3 was fluorinated with 37-2b, giving the precursor 37-4 (R = Boc) in 51% yield and with 57% ee (Scheme 83).
![[1860-5397-17-123-i83]](/bjoc/content/inline/1860-5397-17-123-i83.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 83: Asymmetric fluorination with chiral 37-2b.
Scheme 83: Asymmetric fluorination with chiral 37-2b.
The oxidative aminofluorination of indene (37-5) with 37-2a and -2b gave four diastereomers 37-6a and -6b in 77 and 76% yield, respectively; however, no asymmetric induction was observed (Scheme 84).
![[1860-5397-17-123-i84]](/bjoc/content/inline/1860-5397-17-123-i84.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 84: Reaction of indene with chiral 37-2a,b.
Scheme 84: Reaction of indene with chiral 37-2a,b.
1-38. N-Fluoromethanesulfonimide (Me-NFSI)
In 2016, Shibata et al. reported the synthesis and reactivity of N-fluoromethanesulfonimide (Me-NFSI, 38-2) [111]. Me-NFSI was first reported in a patent in 1994 [112], however, the reported fluorinations were vague. Prior to Shibata's report, Me-NFSI had not appeared in the literature in over 20 years. Although Me-NFSI is vulnerable with acidic protons on the methyl groups, Shibata realized the high atom economy of Me-NFSI and claimed that Me-NFSI was more effective for the fluorination of active methines than (PhSO2)2NF (NFSI, 14-2) under Lewis acid-catalysis and non-catalysis. Me-NFSI was prepared in good yield by the fluorination of methanesulfonimide 38-1 with 10% F2/N2 in acetonitrile at −40 °C in the presence of NaF (Scheme 85).
![[1860-5397-17-123-i85]](/bjoc/content/inline/1860-5397-17-123-i85.svg?scale=2.0&max-width=1024&background=FFFFFF)
Many active methine compounds were fluorinated in high yields using Me-NFSI in the presence of 10 mol % Ti(OiPr)4 in methylene chloride as a solvent at room temperature (Scheme 86). The reactions with Me-NFSI were faster than those with NFSI. In particular, the fluorination of sterically demanding tert-butyl keto ester 38-3b with Me-NFSI gave the product 38-4b in 90% yield within only 5 min (92% in 3 h), while NFSI gave the product in 20% yield after 10 min and the yield gradually increased to 80% over 24 h. The faster reaction of Me-NFSI was explained by the activation of its N-F moiety via the stronger complexation with the Lewis acid Ti(OiPr)4 than NFSI.
![[1860-5397-17-123-i86]](/bjoc/content/inline/1860-5397-17-123-i86.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 86: Fluorination of active methine compounds with Me-NFSI.
Scheme 86: Fluorination of active methine compounds with Me-NFSI.
For the fluorination of malonates, different conditions were needed. Treatment with 2 equiv of Me-NFSI in the presence of 20 mol % Ti(OiPr)4 in toluene at reflux temperature gave the fluorinated malonates in satisfactory yields (Scheme 87).
![[1860-5397-17-123-i87]](/bjoc/content/inline/1860-5397-17-123-i87.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 87: Fluorination of malonates with Me-NFSI.
Scheme 87: Fluorination of malonates with Me-NFSI.
Keto esters were fluorinated in high yields with Me-NFSI employing methanol or water as solvent under catalysis-free conditions (entries 1–3, Scheme 88).
![[1860-5397-17-123-i88]](/bjoc/content/inline/1860-5397-17-123-i88.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 88: Fluorination of keto esters with Me-NFSI.
Scheme 88: Fluorination of keto esters with Me-NFSI.
1-39. An N–F reagent derived from the ethano-Tröger’s base
In 2016, Gouverneur and Cvengroš reported the synthesis of a novel N-F reagent 39-3 derived from the ethylene-bridged Tröger’s base 39-1 [113]. The fluorination of the precursor 39-2, obtained from 39-1, was achieved only after reaction with N-fluoropentachloropyridinium triflate (5-1v) in acetonitrile at −35 °C, giving the N-F reagent 39-3 in higher than 95% conversion (Scheme 89). All other attempts to fluorinate, either with F2, XeF2, Selectfluor, or N-fluoro-2,6-dichloropyridinium triflate (5-1r) failed. This suggested that reagent 39-3 was more reactive than the other known reagents such as Selectfluor, but less active than the N-fluoropentachloropyridinium salts. The salt 39-3 is not stable and decomposition occurred when a solution of 39-3 in acetonitrile was left standing at room temperature for 8 hours or more. Therefore, 39-3 was best prepared with 5-1v immediately before use. The characterization of 39-3 was made by high-resolution mass spectrometry and NMR spectroscopic analyses containing 2D 19F-15N heteronuclear correlation experiments.
![[1860-5397-17-123-i89]](/bjoc/content/inline/1860-5397-17-123-i89.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 89: Synthesis of N-F 39-3 derived from the ethylene-bridged Tröger’s base.
Scheme 89: Synthesis of N-F 39-3 derived from the ethylene-bridged Tröger’s base.
Reagent 39-3 had the ability to formally transfer F+ onto the Selectfluor precursor 39-4 to form a Selectfluor analog 39-5 (Scheme 90). The fluorine transfer of 39-3 to the precursor 39-4 was completed after 5 min at room temperature in acetonitrile.
![[1860-5397-17-123-i90]](/bjoc/content/inline/1860-5397-17-123-i90.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 90: Fluorine transfer from N-F 39-3.
Scheme 90: Fluorine transfer from N-F 39-3.
The ability of 39-3 to fluorinate C-substrates was investigated. The fluorination of benzene, anisole, and fluorobenzene with 39-3 occurred faster than with Selectfluor (Scheme 91). The fluorination of styrenes 39-10 provided the expected addition products 39-11–14 in good yields. However, 39-3 did not react with less-activated alkenes, such as cyclohexene.
![[1860-5397-17-123-i91]](/bjoc/content/inline/1860-5397-17-123-i91.svg?scale=2.0&max-width=1024&background=FFFFFF)
1-40. 1-Cyanomethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bistriflate
In early 2018, Lan and Liu reported the synthesis of 1-cyanomethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bistriflate (SelectfluorCN) employing the same method as for Selectfluor (Scheme 92) [114].
![[1860-5397-17-123-i92]](/bjoc/content/inline/1860-5397-17-123-i92.svg?scale=2.0&max-width=1024&background=FFFFFF)
As shown in Scheme 93, SelectfluorCN was used as an oxidant to improve the Pd-catalyzed bistrifluoromethoxylation of alkenes with AgOCF3. It was shown that the yield of the bis(CF3O) product 40-4 increased as the fluorinating power of the N-F reagent increased in the order of NFSI < 16-3 (R = Me) < Selectfluor (R = CH2Cl) < SelectfluorCN (R = CH2CN). A considerable number of olefin derivatives were bistrifluoromethoxylated using this method with SelectfluorCN [114].
![[1860-5397-17-123-i93]](/bjoc/content/inline/1860-5397-17-123-i93.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 93: Bistrifluoromethoxylation of alkenes using SelectfluorCN.
Scheme 93: Bistrifluoromethoxylation of alkenes using SelectfluorCN.
1-41. N-Fluoro-N-aryl-arenesulfonamides (NFAS)
In 2018, Zipse and Renaud reported the synthesis of a series of N-fluoro-N-aryl-arenesulfonamides (NFAS) 41-2 suitable for radical fluorination, by reacting 41-1 with NFSI in the presence of cesium carbonate in methylene chloride and in moderate to good yields (Figure 13) [115]. They suggested that NFAS belongs to a third generation of radical fluorinating agents.
![[1860-5397-17-123-13]](/bjoc/content/figures/1860-5397-17-123-13.svg?scale=2.0&max-width=1024&background=FFFFFF)
These authors designed NFAS 41-2a–i as having a low N-F homolytic bond-dissociation energy (BDE). They demonstrated experimentally that NFAS 41-2 were more useful for radical fluorinations than N-fluoro-N-alkyl-arenesulfonamides 4-1, N-fluoro-N-alkyl-arylamides, NFSI 14-2, and Selectfluor, all of which have higher N–F homolytic BDEs than NFAS 41-2 (Scheme 94).
![[1860-5397-17-123-i94]](/bjoc/content/inline/1860-5397-17-123-i94.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 94: Radical fluorination with different N-F reagents.
Scheme 94: Radical fluorination with different N-F reagents.
The hydrofluorinated products were obtained in moderate yields along with good stereoselectivities in some cases (Scheme 95).
![[1860-5397-17-123-i95]](/bjoc/content/inline/1860-5397-17-123-i95.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 95: Radical fluorination of alkenes with NFAS 41-2.
Scheme 95: Radical fluorination of alkenes with NFAS 41-2.
As for the less nucleophilic terminal alkenes, remote fluorinated products were favored via the 1,5 H-atom abstraction process (Scheme 96).
![[1860-5397-17-123-i96]](/bjoc/content/inline/1860-5397-17-123-i96.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 96: Radical fluorination of alkenes with NFAS 41-2f.
Scheme 96: Radical fluorination of alkenes with NFAS 41-2f.
Decarboxylative fluorination of tert-butyl peresters also proved that these NFAS (41-2a and -2f) were better choices than NFSI and Selectfluor (Scheme 97).
![[1860-5397-17-123-i97]](/bjoc/content/inline/1860-5397-17-123-i97.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 97: Decarboxylative fluorination with NFAS 41-2a,f.
Scheme 97: Decarboxylative fluorination with NFAS 41-2a,f.
2. Evaluation of fluorinating power of N-F fluorinating agents
It emerges from the overview above that many N-F fluorination reagent variants have been developed, each with a different fluorination power (reactivity). Therefore, the power ordering of N-F reagents has become a matter of interest. Early on, Umemoto et al. revealed that the fluorinating power of N-fluoropyridinium salt systems could be tuned by the electron density at the nitrogen site, a property which is controlled by the electronic nature of the substituents. In order to quantitatively explain the variability of the N-F reagents, many attempts have been made: Theoretical calculations were performed by Hachisuka and Umemoto in 1991 [116,117], Woolf in 1994 [118], and Fainzil’berg in 1994 and 2001 [119,120]; N-F 19-fluorine NMR chemical shift analysis was accessed by Umemoto in 1991 [34]; measurements of peak reduction potentials were carried out by Gilicinski in 1992 [121], Differding in 1992 [122], Evans in 1999 [123], Zhang in 2013 [104], and reported by Umemoto in 2016 [124]; relative fluorination rates (kinetic data) were conducted by Togni in 2004 [125], and electromotive forces with Li, Mg, and Zn metals were reported by Umemoto in 2016 [124].
In a significant advancement in this regard, in 2016 Xue and Cheng published a comprehensive energetic scale for the quantitative estimation of the fluorinating power of N-F reagents [126,127]. They applied the Fluorine Plus Detachment (FPD) energy parameter, introduced by Christe and Dixon in 1992 [128], to electrophilic N-F reagents and calculated the FPD energies of 130 N-F reagents (Scheme 98).
![[1860-5397-17-123-i98]](/bjoc/content/inline/1860-5397-17-123-i98.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 98: Fluorine plus detachment (FPD).
Scheme 98: Fluorine plus detachment (FPD).
As seen in Figure 14, a large number of N-F reagents were clearly arranged in their power order; the smaller the FPD value the stronger the fluorinating power. This energetic scale is useful for an evaluation of the electrophilic fluorinating power of the N-F reagents.
![[1860-5397-17-123-14]](/bjoc/content/figures/1860-5397-17-123-14.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: FPD values of representative N-F reagents in CH2Cl2 and CH3CN (in parentheses). Adapted with permission from ref. [127]. Copyright 2020 American Chemical Society.
Figure 14: FPD values of representative N-F reagents in CH2Cl2 and CH3CN (in parentheses). Adapted with permis...
In addition, since some N-F reagents have been proven to be useful radical fluorinating agents [129-135], Xue and Cheng introduced the N-F homolytic bond-dissociation energy (BDE) parameter for radical fluorination abilities of N-F reagents in 2017 (Scheme 99) [127,136]. They calculated the BDE strength of 88 N-F reagents and clearly arranged them in terms of their radical fluorination power; the smaller the BDE value the stronger the radical fluorinating power (Figure 15). The BDE is useful for identifying and designing new atomic fluorine sources.
![[1860-5397-17-123-i99]](/bjoc/content/inline/1860-5397-17-123-i99.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 99: N-F homolytic bond dissociation energy (BDE).
Scheme 99: N-F homolytic bond dissociation energy (BDE).
![[1860-5397-17-123-15]](/bjoc/content/figures/1860-5397-17-123-15.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: BDE values of representative N-F reagents in CH3CN. Adapted with permission from ref. [127]. Copyright 2020 American Chemical Society.
Figure 15: BDE values of representative N-F reagents in CH3CN. Adapted with permission from ref. [127]. Copyright 2...
In 2018, another significant advancement was reported. Since FPD and BDE are thermodynamic parameters, they cannot be directly used for predicting reaction rates. Mayr et al. [137] and Hodgson et al. [138] independently reported the quantitative reactivity scales for electrophilic fluorinations of popular N-F reagents. They provided detailed kinetic fluorination studies of a series of popular N-F fluorinating agents such as N-fluoropyridinium salt derivatives, NFSI, and SelectfluorTM with enamines, carbanions, or 1,3-diketones. Figure 16 shows the relative reactivity scale of the popular N-F reagents, which covers eight orders of magnitude. These reactivity scales can be used for predicting the actual fluorination rates of N-F fluorinating agents. Hodgson et al. further advanced their kinetic studies with N-F fluorinating agents in 2019 [139] and 2020 [140].
![[1860-5397-17-123-16]](/bjoc/content/figures/1860-5397-17-123-16.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 16: Quantitative reactivity scale for popular N-F reagents. Adapted with permission from ref. [138], published by the Royal Society of Chemistry.
Figure 16: Quantitative reactivity scale for popular N-F reagents. Adapted with permission from ref. [138], publish...
In 2019, Zhang et al. reported detailed theoretical calculations of the fluorination of benzene with 16 different disubstituted N-fluoropyridinum salts in acetonitrile solution and as a result, suggested a 2,6-dinitro-substituted N-fluoropyridinium salt as the most promising fluorinating reagent [141].
3. Reaction mechanisms of N-F fluorinating agents
Another noteworthy issue is the reaction mechanism for the fluorination with N-F reagents. There have been long standing arguments regarding which mechanism is operating in these fluorinations: a single-electron transfer (SET) or a nucleophilic substitution (SN2) mechanism (Scheme 100) [22,142]. In the SET process, a single-electron transfer from a substrate (Nu−) to a N-F reagent [N-F] occurs to form a radical anion [N-F]·− and a Nu· radical, and the former undertakes the homolytic N–F bond cleavage to give a F· radical and a N:− anion, and then the F· radical and the Nu· radical combine to generate the final product Nu–F. In the SN2 mechanism, a nucleophile directly attacks the F atom of the N-F reagent to give the fluorinated product via the heterolytic cleavage of the N–F bond. The key point is whether the N–F bond cleaves homolytically or heterolytically. The prime cause of this argument is based on the fact that the formation of F+ requires extremely high energy (ionization potential of F, 1681.0 kJ/mol) and contrasts significantly with that of other halogenations (Cl, 1251.2 kJ/mol; Br, 1139.9 kJ/mol).
![[1860-5397-17-123-i100]](/bjoc/content/inline/1860-5397-17-123-i100.svg?scale=2.0&max-width=1024&background=FFFFFF)
In 1991, Umemoto et al. explained the observed fluorination reactions of N-fluoropyridinium salts with anionic and neutral substrates by SET processes involving a F· radical species [32]. Soon after, in 1992, Kochi et al. reported fluorination reactions of aromatic compounds with N-fluoropyridinium salts via electron donor–acceptor complexes, with outcomes which also supported the SET mechanism [143,144]. In 1992, DesMarteau et al. proposed a similar SET process for the fluorination with N-fluoroperfluoroalkanesulfonimide [48]. On the other hand, in 1991, Differding and co-worker investigated radical clock experiments to monitor the reaction for the presence of intermediate radicals [145] (Scheme 101).
![[1860-5397-17-123-i101]](/bjoc/content/inline/1860-5397-17-123-i101.svg?scale=2.0&max-width=1024&background=FFFFFF)
As illustrated in Scheme 102, the reactions of potassium enolate I of citronellic ester with the N-F reagents 10-1, 14-2 (NFSI), or 8-1 produced only the non-cyclized products II and III. They also showed that the observed reaction rates were much faster than the calculated electron-transfer rates and therefore they concluded that a SN2 mechanism occurred to give the fluorinated products [146].
![[1860-5397-17-123-i102]](/bjoc/content/inline/1860-5397-17-123-i102.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 102: Reaction of potassium enolate of citronellic ester with N-F reagents, 10-1, NFSI, and 8-1.
Scheme 102: Reaction of potassium enolate of citronellic ester with N-F reagents, 10-1, NFSI, and 8-1.
In 1995, based on the study on the fluorination reactions of the N-F reagent NFOBS 13-2, Davis et al. described that all of their available data supported the SN2 reaction process, but the SET mechanism could not be rigorously excluded [58]. In 1998, Zupan et al. studied the reactions of norbornene with Selectfluor, NFTh 19-2, and N-fluoro-2,6-dichloropyridinium triflate [147]. Although their results ended toward SN2, they too could not completely exclude the SET. Soon after, in 1999, Wong et al. reported their studies using compound VI having a phenyl-cyclopropyl moiety which was the hypersensitive radical probe (rate: 1011 s−1) (Scheme 103) [148].
![[1860-5397-17-123-i103]](/bjoc/content/inline/1860-5397-17-123-i103.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 103: Reaction of compound IV with Selectfluor (OTf) and NFSI.
Scheme 103: Reaction of compound IV with Selectfluor (OTf) and NFSI.
They found that Selectfluor (OTf salt) produced product VII in 45% yield with no rearrangement products despite the complete consumption of VI (entry 1, Scheme 103). This result is aligned with the SN2 mechanism, however, the reaction of VI with Selecfluor was completely blocked by the radical scavenger, 2,2,6,6-tetramethylpiperidinoxyl (TEMPO), which suggested a SET mechanism. The reaction of VI with NFSI gave VII (40%) and a product IX (5%) derived from the rearrangement fluoro product VIII (entry 2 in Scheme 103). This result is consistent with the SET mechanism. In their review published in 2005 [149], Wong discussed these results in more detail. Since the reactivity of the radical (F·) is extremely fast and diffusion-controlled, the combination of F· with any alkyl radical R· should proceed at an equal or greater rate than the cyclization of the alkyl radical as observed in the case of Differding [145] and the ring-opening of the cyclopropyl radical in the case of Wong [148]. Despite extensive research, the mechanism remains unclear as a result of the difficulty of assessing reaction rates. The key may be the lifetime of the intermediate N-F radical or radical anion, [N-F]· or [N-F]·−, which releases the radical F·. Finally, Wong mentioned that, if these radicals react faster than the rates of diffusion, anything beyond that would probably remain unproven [149].
In the meantime, in 2002, Togni and Rothlisberger provided evidence which strongly supported the SET mechanism from their studies on the fluorination mechanism of β-ketoesters with Selectfluor and a chiral TiCl2(TADDOLato) catalyst [150]. Their computational study suggested a one-electron transfer followed by simultaneous N–F bond cleavage and the accompanying experimental outcomes demonstrated that a chlorination side reaction was quenched by a radical scavenger, while the fluorination reaction was unaffected. Since Selectfluor is inert towards chloride, they proposed that any [N-F]· radical formed by the SET process would react with chloride ions (Cl−) present in the reaction solution to form the relatively stable Cl· radical, which caused the side chlorination.
On the other hand, in 2002, Laali and co-worker reported that competitive fluorination studies of mesitylene and durene with Selecfluor in CH3CN and an ionic liquid supported the conventional polar mechanism (SN2) [151]. However, in 2006, Stavber et al. made similar competition studies and proposed a SET process as the dominant mechanism that explained the higher reactivity of durene viz-à-viz mesitylene towards Selectfluor in aq CH3CN [152]. In 2009, Shubin et al. performed competitive studies of mesitylene and durene with NFSI under solvent-free reaction conditions and reported that the reaction rate ratio (kMes/kDur) followed a polar mechanism [153].
Since 2015, many papers supporting either SET or SN2 mechanisms have appeared, including those reported by C. Liu et al. (SET) in 2015 [154], Chen et al. (SN2) in 2016 [155], Tang et al. (SET) in 2016 [156], F. Liu et al. (SET) in 2016 [157] and 2017 [158], Mayr et al. (SN2) in 2018 [137], Hodgson et al. (SN2) in 2018 [138], Li et al. (SET) in 2018 [159], Shi et al. (SET) in 2018 [160], Li et al. (SET) in 2019 [161], Mernyak et al. (SET) in 2019 [162], and Nelson et al. (SN2) in 2019 [163].
TEMPO has been often used as a radical scavenger to explore the reaction mechanisms of N-F fluorinating agents but, in Nelson's paper [163] in 2019, it was revealed that Selectfluor reacted with TEMPO (X) to form the oxoammonium salt XI (Scheme 104). This author concluded that the use of TEMPO as a radical trap in the reactions with Selectfluor was unreliable. This conclusion was subsequently confirmed by Borodkin et al. [164]. The latest review [165] by Hodgson and co-worker in 2021 discussed the reactivities and reaction mechanisms of electrophilic N-F fluorinating agents in detail.
![[1860-5397-17-123-i104]](/bjoc/content/inline/1860-5397-17-123-i104.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 104: Reaction of TEMPO with Selecfluor.
Scheme 104: Reaction of TEMPO with Selecfluor.
Conclusion
A historical timeline on the progress of N-F fluorinating reagents and their fluorinations has been described in this review. Table 1 shows a historical list of all the N-F reagents. The fluorination power and reaction mechanism of the N-F fluorinating agents have also been summarized briefly and from a historical perspective. The fluorination reactions described here for each N-F fluorinating agent are those reported at the time. The synthetic applications of each N-F reagent since then are too numerous to be covered in this review. As many reviews on the reactions and applications of the N-F fluorinating agents have been published [39,133,142,165-181], readers are recommended to consult these reviews.
Table 1: Chronological list of the development of N-F fluorinating agents.
| year | structure | “F” source | yield | references |
| 1964 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-123-i105.svg?max-width=637&scale=1.0)
|
HF | 7.5–13% | [16-22] |
| 1981 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-123-i106.svg?max-width=637&scale=1.0)
|
XeF2 | 50–65% | [23] |
| 1983 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-123-i107.svg?max-width=637&scale=1.0)
|
5% F2/N2 | 63% | [24,25] |
| 1984 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-123-i108.svg?max-width=637&scale=1.0)
R1 = p-tolyl, n-butyl; R2 = methyl, tert-butyl, exo/endo-2-norbornyl, cyclohexyl, neopentyl |
1–5% F2/N2 | 11–71% | [26,27] |
|
1986–
1991 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-123-i109.svg?max-width=637&scale=1.0)
X = OTf, BF4, PF6, AsF6, SbF6, ONf, OMs, etc. R1, R2, R3, R4, R5 = H, Me, Cl, F, COOMe, OMe, CH2OMe, CH2OAc, CF3, CN, NO2, OMenth, etc.; (total 62 examples) |
10% F2/N2 | 15–96% | [28-34,36-41] |
| 1986 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-123-i110.svg?max-width=637&scale=1.0)
|
100% F2 | 86% | [43,44] |
| 1987 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-123-i111.svg?max-width=637&scale=1.0)
Rf1,2 = CF3, C4F9, C6F13 |
100% F2 | 61–96% | [45-49] |
| 1988 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-123-i112.svg?max-width=637&scale=1.0)
|
100% F2 | 80–88% | [50,51] |
| 1988 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-123-i113.svg?max-width=637&scale=1.0)
R = H, CH3 |
10% F2/N2 | 75–80% | [52] |
| 1989 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-123-i114.svg?max-width=637&scale=1.0)
|
10% F2/N2 | 74% | [53] |
| 1990 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-123-i115.svg?max-width=637&scale=1.0)
|
100% F2 | 89% | [54] |
| 1990 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-123-i116.svg?max-width=637&scale=1.0)
|
0.05% 18F2/Ne | 33–79% | [55,56] |
| 1991 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-17-123-i117.svg?max-width=637&scale=1.0)
|
10% F2/N2 | >90% | [57,58] |
| 1991 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-17-123-i118.svg?max-width=637&scale=1.0)
|
10% F2/N2 | 70% | [59] |
| 1991 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-17-123-i119.svg?max-width=637&scale=1.0)
X = SO2, CO |
Cs+-OSO2OF | 48–69% | [60] |
| 1992 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-17-123-i120.svg?max-width=637&scale=1.0)
X = OTf, BF4 R = CH3, CH2Cl, CH2CF3 SelectfluorTM (R = CH2Cl, X = BF4) |
10% F2/N2 | 87–95% | [42,61-74] |
| 1993 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-17-123-i121.svg?max-width=637&scale=1.0)
|
10% F2/N2 | 68% | [75] |
| 1995 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-17-123-i122.svg?max-width=637&scale=1.0)
R3, R4, R5, R6 = H, CH3, CF3, Cl, C2H5, tert-butyl |
10% F2/N2 | 65–95% | [76] |
| 1995 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-17-123-i123.svg?max-width=637&scale=1.0)
NFTh/AccufluorTM |
10% F2/N2 | 75% | [78-80] |
| 1995 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-17-123-i124.svg?max-width=637&scale=1.0)
|
5% F2/N2 | 83% | [81] |
| 1996 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-17-123-i125.svg?max-width=637&scale=1.0)
|
100% F2 | 75% | [82] |
| 1996 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-17-123-i126.svg?max-width=637&scale=1.0)
X1, X2 = OTf, BF4, HSO4, SbF6, PF6, etc. |
10% F2/N2 | 56–87% | [83] |
| 1997 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-17-123-i127.svg?max-width=637&scale=1.0)
R = BF3, F, CH3 |
10% F2/N2 | 62–82% | [85] |
| 1997 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-17-123-i128.svg?max-width=637&scale=1.0)
R1 = CH3, CH2OCOCH3 R2 = CH3, p-tolyl |
FClO3 | 13–52% | [92] |
| 1998 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-17-123-i129.svg?max-width=637&scale=1.0)
X = OTf, BF4, PF6, SbF6 bipyridyl = 2,2', 2,4', 3,3', 4,4'; R = H, CH3, Cl, CF3, Ph, COOCH3 |
10-20% F2/N2 | 64–97% | [86] |
| 1998 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-17-123-i130.svg?max-width=637&scale=1.0)
|
5% F2/N2 | – | [87,88] |
| 1999 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-17-123-i131.svg?max-width=637&scale=1.0)
R = CH3, CF3 |
10% F2/N2 | 65–82% | [89] |
| 1999 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-17-123-i132.svg?max-width=637&scale=1.0)
|
FClO3 | 71% | [90] |
| 1999 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-17-123-i133.svg?max-width=637&scale=1.0)
(R)-(+) (S)-(–) |
15% F2/He | 51–65% | [91] |
| 2000 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-17-123-i134.svg?max-width=637&scale=1.0)
(R), (S) |
FClO3 | 66–83% | [93] |
| 2000 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-17-123-i135.svg?max-width=637&scale=1.0)
(S,R,R), (S,S,R) |
FClO3 | 44–81% | [94] |
| 2000 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-17-123-i136.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-17-123-i137.svg?max-width=637&scale=1.0)
SelectfluorTM |
prepared in situ | [95,96] |
| 2000 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-17-123-i138.svg?max-width=637&scale=1.0)
R = H, OCH3 |
SelectfluorTM | 84% | [97,98] |
| 2003 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-17-123-i139.svg?max-width=637&scale=1.0)
X = BF4, OTf, N(SO2Ph)2 |
Selectfluor (F-TEDA-BF4)
F-TEDA-OTf NFTh NFSI N-F-diCl-pyridinium BF4 |
100% | [99] |
| 2003 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-17-123-i140.svg?max-width=637&scale=1.0)
|
100% F2 | 95% | [100] |
| 2011 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-17-123-i141.svg?max-width=637&scale=1.0)
NFBSI |
10% F2/N2 | 57% | [102] |
| 2012 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-17-123-i142.svg?max-width=637&scale=1.0)
R = F, Cl, Br, CF3, OCF3, t-Bu, Me, OCH3 |
10% F2/N2 | 20.9–89.4% | [103-105] |
| 2013 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-17-123-i143.svg?max-width=637&scale=1.0)
R = 3,5-(CF3)2C6H3, C6F5 |
XeF2 | 29–51% | [106] |
| 2013 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-17-123-i144.svg?max-width=637&scale=1.0)
R1 = H, CF3; R2 = H, CH3 |
10% F2/N2 or
![[Graphic 41]](/bjoc/content/inline/1860-5397-17-123-i145.svg?max-width=637&scale=1.0)
|
>95% | [107] |
| 2013 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-17-123-i146.svg?max-width=637&scale=1.0)
(R), R = H; (S), R = 3,5-(CF3)2C6H3 |
0.2% F2/N2 | 27–67% | [108] |
| 2016 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-17-123-i147.svg?max-width=637&scale=1.0)
Me-NFSI |
10% F2/N2 | 76% | [111] |
| 2016 |
![[Graphic 44]](/bjoc/content/inline/1860-5397-17-123-i148.svg?max-width=637&scale=1.0)
|
![[Graphic 45]](/bjoc/content/inline/1860-5397-17-123-i149.svg?max-width=637&scale=1.0)
|
>95% conversion | [113] |
| 2018 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-17-123-i150.svg?max-width=637&scale=1.0)
|
20% F2/N2 | 50% | [114] |
| 2018 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-17-123-i151.svg?max-width=637&scale=1.0)
Ar1 = C6H5, 4-Cl-C6H4, 2,4,6-triMe-C6H2, etc.; Ar2 = 4-CF3-C6H4, 3-CF3-C6H4, 3,5-diCF3C6H3 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-17-123-i152.svg?max-width=637&scale=1.0)
NFSI |
20–93% | [115] |
The seminal work of perfluoro-N-fluoropiperidine by Banks was reported in 1964 [16]. However, this compound could not be contemplated for two decades because of the very low yields in its production and fluorination reactions. At that time, FClO3 [10] and O-F reagents such as CF3OF and CF2(OF)2 [11] were used as electrophilic fluorinating agents despite their inherent safety issues. The real attention on N-F compounds started in the 1980’s when the first report on a N-F fluorinating agent, N-fluoro-2-pyridone, was published by Purrington in 1983 [24]. The next year, Barnette reported N-fluoro-N-alkylarenesulfonamides that were easy-to-handle, stable crystalline compounds useful for the fluorination of carbanions [26]. Soon after (1986), Umemoto reported a series of N-fluoropyridinium triflates as easy-to-handle, reactive, and widely applicable reagents [28,29].
Since then, many N-F fluorinating agents have been reported, one after another, as follows: powerful N-fluorotrifluoromethanesulfonimide by DesMarteau in 1987 [45], mild N-fluorosultams including first chiral N-fluorosultams by Lang in 1988 [52] and 1989 [53], less reactive N-fluorolactams by Sathyamurthy in 1990 [55], moderate N-fluoro-o-benzenedisulfonimide (NFOBS) [57] and N-fluorobenzenesulfonimide (NFSI) [59] by Davis and Differding, respectively, in 1991, and powerful 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate (SelectfluorTM) by Banks in 1992 [42]. Currently popular N-F fluorinating agents such as N-fluoropyridinium salts, NFSI, and Selectfluor were developed within a decade from the first Purrington’s report (1983).
After this first rush, other notable N-F reagents were developed such as a series of zwitterionic N-fluoropyridinium salts by Umemoto in 1995 [76], another Selectfluor-type reagent, 1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate (NFTh/AccufluorTM) by Poss and Shia in 1995 [78], a more powerful reagent, 1,4-difluoro-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate by Umemoto in 1996 [83], and a powerful and high fluorine-content reagent, N,N’-difluoro-2,2’-bipyridinium bistetrafluoroborate (SynfluorTM) by Umemoto in 1998 [86].
In 2000, practical chiral reagents, N-fluorinated chiral cinchona alkaloids were reported by Shibata and Takeuchi [95] and by Cahard [97]. In 2003, Cahard demonstrated that the chiral N-fluorocinchona alkaloids could be prepared quantitatively with not only Selectfluor but also with other powerful N-F fluorinating agents such as NFTh, NFSI, and N-fluoro-2,6-dichloropyridinium tetrafluoroborate [99]. After a break of a decade, in 2013, chiral dicationic DABCO-based N-F reagents 36-5 and chiral N-fluorobinaphthyldisulfonimides 37-2 were reported by Gouverneur [107] and by Shibata, Ma, and Cahard [108], respectively. In 2016, Shibata reported N-fluoromethanesulfonimide (Me-NFSI) as a high atom-economy reagent [111] and Gouverneur and Cvingroš reported a novel N-F reagent 39-3 derived from the ethylene-bridged Tröger base [113]. More recently, Zipse and Renaud in 2018, disclosed N-fluoro-N-aryl-arenesulfonamides (NFAS) as radical fluorinating agents [115].
A very large number of N-F fluorinating agents have been developed so far and a considerable number of easy-to-handle N-F reagents have been commercialized. In particular, Selectfluor, NFSI, and N-fluoropyridinium salt derivatives are produced in factories on a large scale. This has greatly contributed to the remarkable advancement of organofluorine chemistry. Before the appearance of N-F reagents, electrophilic fluorinating agents were thought dangerous and difficult to handle and special techniques were required. Chemists avoided them. Now, practicing chemists use them without any problem. The N-F reagents have brought such a dramatic change that many chemists nowadays may not realize the magnitude of this change and the contributions of the pioneers.
For practitioners in the field, high thermal stability and non-hygroscopicity of reagents are key factors for easy handling and a long shelf life. Many N-F reagents satisfy this requirement, however, there are limitations as the fluorinating power increases. N-Fluoropentachloropyridinium triflate (5-4v) and tetrafluoroborate (5-4w) rank as the most powerful of the non-hygroscopic and thermally stable N-F agents. However, the more powerful N-fluoro-2,4,6-trichloro-1,3,5-triazinium tetrafluoroborate (31-2) is moisture-sensitive and not easy-to-handle. N,N’-Difluoro-1,4-diazoniabicyclo[2.2.2]octane bistetrafluoroborate (22-1d) ranks top in the easy-to-handle 1,4-diazoniabicyclo[2.2.]octane salt series. Salt 22-1d is more powerful than Selectfluor. Unfortunately, synthetic chemists have had little chance to use it because it has not been commercialized due to patent issues despite its simple one-pot process using cheap materials. However, since all the patents have now expired, it is expected that 22-1d may soon be commercialized.
It is now rare for researchers and laboratories to use molecular fluorine (F2) in universities and institutes, because of strict safety regulations, although 20% or 10% F2 diluted with N2 can be handled without significant problems. As a result, currently reported fluorination reactions have been limited to the use of a few commercially available and inexpensive N-F reagents such as Selectfluor and NFSI. As mentioned in this review, there are many other useful N-F fluorinating agents and the plentiful supply will satisfy the strong demand for fluorinations. That said, there is still room for improvement or refinement. The authors think that further research and development on N-F fluorinating reagents is needed for the advancement of synthetic fluorine chemistry but, for this to happen, it would be highly desirable to develop a safe and convenient F2 generator.
References
-
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons, Ltd: Chichester, United Kingdom, 2009. doi:10.1002/9781444312096
See for a selected book.
Return to citation in text: [1] -
Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833
Return to citation in text: [1] -
Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f
Return to citation in text: [1] -
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] -
Mei, H.; Remete, A. M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2020, 31, 2401–2413. doi:10.1016/j.cclet.2020.03.050
Return to citation in text: [1] -
Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467
Return to citation in text: [1] -
Uneyama, K., Ed. Organofluorine Chemistry; Blackwell Publishing: Oxford, United Kingdom, 2006. doi:10.1002/9780470988589
Return to citation in text: [1] -
Ramachandran, P. V. Future Med. Chem. 2009, 1, 771–772. doi:10.4155/fmc.09.67
Return to citation in text: [1] -
Harper, D. B.; O'Hagan, D. Nat. Prod. Rep. 1994, 11, 123–133. doi:10.1039/np9941100123
Return to citation in text: [1] -
Schlosser, M.; Heinz, G. Chem. Ber. 1969, 102, 1944–1953. doi:10.1002/cber.19691020620
Return to citation in text: [1] [2] -
Barton, D. H. R. Pure Appl. Chem. 1977, 49, 1241–1249. doi:10.1351/pac197749091241
Return to citation in text: [1] [2] [3] -
Appelman, E. H.; Basile, L. J.; Thompson, R. C. J. Am. Chem. Soc. 1979, 101, 3384–3385. doi:10.1021/ja00506a046
Return to citation in text: [1] -
Rozen, S.; Lerman, O. J. Am. Chem. Soc. 1979, 101, 2782–2784. doi:10.1021/ja00504a073
Return to citation in text: [1] -
Rozen, S.; Lerman, O.; Kol, M.; Hebel, D. J. Org. Chem. 1985, 50, 4753–4758. doi:10.1021/jo00224a019
Return to citation in text: [1] -
Filler, R. Isr. J. Chem. 1978, 17, 71–79. doi:10.1002/ijch.197800010
Return to citation in text: [1] -
Banks, R. E.; Williamson, G. E. Chem. Ind. (London) 1964, 1864–1865.
Return to citation in text: [1] [2] [3] -
Banks, R. E.; Cheng, W. M.; Haszeldine, R. N. J. Chem. Soc. 1962, 3407–3416. doi:10.1039/jr9620003407
Return to citation in text: [1] [2] -
Banks, R. E.; Ginsberg, A. E.; Haszeldine, R. N. J. Chem. Soc. 1961, 1740–1743. doi:10.1039/jr9610001740
Return to citation in text: [1] [2] -
Banks, R. E.; Haszeldine, R. N.; Hatton, R. Tetrahedron Lett. 1967, 8, 3993–3996. doi:10.1016/s0040-4039(01)89722-7
Return to citation in text: [1] [2] -
Polishchuk, V. R.; German, L. S. Tetrahedron Lett. 1972, 13, 5169–5172. doi:10.1016/s0040-4039(01)85198-4
Return to citation in text: [1] [2] -
Banks, R. E.; Tsiliopoulos, E. J. Fluorine Chem. 1986, 34, 281–285. doi:10.1016/s0022-1139(00)85081-2
Return to citation in text: [1] [2] -
Banks, R. E.; Murtagh, V.; Tsiliopoulos, E. J. Fluorine Chem. 1991, 52, 389–401. doi:10.1016/s0022-1139(00)80353-x
Return to citation in text: [1] [2] [3] -
Yagupols’kii, Y. L.; Savina, T. I. J. Org. Chem. USSR 1981, 17, 1180–1181.
Return to citation in text: [1] [2] -
Purrington, S. T.; Jones, W. A. J. Org. Chem. 1983, 48, 761–762. doi:10.1021/jo00153a037
Return to citation in text: [1] [2] [3] [4] [5] -
Purrington, S. T.; Jones, W. A. J. Fluorine Chem. 1984, 26, 43–46. doi:10.1016/s0022-1139(00)85117-9
Return to citation in text: [1] [2] [3] -
Barnette, W. E. J. Am. Chem. Soc. 1984, 106, 452–454. doi:10.1021/ja00314a050
Return to citation in text: [1] [2] [3] [4] [5] -
Lee, S. H.; Schwartz, J. J. Am. Chem. Soc. 1986, 108, 2445–2447. doi:10.1021/ja00269a052
Return to citation in text: [1] [2] -
Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Umemoto, T.; Kawada, K.; Tomita, K. Tetrahedron Lett. 1986, 27, 4465–4468. doi:10.1016/s0040-4039(00)84980-1
Return to citation in text: [1] [2] [3] [4] [5] -
Umemoto, T.; Tomizawa, G. Bull. Chem. Soc. Jpn. 1986, 59, 3625–3629. doi:10.1246/bcsj.59.3625
Return to citation in text: [1] [2] [3] [4] -
Umemoto, T.; Tomita, K.; Kawada, K. Org. Synth. 1990, 69, 129–140. doi:10.15227/orgsyn.069.0129
Return to citation in text: [1] [2] [3] [4] -
Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. Bull. Chem. Soc. Jpn. 1991, 64, 1081–1092. doi:10.1246/bcsj.64.1081
Return to citation in text: [1] [2] [3] [4] -
Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. J. Fluorine Chem. 1991, 53, 369–377. doi:10.1016/s0022-1139(00)80960-4
Return to citation in text: [1] [2] [3] -
Meinert, H. Z. Chem. 1965, 5, 64. doi:10.1002/zfch.19650050208
Return to citation in text: [1] -
Umemoto, T.; Tomita, K. N-Fluoropyridinium salt-containing polymer. Jpn. Kokai Tokkyo Koho S63-295610, Dec 2, 1988.
Return to citation in text: [1] [2] -
Nukui, K.; Kamiya, K. Production method of N-fluoropyridinium salts. Jpn. Patent 3038065, May 8, 2000.
Return to citation in text: [1] [2] -
Fukami, S.; Nukui, K.; Kawada, K. Production method of N-fluoropyridinium tetrafluoroborate. Jpn. Kokai Tokkyo Koho H09-255657, Sept 30, 1997.
Return to citation in text: [1] [2] -
Kawada, K. N-Fluoropyridinium Salt Fluorination for Preparing Aryl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2020. doi:10.1007/978-981-10-1855-8_40-2
Return to citation in text: [1] [2] [3] -
Naruta, Y.; Tani, F.; Maruyama, K. Tetrahedron Lett. 1992, 33, 1069–1072. doi:10.1016/s0040-4039(00)91863-x
Return to citation in text: [1] [2] -
Poss, A. J.; Van der Puy, M.; Nalewajek, D.; Shia, G. A.; Wagner, W. J.; Frenette, R. L. J. Org. Chem. 1991, 56, 5962–5964. doi:10.1021/jo00020a051
Return to citation in text: [1] [2] -
Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595
Return to citation in text: [1] [2] [3] [4] [5] -
Banks, R. E.; Du Boisson, R. A.; Tsiliopoulos, E. J. Fluorine Chem. 1986, 32, 461–466. doi:10.1016/s0022-1139(00)81953-3
Return to citation in text: [1] [2] -
Banks, R. E.; Du Boisson, R. A.; Morton, W. D.; Tsiliopoulos, E. J. Chem. Soc., Perkin Trans. 1 1988, 2805–2811. doi:10.1039/p19880002805
Return to citation in text: [1] [2] -
Singh, S.; DesMarteau, D. D.; Zuberi, S. S.; Witz, M.; Huang, H.-N. J. Am. Chem. Soc. 1987, 109, 7194–7196. doi:10.1021/ja00257a051
Return to citation in text: [1] [2] [3] -
Resnati, G.; DesMarteau, D. D. J. Org. Chem. 1991, 56, 4925–4929. doi:10.1021/jo00016a022
Return to citation in text: [1] [2] -
Xu, Z.-Q.; DesMarteau, D. D.; Gotoh, Y. J. Chem. Soc., Chem. Commun. 1991, 179–181. doi:10.1039/c39910000179
Return to citation in text: [1] [2] -
DesMarteau, D. D.; Xu, Z.-Q.; Witz, M. J. Org. Chem. 1992, 57, 629–635. doi:10.1021/jo00028a042
Return to citation in text: [1] [2] [3] -
Pennington, W. T.; Resnati, G.; DesMarteau, D. D. J. Org. Chem. 1992, 57, 1536–1539. doi:10.1021/jo00031a038
Return to citation in text: [1] [2] -
Banks, R. E.; Sharif, I. J. Fluorine Chem. 1988, 41, 297–300. doi:10.1016/s0022-1139(00)81553-5
Return to citation in text: [1] [2] -
Banks, R. E.; Sharif, I. J. Fluorine Chem. 1991, 55, 207–214. doi:10.1016/s0022-1139(00)80123-2
Return to citation in text: [1] [2] -
Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087–6090. doi:10.1016/s0040-4039(00)82271-6
Return to citation in text: [1] [2] [3] -
Differding, E.; Lang, R. W. Helv. Chim. Acta 1989, 72, 1248–1252. doi:10.1002/hlca.19890720609
Return to citation in text: [1] [2] [3] [4] -
Banks, R. E.; Khazaei, A. J. Fluorine Chem. 1990, 46, 297–305. doi:10.1016/s0022-1139(00)80997-5
Return to citation in text: [1] [2] -
Satyamurthy, N.; Bida, G. T.; Phelps, M. E.; Barrio, J. R. J. Org. Chem. 1990, 55, 3373–3374. doi:10.1021/jo00297a071
Return to citation in text: [1] [2] [3] -
Grakauskas, V.; Baum, K. J. Org. Chem. 1970, 35, 1545–1549. doi:10.1021/jo00830a061
Return to citation in text: [1] [2] -
Davis, F. A.; Han, W. Tetrahedron Lett. 1991, 32, 1631–1634. doi:10.1016/s0040-4039(00)74290-0
Return to citation in text: [1] [2] [3] -
Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730–4737. doi:10.1021/jo00120a014
Return to citation in text: [1] [2] [3] -
Differding, E.; Ofner, H. Synlett 1991, 187–189. doi:10.1055/s-1991-20673
Return to citation in text: [1] [2] [3] -
Gakh, A. A.; Romaniko, S. V.; Ugrak, B. I.; Fainzilberg, A. A. Tetrahedron 1991, 47, 7447–7458. doi:10.1016/s0040-4020(01)89746-5
Return to citation in text: [1] [2] -
Banks, R. E.; Lawrence, N. J.; Popplewell, A. L. J. Chem. Soc., Chem. Commun. 1994, 343–344. doi:10.1039/c39940000343
Return to citation in text: [1] [2] -
Abdul-Ghani, M.; Banks, E. R.; Besheesh, M. K.; Sharif, I.; Syvret, R. G. J. Fluorine Chem. 1995, 73, 255–257. doi:10.1016/0022-1139(94)03225-o
Return to citation in text: [1] [2] [3] [4] -
Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Chem. Soc., Perkin Trans. 1 1996, 2069–2076. doi:10.1039/p19960002069
Return to citation in text: [1] [2] -
Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Fluorine Chem. 1996, 78, 43–50. doi:10.1016/0022-1139(96)03395-7
Return to citation in text: [1] [2] -
Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Stavber, S.; Sotler, T.; Zupan, M. Tetrahedron Lett. 1994, 35, 1105–1108. doi:10.1016/s0040-4039(00)79977-1
Return to citation in text: [1] [2] -
Lal, G. S. Synth. Commun. 1995, 25, 725–737. doi:10.1080/00397919508011410
Return to citation in text: [1] [2] [3] -
Brown, D. S.; Marples, B. A.; Smith, P.; Walton, L. Tetrahedron 1995, 51, 3587–3606. doi:10.1016/0040-4020(95)00075-j
Return to citation in text: [1] [2] -
Lal, G. S.; Pastore, W.; Pesaresi, R. J. Org. Chem. 1995, 60, 7340–7342. doi:10.1021/jo00127a046
Return to citation in text: [1] [2] -
Zupan, M.; Iskra, J.; Stavber, S. J. Org. Chem. 1995, 60, 259–260. doi:10.1021/jo00106a045
Return to citation in text: [1] [2] -
Brunavs, M.; Dell, C. P.; Owton, W. M. J. Fluorine Chem. 1994, 68, 201–203. doi:10.1016/0022-1139(93)03042-k
Return to citation in text: [1] [2] -
Matthews, D. P.; Miller, S. C.; Jarvi, E. T.; Sabol, J. S.; McCarthy, J. R. Tetrahedron Lett. 1993, 34, 3057–3060. doi:10.1016/s0040-4039(00)93378-1
Return to citation in text: [1] [2] -
Hodson, H. F.; Madge, D. J.; Slawin, A. N. Z.; Widdowson, D. A.; Williams, D. J. Tetrahedron 1994, 50, 1899–1906. doi:10.1016/s0040-4020(01)80862-0
Return to citation in text: [1] [2] -
McClinton, M. A.; Sik, V. J. Chem. Soc., Perkin Trans. 1 1992, 1891–1895. doi:10.1039/p19920001891
Return to citation in text: [1] [2] -
Davis, F. A.; Zhou, P.; Murphy, C. K. Tetrahedron Lett. 1993, 34, 3971–3974. doi:10.1016/s0040-4039(00)60592-0
Return to citation in text: [1] [2] -
Umemoto, T.; Tomizawa, G. J. Org. Chem. 1995, 60, 6563–6570. doi:10.1021/jo00125a049
Return to citation in text: [1] [2] [3] -
MEC Reagents: Fluorinating agents and Perfluoroalkylating agents; Daikin Chemical Sales company Brochure, 1997.
Return to citation in text: [1] -
Poss, A. J.; Shia, G. 1-Substituted-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts and their application as fluorinating agents. U.S. Patent US5,459,267, Oct 17, 1995.
Return to citation in text: [1] [2] [3] -
Stavber, S.; Zupan, M. Tetrahedron Lett. 1996, 37, 3591–3594. doi:10.1016/0040-4039(96)00630-2
Return to citation in text: [1] [2] -
Poss, A. J.; Shia, G. A. Tetrahedron Lett. 1999, 40, 2673–2676. doi:10.1016/s0040-4039(99)00303-2
Return to citation in text: [1] [2] -
Cabrera, I.; Appel, W. K. Tetrahedron 1995, 51, 10205–10208. doi:10.1016/0040-4020(95)00597-2
Return to citation in text: [1] [2] -
Banks, R. E.; Besheesh, M. K.; Tsiliopoulos, E. J. Fluorine Chem. 1996, 78, 39–42. doi:10.1016/0022-1139(95)03380-7
Return to citation in text: [1] [2] -
Umemoto, T.; Nagayoshi, M. Bull. Chem. Soc. Jpn. 1996, 69, 2287–2295. doi:10.1246/bcsj.69.2287
Return to citation in text: [1] [2] [3] -
Banks, R. E.; Besheesh, M. K. J. Fluorine Chem. 1995, 74, 165–166. doi:10.1016/0022-1139(95)03295-o
Return to citation in text: [1] -
Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Fluorine Chem. 1997, 81, 157–161. doi:10.1016/s0022-1139(96)03521-x
Return to citation in text: [1] [2] -
Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. J. Org. Chem. 1998, 63, 3379–3385. doi:10.1021/jo972338q
Return to citation in text: [1] [2] [3] -
Laali, K. K.; Tanaka, M.; Forohar, F.; Cheng, M.; Fetzer, J. C. J. Fluorine Chem. 1998, 91, 185–190. doi:10.1016/s0022-1139(98)00224-3
Return to citation in text: [1] [2] [3] -
Forohar, F.; Damavarapu, R.; Jayasuriya, K.; Kwok, T. J. 13th Winter Fluorine Conference; St. Petersburg, Florida, USA, 1997; p 39.
Return to citation in text: [1] [2] -
Banks, R. E.; Besheesh, M. K.; Lawrence, N. J.; Tovell, D. J. J. Fluorine Chem. 1999, 97, 79–84. doi:10.1016/s0022-1139(99)00032-9
Return to citation in text: [1] [2] -
Takeuchi, Y.; Liu, Z.; Suzuki, E.; Shibata, N.; Kirk, K. L. J. Fluorine Chem. 1999, 97, 65–67. doi:10.1016/s0022-1139(99)00028-7
Return to citation in text: [1] [2] [3] -
Takeuchi, Y.; Suzuki, T.; Satoh, A.; Shiragami, T.; Shibata, N. J. Org. Chem. 1999, 64, 5708–5711. doi:10.1021/jo9904011
Return to citation in text: [1] [2] -
Takeuchi, Y.; Satoh, A.; Suzuki, T.; Kameda, A.; Dohrin, M.; Satoh, T.; Koizumi, T.; Kirk, K. L. Chem. Pharm. Bull. 1997, 45, 1085–1088. doi:10.1248/cpb.45.1085
Return to citation in text: [1] [2] -
Shibata, N.; Liu, Z.; Takeuchi, Y. Chem. Pharm. Bull. 2000, 48, 1954–1958. doi:10.1248/cpb.48.1954
Return to citation in text: [1] [2] -
Liu, Z.; Shibata, N.; Takeuchi, Y. J. Org. Chem. 2000, 65, 7583–7587. doi:10.1021/jo0055584
Return to citation in text: [1] [2] -
Shibata, N.; Suzuki, E.; Takeuchi, Y. J. Am. Chem. Soc. 2000, 122, 10728–10729. doi:10.1021/ja002732x
Return to citation in text: [1] [2] [3] -
Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t
Return to citation in text: [1] [2] -
Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Roques, N. Org. Lett. 2000, 2, 3699–3701. doi:10.1021/ol006610l
Return to citation in text: [1] [2] [3] -
Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Toupet, L.; Roques, N. Tetrahedron Lett. 2001, 42, 1867–1869. doi:10.1016/s0040-4039(01)00017-x
Return to citation in text: [1] [2] -
Baudequin, C.; Loubassou, J.-F.; Plaquevent, J.-C.; Cahard, D. J. Fluorine Chem. 2003, 122, 189–193. doi:10.1016/s0022-1139(03)00085-x
Return to citation in text: [1] [2] [3] -
Banks, R. E.; Besheesh, M. K.; Fraenk, W.; Klapötke, T. M. J. Fluorine Chem. 2003, 124, 229–232. doi:10.1016/j.jfluchem.2003.08.010
Return to citation in text: [1] [2] -
Schleyer, P. v. R.; Buzek, P.; Klapötke, T. M.; Tornieporth-Oetting, I. C.; Broschag, M.; Pickardt, J. Inorg. Chem. 1993, 32, 1523–1524. doi:10.1021/ic00060a033
Return to citation in text: [1] -
Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N. J. Fluorine Chem. 2011, 132, 222–225. doi:10.1016/j.jfluchem.2011.01.005
Return to citation in text: [1] [2] -
Chen, G.; Chen, F.; Zhang, Y.; Yang, X.; Yuan, X.; Wu, F.; Yang, X. J. Fluorine Chem. 2012, 133, 155–159. doi:10.1016/j.jfluchem.2011.10.012
Return to citation in text: [1] [2] -
Zhang, Y.; Yang, X.-J.; Xie, T.; Chen, G.-L.; Zhu, W.-H.; Zhang, X.-Q.; Yang, X.-Y.; Wu, X.-Y.; He, X.-P.; He, H.-M. Tetrahedron 2013, 69, 4933–4937. doi:10.1016/j.tet.2013.04.037
Return to citation in text: [1] [2] [3] -
Wang, F.; Li, J.; Hu, Q.; Yang, X.; Wu, X.-Y.; He, H. Eur. J. Org. Chem. 2014, 3607–3613. doi:10.1002/ejoc.201402072
Return to citation in text: [1] [2] -
Shunatona, H. P.; Früh, N.; Wang, Y.-M.; Rauniyar, V.; Toste, F. D. Angew. Chem., Int. Ed. 2013, 52, 7724–7727. doi:10.1002/anie.201302002
Return to citation in text: [1] [2] [3] -
Wolstenhulme, J. R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K. M.; Pidgeon, G. W.; Moore, P. R.; Sandford, G.; Gouverneur, V. Angew. Chem., Int. Ed. 2013, 52, 9796–9800. doi:10.1002/anie.201304845
Return to citation in text: [1] [2] [3] -
Zhu, C.-L.; Maeno, M.; Zhang, F.-G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.-A.; Cahard, D. Eur. J. Org. Chem. 2013, 6501–6505. doi:10.1002/ejoc.201300956
Return to citation in text: [1] [2] [3] -
Ratjen, L.; García-García, P.; Lay, F.; Beck, M. E.; List, B. Angew. Chem., Int. Ed. 2011, 50, 754–758. doi:10.1002/anie.201005954
Return to citation in text: [1] -
Treskow, M.; Neudörfl, J.; Giernoth, R. Eur. J. Org. Chem. 2009, 3693–3697. doi:10.1002/ejoc.200900548
Return to citation in text: [1] -
Fukushi, K.; Suzuki, S.; Kamo, T.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Kawada, K.; Shibata, N. Green Chem. 2016, 18, 1864–1868. doi:10.1039/c5gc02612a
Return to citation in text: [1] [2] [3] -
Bohlmann, R. N-Fluorosulfonimides, process for their production, as well as their use as fluorinating agents. WO Patent WO94/24098, Oct 27, 1994.
Return to citation in text: [1] -
Pereira, R.; Wolstenhulme, J.; Sandford, G.; Claridge, T. D. W.; Gouverneur, V.; Cvengroš, J. Chem. Commun. 2016, 52, 1606–1609. doi:10.1039/c5cc08375c
Return to citation in text: [1] [2] [3] -
Chen, C.; Luo, Y.; Fu, L.; Chen, P.; Lan, Y.; Liu, G. J. Am. Chem. Soc. 2018, 140, 1207–1210. doi:10.1021/jacs.7b11470
Return to citation in text: [1] [2] [3] -
Meyer, D.; Jangra, H.; Walther, F.; Zipse, H.; Renaud, P. Nat. Commun. 2018, 9, 4888. doi:10.1038/s41467-018-07196-9
Return to citation in text: [1] [2] [3] -
Hachisuka, H.; Kitano, M. J. Fluorine Chem. 1991, 54, 205. doi:10.1016/s0022-1139(00)83715-x
Return to citation in text: [1] -
Kitano, M.; Hachisuka, H.; Umemoto, T. 16th National Meeting on Fluorine Chemistry, Oct 21, 1991; Nagoya, Japan; pp 101–102.
Return to citation in text: [1] -
Sudlow, K.; Woolf, A. A. J. Fluorine Chem. 1994, 66, 9–11. doi:10.1016/0022-1139(93)02929-9
Return to citation in text: [1] -
Solkan, V. N.; Fainzil’berg, A. A. Russ. J. Org. Chem. 1994, 30, 1200.
Return to citation in text: [1] -
Fainzil'berg, A. A.; Faustov, V. I. Russ. J. Org. Chem. 2001, 37, 755–756. doi:10.1023/a:1012436925944
Return to citation in text: [1] -
Gilicinski, A. G.; Pez, G. P.; Syvret, R. G.; Lal, G. S. J. Fluorine Chem. 1992, 59, 157–162. doi:10.1016/s0022-1139(00)80214-6
Return to citation in text: [1] -
Differding, E.; Bersier, P. M. Tetrahedron 1992, 48, 1595–1604. doi:10.1016/s0040-4020(01)88718-4
Return to citation in text: [1] -
Oliver, E. W.; Evans, D. H. J. Electroanal. Chem. 1999, 474, 1–8. doi:10.1016/s0022-0728(99)00296-x
Return to citation in text: [1] -
Adachi, K.; Takahashi, I.; Harada, M.; Umemoto, T. J. Fluorine Chem. 2016, 183, 100–111. doi:10.1016/j.jfluchem.2015.09.013
Return to citation in text: [1] [2] -
Toullec, P. Y.; Devillers, I.; Frantz, R.; Togni, A. Helv. Chim. Acta 2004, 87, 2706–2711. doi:10.1002/hlca.200490240
Return to citation in text: [1] -
Xue, X.-S.; Wang, Y.; Li, M.; Cheng, J.-P. J. Org. Chem. 2016, 81, 4280–4289. doi:10.1021/acs.joc.6b00683
Return to citation in text: [1] -
Li, M.; Xue, X.-S.; Cheng, J.-P. Acc. Chem. Res. 2020, 53, 182–197. doi:10.1021/acs.accounts.9b00393
Return to citation in text: [1] [2] [3] [4] -
Christe, K. O.; Dixon, D. A. J. Am. Chem. Soc. 1992, 114, 2978–2985. doi:10.1021/ja00034a033
Return to citation in text: [1] -
Rueda-Becerril, M.; Chatalova Sazepin, C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.; Paquin, J.-F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026–4029. doi:10.1021/ja211679v
Return to citation in text: [1] -
Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583
Return to citation in text: [1] -
Shigehisa, H.; Aoki, T.; Yamaguchi, S.; Shimizu, N.; Hiroya, K. J. Am. Chem. Soc. 2013, 135, 10306–10309. doi:10.1021/ja405219f
Return to citation in text: [1] -
Shigehisa, H.; Nishi, E.; Fujisawa, M.; Hiroya, K. Org. Lett. 2013, 15, 5158–5161. doi:10.1021/ol402696h
Return to citation in text: [1] -
Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.-F.; Sammis, G. M. Synthesis 2015, 47, 2554–2569. doi:10.1055/s-0034-1378824
Return to citation in text: [1] [2] -
Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a
Return to citation in text: [1] -
Szpera, R.; Moseley, D. F. J.; Smith, L. B.; Sterling, A. J.; Gouverneur, V. Angew. Chem., Int. Ed. 2019, 58, 14824–14848. doi:10.1002/anie.201814457
Return to citation in text: [1] -
Yang, J.-D.; Wang, Y.; Xue, X.-S.; Cheng, J.-P. J. Org. Chem. 2017, 82, 4129–4135. doi:10.1021/acs.joc.7b00036
Return to citation in text: [1] -
Timofeeva, D. S.; Ofial, A. R.; Mayr, H. J. Am. Chem. Soc. 2018, 140, 11474–11486. doi:10.1021/jacs.8b07147
Return to citation in text: [1] [2] -
Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b
Return to citation in text: [1] [2] [3] -
Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k
Return to citation in text: [1] -
Rozatian, N.; Harsanyi, A.; Murray, B. J.; Hampton, A. S.; Chin, E. J.; Cook, A. S.; Hodgson, D. R. W.; Sandford, G. Chem. – Eur. J. 2020, 26, 12027–12035. doi:10.1002/chem.202001120
Return to citation in text: [1] -
Du, X.; Zhang, H.; Yao, Y.; Lu, Y.; Wang, A.; Wang, Y.; Li, Z. Theor. Chem. Acc. 2019, 138, No. 28. doi:10.1007/s00214-019-2417-2
Return to citation in text: [1] -
Lal, G. S.; Pez, G. P.; Syvret, R. G. Chem. Rev. 1996, 96, 1737–1756. doi:10.1021/cr941145p
Return to citation in text: [1] [2] -
Lee, K. Y.; Kochi, J. K. J. Chem. Soc., Perkin Trans. 2 1992, 1011–1017. doi:10.1039/p29920001011
Return to citation in text: [1] -
Bockman, T. M.; Lee, K. Y.; Kochi, J. K. J. Chem. Soc., Perkin Trans. 2 1992, 1581–1594. doi:10.1039/p29920001581
Return to citation in text: [1] -
Differding, E.; Rüegg, G. M. Tetrahedron Lett. 1991, 32, 3815–3818. doi:10.1016/s0040-4039(00)79383-x
Return to citation in text: [1] [2] -
Differring, E.; Wehrli, M. Tetrahedron Lett. 1991, 32, 3819–3822. doi:10.1016/s0040-4039(00)79384-1
Return to citation in text: [1] -
Zupan, M.; Škulj, P.; Stavber, S. Chem. Lett. 1998, 27, 641–642. doi:10.1246/cl.1998.641
Return to citation in text: [1] -
Vincent, S. P.; Burkart, M. D.; Tsai, C.-Y.; Zhang, Z.; Wong, C.-H. J. Org. Chem. 1999, 64, 5264–5279. doi:10.1021/jo990686h
Return to citation in text: [1] [2] -
Nyffeler, P. T.; Durón, S. G.; Burkart, M. D.; Vincent, S. P.; Wong, C.-H. Angew. Chem., Int. Ed. 2005, 44, 192–212. doi:10.1002/anie.200400648
Return to citation in text: [1] [2] -
Piana, S.; Devillers, I.; Togni, A.; Rothlisberger, U. Angew. Chem., Int. Ed. 2002, 41, 979–982. doi:10.1002/1521-3773(20020315)41:6<979::aid-anie979>3.0.co;2-e
Return to citation in text: [1] -
Laali, K. K.; Borodkin, G. I. J. Chem. Soc., Perkin Trans. 2 2002, 953–957. doi:10.1039/b111725d
Return to citation in text: [1] -
Kralj, P.; Zupan, M.; Stavber, S. J. Org. Chem. 2006, 71, 3880–3888. doi:10.1021/jo060213s
Return to citation in text: [1] -
Andreev, R. V.; Borodkin, G. I.; Shubin, V. G. Russ. J. Org. Chem. 2009, 45, 1468–1473. doi:10.1134/s107042800910008x
Return to citation in text: [1] -
Geng, C.; Du, L.; Liu, F.; Zhu, R.; Liu, C. RSC Adv. 2015, 5, 33385–33391. doi:10.1039/c4ra15202f
Return to citation in text: [1] -
Chen, Q.; Zeng, J.; Yan, X.; Huang, Y.; Wen, C.; Liu, X.; Zhang, K. J. Org. Chem. 2016, 81, 10043–10048. doi:10.1021/acs.joc.6b01932
Return to citation in text: [1] -
Li, C.-T.; Yuan, X.; Tang, Z.-Y. Tetrahedron Lett. 2016, 57, 5624–5627. doi:10.1016/j.tetlet.2016.11.003
Return to citation in text: [1] -
Wu, S.-W.; Liu, F. Org. Lett. 2016, 18, 3642–3645. doi:10.1021/acs.orglett.6b01691
Return to citation in text: [1] -
Wu, S.-W.; Liu, J.-L.; Liu, F. Chem. Commun. 2017, 53, 12321–12324. doi:10.1039/c7cc07165e
Return to citation in text: [1] -
Zhang, Y.; Wen, C.; Li, J. Org. Biomol. Chem. 2018, 16, 1912–1920. doi:10.1039/c7ob03059b
Return to citation in text: [1] -
Fan, X.; Wang, Q.; Wei, Y.; Shi, M. Chem. Commun. 2018, 54, 10503–10506. doi:10.1039/c8cc05634j
Return to citation in text: [1] -
Zhou, J.; Fang, Y.; Wang, F.; Li, J. Org. Biomol. Chem. 2019, 17, 4470–4474. doi:10.1039/c9ob00467j
Return to citation in text: [1] -
Jójárt, R.; Traj, P.; Kovács, É.; Horváth, Á.; Schneider, G.; Szécsi, M.; Pál, A.; Paragi, G.; Mernyák, E. Molecules 2019, 24, 1783. doi:10.3390/molecules24091783
Return to citation in text: [1] -
Wood, S. H.; Etridge, S.; Kennedy, A. R.; Percy, J. M.; Nelson, D. J. Chem. – Eur. J. 2019, 25, 5574–5585. doi:10.1002/chem.201900029
Return to citation in text: [1] [2] -
Borodkin, G. I.; Elanov, I. R.; Gatilov, Y. V.; Shubin, V. G. J. Fluorine Chem. 2019, 228, 109412. doi:10.1016/j.jfluchem.2019.109412
Return to citation in text: [1] -
Rozatian, N.; Hodgson, D. R. W. Chem. Commun. 2021, 57, 683–712. doi:10.1039/d0cc06339h
Return to citation in text: [1] [2] -
Furin, G. G. In Methods of Organic Chemistry (Houben-Weyl), Organo-fluorine Compounds Vol E10a; Baasner, B.; Hagemann, H.; Tatlow, J. C., Eds.; Thieme: Stuttgart, New York, 1999; pp 432–499.
Return to citation in text: [1] -
Banks, R. E. J. Fluorine Chem. 1998, 87, 1–17. doi:10.1016/s0022-1139(97)00127-9
Return to citation in text: [1] -
Singh, R. P.; Shreeve, J. M. Acc. Chem. Res. 2004, 37, 31–44. doi:10.1021/ar030043v
Return to citation in text: [1] -
Kiselyov, A. S. Chem. Soc. Rev. 2005, 34, 1031–1037. doi:10.1039/b509217p
Return to citation in text: [1] -
Umemoto, T. N-Fluoropyridinium Salts, Synthesis and Fluorination Chemistry. In Advances in Organic Synthesis; Laali, K. K., Ed.; Bentham Science Publishers Ltd.: Netherlands, 2006; Vol. 2, pp 159–181. doi:10.2174/978160805198410602010159
Return to citation in text: [1] -
Umemoto, T. Preparation, Reactivity, and Applications of N-Fluoropyridinium Salts. In Fluorinated Heterocycles; Gakh, A. A.; Kirk, K. L., Eds.; ACS Symposium Series, Vol. 1003; American Chemical Society: Washington, DC, USA, 2009; pp 37–58. doi:10.1021/bk-2009-1003.ch003
Return to citation in text: [1] -
Baudoux, J.; Cahard, D. Electrophilic Fluorination with N–F Reagents. In Organic Reactions; Denmark, S. E., Ed.; John Wiley & Sons, 2008; pp 347–672. doi:10.1002/0471264180.or069.02
Return to citation in text: [1] -
de Azambuja, F.; Altman, R. A. NFSI and Its Analogs Fluorination for Preparing Alkenyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_20-1
Return to citation in text: [1] -
Yan, H.; Zhu, C. NFSI Radical Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_1-1
Return to citation in text: [1] -
Guyon, H.; Cahard, D. Selectfluor and Its Analogs Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_8-1
Return to citation in text: [1] -
de Azambuja, F.; Altman, R. A. SelectFluor and Its Analogs Fluorination for Preparing Alkenyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_19-1
Return to citation in text: [1] -
Yan, H.; Zhu, C. SelectFluor Radical Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_2-1
Return to citation in text: [1] -
Chen, C.; Chen, P.; Liu, G. N,N′-Difluoro-1,4-Diazoniabicyclo[2.2.2]octane Salt Electrophilic Fluorination. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_47-1
Return to citation in text: [1] -
Kawada, K. N-Fluoropyridinium Salt Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_21-2
Return to citation in text: [1] -
Kawada, K. N-Fluoropyridinium Salt Radical Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_41-1
Return to citation in text: [1] -
Wang, G.; Ma, J.-A. NFSI and Its Analogs Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_6-1
Return to citation in text: [1]
| 65. | Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023 |
| 66. | Stavber, S.; Sotler, T.; Zupan, M. Tetrahedron Lett. 1994, 35, 1105–1108. doi:10.1016/s0040-4039(00)79977-1 |
| 65. | Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023 |
| 67. | Lal, G. S. Synth. Commun. 1995, 25, 725–737. doi:10.1080/00397919508011410 |
| 138. | Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b |
| 63. | Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Chem. Soc., Perkin Trans. 1 1996, 2069–2076. doi:10.1039/p19960002069 |
| 64. | Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Fluorine Chem. 1996, 78, 43–50. doi:10.1016/0022-1139(96)03395-7 |
| 137. | Timofeeva, D. S.; Ofial, A. R.; Mayr, H. J. Am. Chem. Soc. 2018, 140, 11474–11486. doi:10.1021/jacs.8b07147 |
| 156. | Li, C.-T.; Yuan, X.; Tang, Z.-Y. Tetrahedron Lett. 2016, 57, 5624–5627. doi:10.1016/j.tetlet.2016.11.003 |
| 155. | Chen, Q.; Zeng, J.; Yan, X.; Huang, Y.; Wen, C.; Liu, X.; Zhang, K. J. Org. Chem. 2016, 81, 10043–10048. doi:10.1021/acs.joc.6b01932 |
| 70. | Zupan, M.; Iskra, J.; Stavber, S. J. Org. Chem. 1995, 60, 259–260. doi:10.1021/jo00106a045 |
| 158. | Wu, S.-W.; Liu, J.-L.; Liu, F. Chem. Commun. 2017, 53, 12321–12324. doi:10.1039/c7cc07165e |
| 157. | Wu, S.-W.; Liu, F. Org. Lett. 2016, 18, 3642–3645. doi:10.1021/acs.orglett.6b01691 |
| 152. | Kralj, P.; Zupan, M.; Stavber, S. J. Org. Chem. 2006, 71, 3880–3888. doi:10.1021/jo060213s |
| 67. | Lal, G. S. Synth. Commun. 1995, 25, 725–737. doi:10.1080/00397919508011410 |
| 69. | Lal, G. S.; Pastore, W.; Pesaresi, R. J. Org. Chem. 1995, 60, 7340–7342. doi:10.1021/jo00127a046 |
| 151. | Laali, K. K.; Borodkin, G. I. J. Chem. Soc., Perkin Trans. 2 2002, 953–957. doi:10.1039/b111725d |
| 65. | Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023 |
| 68. | Brown, D. S.; Marples, B. A.; Smith, P.; Walton, L. Tetrahedron 1995, 51, 3587–3606. doi:10.1016/0040-4020(95)00075-j |
| 154. | Geng, C.; Du, L.; Liu, F.; Zhu, R.; Liu, C. RSC Adv. 2015, 5, 33385–33391. doi:10.1039/c4ra15202f |
| 153. | Andreev, R. V.; Borodkin, G. I.; Shubin, V. G. Russ. J. Org. Chem. 2009, 45, 1468–1473. doi:10.1134/s107042800910008x |
| 159. | Zhang, Y.; Wen, C.; Li, J. Org. Biomol. Chem. 2018, 16, 1912–1920. doi:10.1039/c7ob03059b |
| 75. | Davis, F. A.; Zhou, P.; Murphy, C. K. Tetrahedron Lett. 1993, 34, 3971–3974. doi:10.1016/s0040-4039(00)60592-0 |
| 76. | Umemoto, T.; Tomizawa, G. J. Org. Chem. 1995, 60, 6563–6570. doi:10.1021/jo00125a049 |
| 73. | Hodson, H. F.; Madge, D. J.; Slawin, A. N. Z.; Widdowson, D. A.; Williams, D. J. Tetrahedron 1994, 50, 1899–1906. doi:10.1016/s0040-4020(01)80862-0 |
| 74. | McClinton, M. A.; Sik, V. J. Chem. Soc., Perkin Trans. 1 1992, 1891–1895. doi:10.1039/p19920001891 |
| 71. | Brunavs, M.; Dell, C. P.; Owton, W. M. J. Fluorine Chem. 1994, 68, 201–203. doi:10.1016/0022-1139(93)03042-k |
| 72. | Matthews, D. P.; Miller, S. C.; Jarvi, E. T.; Sabol, J. S.; McCarthy, J. R. Tetrahedron Lett. 1993, 34, 3057–3060. doi:10.1016/s0040-4039(00)93378-1 |
| 16. | Banks, R. E.; Williamson, G. E. Chem. Ind. (London) 1964, 1864–1865. |
| 17. | Banks, R. E.; Cheng, W. M.; Haszeldine, R. N. J. Chem. Soc. 1962, 3407–3416. doi:10.1039/jr9620003407 |
| 18. | Banks, R. E.; Ginsberg, A. E.; Haszeldine, R. N. J. Chem. Soc. 1961, 1740–1743. doi:10.1039/jr9610001740 |
| 19. | Banks, R. E.; Haszeldine, R. N.; Hatton, R. Tetrahedron Lett. 1967, 8, 3993–3996. doi:10.1016/s0040-4039(01)89722-7 |
| 20. | Polishchuk, V. R.; German, L. S. Tetrahedron Lett. 1972, 13, 5169–5172. doi:10.1016/s0040-4039(01)85198-4 |
| 21. | Banks, R. E.; Tsiliopoulos, E. J. Fluorine Chem. 1986, 34, 281–285. doi:10.1016/s0022-1139(00)85081-2 |
| 22. | Banks, R. E.; Murtagh, V.; Tsiliopoulos, E. J. Fluorine Chem. 1991, 52, 389–401. doi:10.1016/s0022-1139(00)80353-x |
| 164. | Borodkin, G. I.; Elanov, I. R.; Gatilov, Y. V.; Shubin, V. G. J. Fluorine Chem. 2019, 228, 109412. doi:10.1016/j.jfluchem.2019.109412 |
| 163. | Wood, S. H.; Etridge, S.; Kennedy, A. R.; Percy, J. M.; Nelson, D. J. Chem. – Eur. J. 2019, 25, 5574–5585. doi:10.1002/chem.201900029 |
| 39. | Kawada, K. N-Fluoropyridinium Salt Fluorination for Preparing Aryl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2020. doi:10.1007/978-981-10-1855-8_40-2 |
| 133. | Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.-F.; Sammis, G. M. Synthesis 2015, 47, 2554–2569. doi:10.1055/s-0034-1378824 |
| 142. | Lal, G. S.; Pez, G. P.; Syvret, R. G. Chem. Rev. 1996, 96, 1737–1756. doi:10.1021/cr941145p |
| 165. | Rozatian, N.; Hodgson, D. R. W. Chem. Commun. 2021, 57, 683–712. doi:10.1039/d0cc06339h |
| 166. | Furin, G. G. In Methods of Organic Chemistry (Houben-Weyl), Organo-fluorine Compounds Vol E10a; Baasner, B.; Hagemann, H.; Tatlow, J. C., Eds.; Thieme: Stuttgart, New York, 1999; pp 432–499. |
| 167. | Banks, R. E. J. Fluorine Chem. 1998, 87, 1–17. doi:10.1016/s0022-1139(97)00127-9 |
| 168. | Singh, R. P.; Shreeve, J. M. Acc. Chem. Res. 2004, 37, 31–44. doi:10.1021/ar030043v |
| 169. | Kiselyov, A. S. Chem. Soc. Rev. 2005, 34, 1031–1037. doi:10.1039/b509217p |
| 170. | Umemoto, T. N-Fluoropyridinium Salts, Synthesis and Fluorination Chemistry. In Advances in Organic Synthesis; Laali, K. K., Ed.; Bentham Science Publishers Ltd.: Netherlands, 2006; Vol. 2, pp 159–181. doi:10.2174/978160805198410602010159 |
| 171. | Umemoto, T. Preparation, Reactivity, and Applications of N-Fluoropyridinium Salts. In Fluorinated Heterocycles; Gakh, A. A.; Kirk, K. L., Eds.; ACS Symposium Series, Vol. 1003; American Chemical Society: Washington, DC, USA, 2009; pp 37–58. doi:10.1021/bk-2009-1003.ch003 |
| 172. | Baudoux, J.; Cahard, D. Electrophilic Fluorination with N–F Reagents. In Organic Reactions; Denmark, S. E., Ed.; John Wiley & Sons, 2008; pp 347–672. doi:10.1002/0471264180.or069.02 |
| 173. | de Azambuja, F.; Altman, R. A. NFSI and Its Analogs Fluorination for Preparing Alkenyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_20-1 |
| 174. | Yan, H.; Zhu, C. NFSI Radical Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_1-1 |
| 175. | Guyon, H.; Cahard, D. Selectfluor and Its Analogs Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_8-1 |
| 176. | de Azambuja, F.; Altman, R. A. SelectFluor and Its Analogs Fluorination for Preparing Alkenyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_19-1 |
| 177. | Yan, H.; Zhu, C. SelectFluor Radical Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_2-1 |
| 178. | Chen, C.; Chen, P.; Liu, G. N,N′-Difluoro-1,4-Diazoniabicyclo[2.2.2]octane Salt Electrophilic Fluorination. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_47-1 |
| 179. | Kawada, K. N-Fluoropyridinium Salt Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_21-2 |
| 180. | Kawada, K. N-Fluoropyridinium Salt Radical Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_41-1 |
| 181. | Wang, G.; Ma, J.-A. NFSI and Its Analogs Electrophilic Fluorination for Preparing Alkyl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2018. doi:10.1007/978-981-10-1855-8_6-1 |
| 165. | Rozatian, N.; Hodgson, D. R. W. Chem. Commun. 2021, 57, 683–712. doi:10.1039/d0cc06339h |
| 78. | Poss, A. J.; Shia, G. 1-Substituted-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts and their application as fluorinating agents. U.S. Patent US5,459,267, Oct 17, 1995. |
| 161. | Zhou, J.; Fang, Y.; Wang, F.; Li, J. Org. Biomol. Chem. 2019, 17, 4470–4474. doi:10.1039/c9ob00467j |
| 79. | Stavber, S.; Zupan, M. Tetrahedron Lett. 1996, 37, 3591–3594. doi:10.1016/0040-4039(96)00630-2 |
| 80. | Poss, A. J.; Shia, G. A. Tetrahedron Lett. 1999, 40, 2673–2676. doi:10.1016/s0040-4039(99)00303-2 |
| 160. | Fan, X.; Wang, Q.; Wei, Y.; Shi, M. Chem. Commun. 2018, 54, 10503–10506. doi:10.1039/c8cc05634j |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 163. | Wood, S. H.; Etridge, S.; Kennedy, A. R.; Percy, J. M.; Nelson, D. J. Chem. – Eur. J. 2019, 25, 5574–5585. doi:10.1002/chem.201900029 |
| 77. | MEC Reagents: Fluorinating agents and Perfluoroalkylating agents; Daikin Chemical Sales company Brochure, 1997. |
| 162. | Jójárt, R.; Traj, P.; Kovács, É.; Horváth, Á.; Schneider, G.; Szécsi, M.; Pál, A.; Paragi, G.; Mernyák, E. Molecules 2019, 24, 1783. doi:10.3390/molecules24091783 |
| 24. | Purrington, S. T.; Jones, W. A. J. Org. Chem. 1983, 48, 761–762. doi:10.1021/jo00153a037 |
| 25. | Purrington, S. T.; Jones, W. A. J. Fluorine Chem. 1984, 26, 43–46. doi:10.1016/s0022-1139(00)85117-9 |
| 29. | Umemoto, T.; Kawada, K.; Tomita, K. Tetrahedron Lett. 1986, 27, 4465–4468. doi:10.1016/s0040-4039(00)84980-1 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 45. | Singh, S.; DesMarteau, D. D.; Zuberi, S. S.; Witz, M.; Huang, H.-N. J. Am. Chem. Soc. 1987, 109, 7194–7196. doi:10.1021/ja00257a051 |
| 26. | Barnette, W. E. J. Am. Chem. Soc. 1984, 106, 452–454. doi:10.1021/ja00314a050 |
| 26. | Barnette, W. E. J. Am. Chem. Soc. 1984, 106, 452–454. doi:10.1021/ja00314a050 |
| 52. | Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087–6090. doi:10.1016/s0040-4039(00)82271-6 |
| 50. | Banks, R. E.; Sharif, I. J. Fluorine Chem. 1988, 41, 297–300. doi:10.1016/s0022-1139(00)81553-5 |
| 51. | Banks, R. E.; Sharif, I. J. Fluorine Chem. 1991, 55, 207–214. doi:10.1016/s0022-1139(00)80123-2 |
| 52. | Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087–6090. doi:10.1016/s0040-4039(00)82271-6 |
| 54. | Banks, R. E.; Khazaei, A. J. Fluorine Chem. 1990, 46, 297–305. doi:10.1016/s0022-1139(00)80997-5 |
| 53. | Differding, E.; Lang, R. W. Helv. Chim. Acta 1989, 72, 1248–1252. doi:10.1002/hlca.19890720609 |
| 53. | Differding, E.; Lang, R. W. Helv. Chim. Acta 1989, 72, 1248–1252. doi:10.1002/hlca.19890720609 |
| 28. | Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3 |
| 28. | Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3 |
| 29. | Umemoto, T.; Kawada, K.; Tomita, K. Tetrahedron Lett. 1986, 27, 4465–4468. doi:10.1016/s0040-4039(00)84980-1 |
| 30. | Umemoto, T.; Tomizawa, G. Bull. Chem. Soc. Jpn. 1986, 59, 3625–3629. doi:10.1246/bcsj.59.3625 |
| 31. | Umemoto, T.; Tomita, K.; Kawada, K. Org. Synth. 1990, 69, 129–140. doi:10.15227/orgsyn.069.0129 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 33. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. Bull. Chem. Soc. Jpn. 1991, 64, 1081–1092. doi:10.1246/bcsj.64.1081 |
| 34. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. J. Fluorine Chem. 1991, 53, 369–377. doi:10.1016/s0022-1139(00)80960-4 |
| 36. | Umemoto, T.; Tomita, K. N-Fluoropyridinium salt-containing polymer. Jpn. Kokai Tokkyo Koho S63-295610, Dec 2, 1988. |
| 37. | Nukui, K.; Kamiya, K. Production method of N-fluoropyridinium salts. Jpn. Patent 3038065, May 8, 2000. |
| 38. | Fukami, S.; Nukui, K.; Kawada, K. Production method of N-fluoropyridinium tetrafluoroborate. Jpn. Kokai Tokkyo Koho H09-255657, Sept 30, 1997. |
| 39. | Kawada, K. N-Fluoropyridinium Salt Fluorination for Preparing Aryl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2020. doi:10.1007/978-981-10-1855-8_40-2 |
| 40. | Naruta, Y.; Tani, F.; Maruyama, K. Tetrahedron Lett. 1992, 33, 1069–1072. doi:10.1016/s0040-4039(00)91863-x |
| 41. | Poss, A. J.; Van der Puy, M.; Nalewajek, D.; Shia, G. A.; Wagner, W. J.; Frenette, R. L. J. Org. Chem. 1991, 56, 5962–5964. doi:10.1021/jo00020a051 |
| 51. | Banks, R. E.; Sharif, I. J. Fluorine Chem. 1991, 55, 207–214. doi:10.1016/s0022-1139(00)80123-2 |
| 26. | Barnette, W. E. J. Am. Chem. Soc. 1984, 106, 452–454. doi:10.1021/ja00314a050 |
| 27. | Lee, S. H.; Schwartz, J. J. Am. Chem. Soc. 1986, 108, 2445–2447. doi:10.1021/ja00269a052 |
| 46. | Resnati, G.; DesMarteau, D. D. J. Org. Chem. 1991, 56, 4925–4929. doi:10.1021/jo00016a022 |
| 47. | Xu, Z.-Q.; DesMarteau, D. D.; Gotoh, Y. J. Chem. Soc., Chem. Commun. 1991, 179–181. doi:10.1039/c39910000179 |
| 48. | DesMarteau, D. D.; Xu, Z.-Q.; Witz, M. J. Org. Chem. 1992, 57, 629–635. doi:10.1021/jo00028a042 |
| 49. | Pennington, W. T.; Resnati, G.; DesMarteau, D. D. J. Org. Chem. 1992, 57, 1536–1539. doi:10.1021/jo00031a038 |
| 45. | Singh, S.; DesMarteau, D. D.; Zuberi, S. S.; Witz, M.; Huang, H.-N. J. Am. Chem. Soc. 1987, 109, 7194–7196. doi:10.1021/ja00257a051 |
| 46. | Resnati, G.; DesMarteau, D. D. J. Org. Chem. 1991, 56, 4925–4929. doi:10.1021/jo00016a022 |
| 47. | Xu, Z.-Q.; DesMarteau, D. D.; Gotoh, Y. J. Chem. Soc., Chem. Commun. 1991, 179–181. doi:10.1039/c39910000179 |
| 48. | DesMarteau, D. D.; Xu, Z.-Q.; Witz, M. J. Org. Chem. 1992, 57, 629–635. doi:10.1021/jo00028a042 |
| 49. | Pennington, W. T.; Resnati, G.; DesMarteau, D. D. J. Org. Chem. 1992, 57, 1536–1539. doi:10.1021/jo00031a038 |
| 50. | Banks, R. E.; Sharif, I. J. Fluorine Chem. 1988, 41, 297–300. doi:10.1016/s0022-1139(00)81553-5 |
| 43. | Banks, R. E.; Du Boisson, R. A.; Tsiliopoulos, E. J. Fluorine Chem. 1986, 32, 461–466. doi:10.1016/s0022-1139(00)81953-3 |
| 44. | Banks, R. E.; Du Boisson, R. A.; Morton, W. D.; Tsiliopoulos, E. J. Chem. Soc., Perkin Trans. 1 1988, 2805–2811. doi:10.1039/p19880002805 |
| 56. | Grakauskas, V.; Baum, K. J. Org. Chem. 1970, 35, 1545–1549. doi:10.1021/jo00830a061 |
| 57. | Davis, F. A.; Han, W. Tetrahedron Lett. 1991, 32, 1631–1634. doi:10.1016/s0040-4039(00)74290-0 |
| 54. | Banks, R. E.; Khazaei, A. J. Fluorine Chem. 1990, 46, 297–305. doi:10.1016/s0022-1139(00)80997-5 |
| 55. | Satyamurthy, N.; Bida, G. T.; Phelps, M. E.; Barrio, J. R. J. Org. Chem. 1990, 55, 3373–3374. doi:10.1021/jo00297a071 |
| 61. | Banks, R. E.; Lawrence, N. J.; Popplewell, A. L. J. Chem. Soc., Chem. Commun. 1994, 343–344. doi:10.1039/c39940000343 |
| 62. | Abdul-Ghani, M.; Banks, E. R.; Besheesh, M. K.; Sharif, I.; Syvret, R. G. J. Fluorine Chem. 1995, 73, 255–257. doi:10.1016/0022-1139(94)03225-o |
| 60. | Gakh, A. A.; Romaniko, S. V.; Ugrak, B. I.; Fainzilberg, A. A. Tetrahedron 1991, 47, 7447–7458. doi:10.1016/s0040-4020(01)89746-5 |
| 42. | Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595 |
| 58. | Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730–4737. doi:10.1021/jo00120a014 |
| 123. | Oliver, E. W.; Evans, D. H. J. Electroanal. Chem. 1999, 474, 1–8. doi:10.1016/s0022-0728(99)00296-x |
| 122. | Differding, E.; Bersier, P. M. Tetrahedron 1992, 48, 1595–1604. doi:10.1016/s0040-4020(01)88718-4 |
| 119. | Solkan, V. N.; Fainzil’berg, A. A. Russ. J. Org. Chem. 1994, 30, 1200. |
| 120. | Fainzil'berg, A. A.; Faustov, V. I. Russ. J. Org. Chem. 2001, 37, 755–756. doi:10.1023/a:1012436925944 |
| 113. | Pereira, R.; Wolstenhulme, J.; Sandford, G.; Claridge, T. D. W.; Gouverneur, V.; Cvengroš, J. Chem. Commun. 2016, 52, 1606–1609. doi:10.1039/c5cc08375c |
| 118. | Sudlow, K.; Woolf, A. A. J. Fluorine Chem. 1994, 66, 9–11. doi:10.1016/0022-1139(93)02929-9 |
| 111. | Fukushi, K.; Suzuki, S.; Kamo, T.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Kawada, K.; Shibata, N. Green Chem. 2016, 18, 1864–1868. doi:10.1039/c5gc02612a |
| 121. | Gilicinski, A. G.; Pez, G. P.; Syvret, R. G.; Lal, G. S. J. Fluorine Chem. 1992, 59, 157–162. doi:10.1016/s0022-1139(00)80214-6 |
| 34. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. J. Fluorine Chem. 1991, 53, 369–377. doi:10.1016/s0022-1139(00)80960-4 |
| 115. | Meyer, D.; Jangra, H.; Walther, F.; Zipse, H.; Renaud, P. Nat. Commun. 2018, 9, 4888. doi:10.1038/s41467-018-07196-9 |
| 114. | Chen, C.; Luo, Y.; Fu, L.; Chen, P.; Lan, Y.; Liu, G. J. Am. Chem. Soc. 2018, 140, 1207–1210. doi:10.1021/jacs.7b11470 |
| 99. | Baudequin, C.; Loubassou, J.-F.; Plaquevent, J.-C.; Cahard, D. J. Fluorine Chem. 2003, 122, 189–193. doi:10.1016/s0022-1139(03)00085-x |
| 114. | Chen, C.; Luo, Y.; Fu, L.; Chen, P.; Lan, Y.; Liu, G. J. Am. Chem. Soc. 2018, 140, 1207–1210. doi:10.1021/jacs.7b11470 |
| 97. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Roques, N. Org. Lett. 2000, 2, 3699–3701. doi:10.1021/ol006610l |
| 116. | Hachisuka, H.; Kitano, M. J. Fluorine Chem. 1991, 54, 205. doi:10.1016/s0022-1139(00)83715-x |
| 117. | Kitano, M.; Hachisuka, H.; Umemoto, T. 16th National Meeting on Fluorine Chemistry, Oct 21, 1991; Nagoya, Japan; pp 101–102. |
| 108. | Zhu, C.-L.; Maeno, M.; Zhang, F.-G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.-A.; Cahard, D. Eur. J. Org. Chem. 2013, 6501–6505. doi:10.1002/ejoc.201300956 |
| 115. | Meyer, D.; Jangra, H.; Walther, F.; Zipse, H.; Renaud, P. Nat. Commun. 2018, 9, 4888. doi:10.1038/s41467-018-07196-9 |
| 107. | Wolstenhulme, J. R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K. M.; Pidgeon, G. W.; Moore, P. R.; Sandford, G.; Gouverneur, V. Angew. Chem., Int. Ed. 2013, 52, 9796–9800. doi:10.1002/anie.201304845 |
| 95. | Shibata, N.; Suzuki, E.; Takeuchi, Y. J. Am. Chem. Soc. 2000, 122, 10728–10729. doi:10.1021/ja002732x |
| 127. | Li, M.; Xue, X.-S.; Cheng, J.-P. Acc. Chem. Res. 2020, 53, 182–197. doi:10.1021/acs.accounts.9b00393 |
| 127. | Li, M.; Xue, X.-S.; Cheng, J.-P. Acc. Chem. Res. 2020, 53, 182–197. doi:10.1021/acs.accounts.9b00393 |
| 128. | Christe, K. O.; Dixon, D. A. J. Am. Chem. Soc. 1992, 114, 2978–2985. doi:10.1021/ja00034a033 |
| 127. | Li, M.; Xue, X.-S.; Cheng, J.-P. Acc. Chem. Res. 2020, 53, 182–197. doi:10.1021/acs.accounts.9b00393 |
| 136. | Yang, J.-D.; Wang, Y.; Xue, X.-S.; Cheng, J.-P. J. Org. Chem. 2017, 82, 4129–4135. doi:10.1021/acs.joc.7b00036 |
| 129. | Rueda-Becerril, M.; Chatalova Sazepin, C.; Leung, J. C. T.; Okbinoglu, T.; Kennepohl, P.; Paquin, J.-F.; Sammis, G. M. J. Am. Chem. Soc. 2012, 134, 4026–4029. doi:10.1021/ja211679v |
| 130. | Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583 |
| 131. | Shigehisa, H.; Aoki, T.; Yamaguchi, S.; Shimizu, N.; Hiroya, K. J. Am. Chem. Soc. 2013, 135, 10306–10309. doi:10.1021/ja405219f |
| 132. | Shigehisa, H.; Nishi, E.; Fujisawa, M.; Hiroya, K. Org. Lett. 2013, 15, 5158–5161. doi:10.1021/ol402696h |
| 133. | Chatalova-Sazepin, C.; Hemelaere, R.; Paquin, J.-F.; Sammis, G. M. Synthesis 2015, 47, 2554–2569. doi:10.1055/s-0034-1378824 |
| 134. | Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a |
| 135. | Szpera, R.; Moseley, D. F. J.; Smith, L. B.; Sterling, A. J.; Gouverneur, V. Angew. Chem., Int. Ed. 2019, 58, 14824–14848. doi:10.1002/anie.201814457 |
| 125. | Toullec, P. Y.; Devillers, I.; Frantz, R.; Togni, A. Helv. Chim. Acta 2004, 87, 2706–2711. doi:10.1002/hlca.200490240 |
| 124. | Adachi, K.; Takahashi, I.; Harada, M.; Umemoto, T. J. Fluorine Chem. 2016, 183, 100–111. doi:10.1016/j.jfluchem.2015.09.013 |
| 126. | Xue, X.-S.; Wang, Y.; Li, M.; Cheng, J.-P. J. Org. Chem. 2016, 81, 4280–4289. doi:10.1021/acs.joc.6b00683 |
| 127. | Li, M.; Xue, X.-S.; Cheng, J.-P. Acc. Chem. Res. 2020, 53, 182–197. doi:10.1021/acs.accounts.9b00393 |
| 124. | Adachi, K.; Takahashi, I.; Harada, M.; Umemoto, T. J. Fluorine Chem. 2016, 183, 100–111. doi:10.1016/j.jfluchem.2015.09.013 |
| 104. | Zhang, Y.; Yang, X.-J.; Xie, T.; Chen, G.-L.; Zhu, W.-H.; Zhang, X.-Q.; Yang, X.-Y.; Wu, X.-Y.; He, X.-P.; He, H.-M. Tetrahedron 2013, 69, 4933–4937. doi:10.1016/j.tet.2013.04.037 |
| 85. | Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Fluorine Chem. 1997, 81, 157–161. doi:10.1016/s0022-1139(96)03521-x |
| 86. | Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. J. Org. Chem. 1998, 63, 3379–3385. doi:10.1021/jo972338q |
| 42. | Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595 |
| 84. | Banks, R. E.; Besheesh, M. K. J. Fluorine Chem. 1995, 74, 165–166. doi:10.1016/0022-1139(95)03295-o |
| 82. | Banks, R. E.; Besheesh, M. K.; Tsiliopoulos, E. J. Fluorine Chem. 1996, 78, 39–42. doi:10.1016/0022-1139(95)03380-7 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 83. | Umemoto, T.; Nagayoshi, M. Bull. Chem. Soc. Jpn. 1996, 69, 2287–2295. doi:10.1246/bcsj.69.2287 |
| 22. | Banks, R. E.; Murtagh, V.; Tsiliopoulos, E. J. Fluorine Chem. 1991, 52, 389–401. doi:10.1016/s0022-1139(00)80353-x |
| 142. | Lal, G. S.; Pez, G. P.; Syvret, R. G. Chem. Rev. 1996, 96, 1737–1756. doi:10.1021/cr941145p |
| 48. | DesMarteau, D. D.; Xu, Z.-Q.; Witz, M. J. Org. Chem. 1992, 57, 629–635. doi:10.1021/jo00028a042 |
| 81. | Cabrera, I.; Appel, W. K. Tetrahedron 1995, 51, 10205–10208. doi:10.1016/0040-4020(95)00597-2 |
| 143. | Lee, K. Y.; Kochi, J. K. J. Chem. Soc., Perkin Trans. 2 1992, 1011–1017. doi:10.1039/p29920001011 |
| 144. | Bockman, T. M.; Lee, K. Y.; Kochi, J. K. J. Chem. Soc., Perkin Trans. 2 1992, 1581–1594. doi:10.1039/p29920001581 |
| 140. | Rozatian, N.; Harsanyi, A.; Murray, B. J.; Hampton, A. S.; Chin, E. J.; Cook, A. S.; Hodgson, D. R. W.; Sandford, G. Chem. – Eur. J. 2020, 26, 12027–12035. doi:10.1002/chem.202001120 |
| 139. | Rozatian, N.; Beeby, A.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2019, 10, 10318–10330. doi:10.1039/c9sc04185k |
| 141. | Du, X.; Zhang, H.; Yao, Y.; Lu, Y.; Wang, A.; Wang, Y.; Li, Z. Theor. Chem. Acc. 2019, 138, No. 28. doi:10.1007/s00214-019-2417-2 |
| 138. | Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b |
| 87. | Laali, K. K.; Tanaka, M.; Forohar, F.; Cheng, M.; Fetzer, J. C. J. Fluorine Chem. 1998, 91, 185–190. doi:10.1016/s0022-1139(98)00224-3 |
| 87. | Laali, K. K.; Tanaka, M.; Forohar, F.; Cheng, M.; Fetzer, J. C. J. Fluorine Chem. 1998, 91, 185–190. doi:10.1016/s0022-1139(98)00224-3 |
| 138. | Rozatian, N.; Ashworth, I. W.; Sandford, G.; Hodgson, D. R. W. Chem. Sci. 2018, 9, 8692–8702. doi:10.1039/c8sc03596b |
| 88. | Forohar, F.; Damavarapu, R.; Jayasuriya, K.; Kwok, T. J. 13th Winter Fluorine Conference; St. Petersburg, Florida, USA, 1997; p 39. |
| 137. | Timofeeva, D. S.; Ofial, A. R.; Mayr, H. J. Am. Chem. Soc. 2018, 140, 11474–11486. doi:10.1021/jacs.8b07147 |
| 149. | Nyffeler, P. T.; Durón, S. G.; Burkart, M. D.; Vincent, S. P.; Wong, C.-H. Angew. Chem., Int. Ed. 2005, 44, 192–212. doi:10.1002/anie.200400648 |
| 148. | Vincent, S. P.; Burkart, M. D.; Tsai, C.-Y.; Zhang, Z.; Wong, C.-H. J. Org. Chem. 1999, 64, 5264–5279. doi:10.1021/jo990686h |
| 150. | Piana, S.; Devillers, I.; Togni, A.; Rothlisberger, U. Angew. Chem., Int. Ed. 2002, 41, 979–982. doi:10.1002/1521-3773(20020315)41:6<979::aid-anie979>3.0.co;2-e |
| 148. | Vincent, S. P.; Burkart, M. D.; Tsai, C.-Y.; Zhang, Z.; Wong, C.-H. J. Org. Chem. 1999, 64, 5264–5279. doi:10.1021/jo990686h |
| 147. | Zupan, M.; Škulj, P.; Stavber, S. Chem. Lett. 1998, 27, 641–642. doi:10.1246/cl.1998.641 |
| 145. | Differding, E.; Rüegg, G. M. Tetrahedron Lett. 1991, 32, 3815–3818. doi:10.1016/s0040-4039(00)79383-x |
| 149. | Nyffeler, P. T.; Durón, S. G.; Burkart, M. D.; Vincent, S. P.; Wong, C.-H. Angew. Chem., Int. Ed. 2005, 44, 192–212. doi:10.1002/anie.200400648 |
| 145. | Differding, E.; Rüegg, G. M. Tetrahedron Lett. 1991, 32, 3815–3818. doi:10.1016/s0040-4039(00)79383-x |
| 58. | Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730–4737. doi:10.1021/jo00120a014 |
| 146. | Differring, E.; Wehrli, M. Tetrahedron Lett. 1991, 32, 3819–3822. doi:10.1016/s0040-4039(00)79384-1 |
| 82. | Banks, R. E.; Besheesh, M. K.; Tsiliopoulos, E. J. Fluorine Chem. 1996, 78, 39–42. doi:10.1016/0022-1139(95)03380-7 |
| 81. | Cabrera, I.; Appel, W. K. Tetrahedron 1995, 51, 10205–10208. doi:10.1016/0040-4020(95)00597-2 |
| 90. | Takeuchi, Y.; Liu, Z.; Suzuki, E.; Shibata, N.; Kirk, K. L. J. Fluorine Chem. 1999, 97, 65–67. doi:10.1016/s0022-1139(99)00028-7 |
| 89. | Banks, R. E.; Besheesh, M. K.; Lawrence, N. J.; Tovell, D. J. J. Fluorine Chem. 1999, 97, 79–84. doi:10.1016/s0022-1139(99)00032-9 |
| 93. | Shibata, N.; Liu, Z.; Takeuchi, Y. Chem. Pharm. Bull. 2000, 48, 1954–1958. doi:10.1248/cpb.48.1954 |
| 91. | Takeuchi, Y.; Suzuki, T.; Satoh, A.; Shiragami, T.; Shibata, N. J. Org. Chem. 1999, 64, 5708–5711. doi:10.1021/jo9904011 |
| 92. | Takeuchi, Y.; Satoh, A.; Suzuki, T.; Kameda, A.; Dohrin, M.; Satoh, T.; Koizumi, T.; Kirk, K. L. Chem. Pharm. Bull. 1997, 45, 1085–1088. doi:10.1248/cpb.45.1085 |
| 85. | Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Fluorine Chem. 1997, 81, 157–161. doi:10.1016/s0022-1139(96)03521-x |
| 87. | Laali, K. K.; Tanaka, M.; Forohar, F.; Cheng, M.; Fetzer, J. C. J. Fluorine Chem. 1998, 91, 185–190. doi:10.1016/s0022-1139(98)00224-3 |
| 88. | Forohar, F.; Damavarapu, R.; Jayasuriya, K.; Kwok, T. J. 13th Winter Fluorine Conference; St. Petersburg, Florida, USA, 1997; p 39. |
| 86. | Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. J. Org. Chem. 1998, 63, 3379–3385. doi:10.1021/jo972338q |
| 83. | Umemoto, T.; Nagayoshi, M. Bull. Chem. Soc. Jpn. 1996, 69, 2287–2295. doi:10.1246/bcsj.69.2287 |
| 94. | Liu, Z.; Shibata, N.; Takeuchi, Y. J. Org. Chem. 2000, 65, 7583–7587. doi:10.1021/jo0055584 |
| 93. | Shibata, N.; Liu, Z.; Takeuchi, Y. Chem. Pharm. Bull. 2000, 48, 1954–1958. doi:10.1248/cpb.48.1954 |
| 107. | Wolstenhulme, J. R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K. M.; Pidgeon, G. W.; Moore, P. R.; Sandford, G.; Gouverneur, V. Angew. Chem., Int. Ed. 2013, 52, 9796–9800. doi:10.1002/anie.201304845 |
| 53. | Differding, E.; Lang, R. W. Helv. Chim. Acta 1989, 72, 1248–1252. doi:10.1002/hlca.19890720609 |
| 106. | Shunatona, H. P.; Früh, N.; Wang, Y.-M.; Rauniyar, V.; Toste, F. D. Angew. Chem., Int. Ed. 2013, 52, 7724–7727. doi:10.1002/anie.201302002 |
| 95. | Shibata, N.; Suzuki, E.; Takeuchi, Y. J. Am. Chem. Soc. 2000, 122, 10728–10729. doi:10.1021/ja002732x |
| 111. | Fukushi, K.; Suzuki, S.; Kamo, T.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Kawada, K.; Shibata, N. Green Chem. 2016, 18, 1864–1868. doi:10.1039/c5gc02612a |
| 94. | Liu, Z.; Shibata, N.; Takeuchi, Y. J. Org. Chem. 2000, 65, 7583–7587. doi:10.1021/jo0055584 |
| 108. | Zhu, C.-L.; Maeno, M.; Zhang, F.-G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.-A.; Cahard, D. Eur. J. Org. Chem. 2013, 6501–6505. doi:10.1002/ejoc.201300956 |
| 90. | Takeuchi, Y.; Liu, Z.; Suzuki, E.; Shibata, N.; Kirk, K. L. J. Fluorine Chem. 1999, 97, 65–67. doi:10.1016/s0022-1139(99)00028-7 |
| 100. | Banks, R. E.; Besheesh, M. K.; Fraenk, W.; Klapötke, T. M. J. Fluorine Chem. 2003, 124, 229–232. doi:10.1016/j.jfluchem.2003.08.010 |
| 90. | Takeuchi, Y.; Liu, Z.; Suzuki, E.; Shibata, N.; Kirk, K. L. J. Fluorine Chem. 1999, 97, 65–67. doi:10.1016/s0022-1139(99)00028-7 |
| 99. | Baudequin, C.; Loubassou, J.-F.; Plaquevent, J.-C.; Cahard, D. J. Fluorine Chem. 2003, 122, 189–193. doi:10.1016/s0022-1139(03)00085-x |
| 92. | Takeuchi, Y.; Satoh, A.; Suzuki, T.; Kameda, A.; Dohrin, M.; Satoh, T.; Koizumi, T.; Kirk, K. L. Chem. Pharm. Bull. 1997, 45, 1085–1088. doi:10.1248/cpb.45.1085 |
| 103. | Chen, G.; Chen, F.; Zhang, Y.; Yang, X.; Yuan, X.; Wu, F.; Yang, X. J. Fluorine Chem. 2012, 133, 155–159. doi:10.1016/j.jfluchem.2011.10.012 |
| 104. | Zhang, Y.; Yang, X.-J.; Xie, T.; Chen, G.-L.; Zhu, W.-H.; Zhang, X.-Q.; Yang, X.-Y.; Wu, X.-Y.; He, X.-P.; He, H.-M. Tetrahedron 2013, 69, 4933–4937. doi:10.1016/j.tet.2013.04.037 |
| 105. | Wang, F.; Li, J.; Hu, Q.; Yang, X.; Wu, X.-Y.; He, H. Eur. J. Org. Chem. 2014, 3607–3613. doi:10.1002/ejoc.201402072 |
| 91. | Takeuchi, Y.; Suzuki, T.; Satoh, A.; Shiragami, T.; Shibata, N. J. Org. Chem. 1999, 64, 5708–5711. doi:10.1021/jo9904011 |
| 102. | Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N. J. Fluorine Chem. 2011, 132, 222–225. doi:10.1016/j.jfluchem.2011.01.005 |
| 89. | Banks, R. E.; Besheesh, M. K.; Lawrence, N. J.; Tovell, D. J. J. Fluorine Chem. 1999, 97, 79–84. doi:10.1016/s0022-1139(99)00032-9 |
| 97. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Roques, N. Org. Lett. 2000, 2, 3699–3701. doi:10.1021/ol006610l |
| 98. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Toupet, L.; Roques, N. Tetrahedron Lett. 2001, 42, 1867–1869. doi:10.1016/s0040-4039(01)00017-x |
| 95. | Shibata, N.; Suzuki, E.; Takeuchi, Y. J. Am. Chem. Soc. 2000, 122, 10728–10729. doi:10.1021/ja002732x |
| 96. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 62. | Abdul-Ghani, M.; Banks, E. R.; Besheesh, M. K.; Sharif, I.; Syvret, R. G. J. Fluorine Chem. 1995, 73, 255–257. doi:10.1016/0022-1139(94)03225-o |
| 102. | Yasui, H.; Yamamoto, T.; Ishimaru, T.; Fukuzumi, T.; Tokunaga, E.; Akikazu, K.; Shiro, M.; Shibata, N. J. Fluorine Chem. 2011, 132, 222–225. doi:10.1016/j.jfluchem.2011.01.005 |
| 28. | Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3 |
| 29. | Umemoto, T.; Kawada, K.; Tomita, K. Tetrahedron Lett. 1986, 27, 4465–4468. doi:10.1016/s0040-4039(00)84980-1 |
| 101. | Schleyer, P. v. R.; Buzek, P.; Klapötke, T. M.; Tornieporth-Oetting, I. C.; Broschag, M.; Pickardt, J. Inorg. Chem. 1993, 32, 1523–1524. doi:10.1021/ic00060a033 |
| 26. | Barnette, W. E. J. Am. Chem. Soc. 1984, 106, 452–454. doi:10.1021/ja00314a050 |
| 104. | Zhang, Y.; Yang, X.-J.; Xie, T.; Chen, G.-L.; Zhu, W.-H.; Zhang, X.-Q.; Yang, X.-Y.; Wu, X.-Y.; He, X.-P.; He, H.-M. Tetrahedron 2013, 69, 4933–4937. doi:10.1016/j.tet.2013.04.037 |
| 103. | Chen, G.; Chen, F.; Zhang, Y.; Yang, X.; Yuan, X.; Wu, F.; Yang, X. J. Fluorine Chem. 2012, 133, 155–159. doi:10.1016/j.jfluchem.2011.10.012 |
| 45. | Singh, S.; DesMarteau, D. D.; Zuberi, S. S.; Witz, M.; Huang, H.-N. J. Am. Chem. Soc. 1987, 109, 7194–7196. doi:10.1021/ja00257a051 |
| 1. |
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons, Ltd: Chichester, United Kingdom, 2009. doi:10.1002/9781444312096
See for a selected book. |
| 98. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Toupet, L.; Roques, N. Tetrahedron Lett. 2001, 42, 1867–1869. doi:10.1016/s0040-4039(01)00017-x |
| 10. | Schlosser, M.; Heinz, G. Chem. Ber. 1969, 102, 1944–1953. doi:10.1002/cber.19691020620 |
| 62. | Abdul-Ghani, M.; Banks, E. R.; Besheesh, M. K.; Sharif, I.; Syvret, R. G. J. Fluorine Chem. 1995, 73, 255–257. doi:10.1016/0022-1139(94)03225-o |
| 100. | Banks, R. E.; Besheesh, M. K.; Fraenk, W.; Klapötke, T. M. J. Fluorine Chem. 2003, 124, 229–232. doi:10.1016/j.jfluchem.2003.08.010 |
| 24. | Purrington, S. T.; Jones, W. A. J. Org. Chem. 1983, 48, 761–762. doi:10.1021/jo00153a037 |
| 99. | Baudequin, C.; Loubassou, J.-F.; Plaquevent, J.-C.; Cahard, D. J. Fluorine Chem. 2003, 122, 189–193. doi:10.1016/s0022-1139(03)00085-x |
| 11. | Barton, D. H. R. Pure Appl. Chem. 1977, 49, 1241–1249. doi:10.1351/pac197749091241 |
| 6. | Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467 |
| 113. | Pereira, R.; Wolstenhulme, J.; Sandford, G.; Claridge, T. D. W.; Gouverneur, V.; Cvengroš, J. Chem. Commun. 2016, 52, 1606–1609. doi:10.1039/c5cc08375c |
| 4. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 5. | Mei, H.; Remete, A. M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2020, 31, 2401–2413. doi:10.1016/j.cclet.2020.03.050 |
| 3. | Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f |
| 97. | Cahard, D.; Audouard, C.; Plaquevent, J.-C.; Roques, N. Org. Lett. 2000, 2, 3699–3701. doi:10.1021/ol006610l |
| 115. | Meyer, D.; Jangra, H.; Walther, F.; Zipse, H.; Renaud, P. Nat. Commun. 2018, 9, 4888. doi:10.1038/s41467-018-07196-9 |
| 96. | Shibata, N.; Suzuki, E.; Asahi, T.; Shiro, M. J. Am. Chem. Soc. 2001, 123, 7001–7009. doi:10.1021/ja010789t |
| 114. | Chen, C.; Luo, Y.; Fu, L.; Chen, P.; Lan, Y.; Liu, G. J. Am. Chem. Soc. 2018, 140, 1207–1210. doi:10.1021/jacs.7b11470 |
| 10. | Schlosser, M.; Heinz, G. Chem. Ber. 1969, 102, 1944–1953. doi:10.1002/cber.19691020620 |
| 9. | Harper, D. B.; O'Hagan, D. Nat. Prod. Rep. 1994, 11, 123–133. doi:10.1039/np9941100123 |
| 7. | Uneyama, K., Ed. Organofluorine Chemistry; Blackwell Publishing: Oxford, United Kingdom, 2006. doi:10.1002/9780470988589 |
| 112. | Bohlmann, R. N-Fluorosulfonimides, process for their production, as well as their use as fluorinating agents. WO Patent WO94/24098, Oct 27, 1994. |
| 86. | Umemoto, T.; Nagayoshi, M.; Adachi, K.; Tomizawa, G. J. Org. Chem. 1998, 63, 3379–3385. doi:10.1021/jo972338q |
| 111. | Fukushi, K.; Suzuki, S.; Kamo, T.; Tokunaga, E.; Sumii, Y.; Kagawa, T.; Kawada, K.; Shibata, N. Green Chem. 2016, 18, 1864–1868. doi:10.1039/c5gc02612a |
| 83. | Umemoto, T.; Nagayoshi, M. Bull. Chem. Soc. Jpn. 1996, 69, 2287–2295. doi:10.1246/bcsj.69.2287 |
| 113. | Pereira, R.; Wolstenhulme, J.; Sandford, G.; Claridge, T. D. W.; Gouverneur, V.; Cvengroš, J. Chem. Commun. 2016, 52, 1606–1609. doi:10.1039/c5cc08375c |
| 108. | Zhu, C.-L.; Maeno, M.; Zhang, F.-G.; Shigehiro, T.; Kagawa, T.; Kawada, K.; Shibata, N.; Ma, J.-A.; Cahard, D. Eur. J. Org. Chem. 2013, 6501–6505. doi:10.1002/ejoc.201300956 |
| 42. | Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595 |
| 107. | Wolstenhulme, J. R.; Rosenqvist, J.; Lozano, O.; Ilupeju, J.; Wurz, N.; Engle, K. M.; Pidgeon, G. W.; Moore, P. R.; Sandford, G.; Gouverneur, V. Angew. Chem., Int. Ed. 2013, 52, 9796–9800. doi:10.1002/anie.201304845 |
| 110. | Treskow, M.; Neudörfl, J.; Giernoth, R. Eur. J. Org. Chem. 2009, 3693–3697. doi:10.1002/ejoc.200900548 |
| 78. | Poss, A. J.; Shia, G. 1-Substituted-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts and their application as fluorinating agents. U.S. Patent US5,459,267, Oct 17, 1995. |
| 109. | Ratjen, L.; García-García, P.; Lay, F.; Beck, M. E.; List, B. Angew. Chem., Int. Ed. 2011, 50, 754–758. doi:10.1002/anie.201005954 |
| 76. | Umemoto, T.; Tomizawa, G. J. Org. Chem. 1995, 60, 6563–6570. doi:10.1021/jo00125a049 |
| 105. | Wang, F.; Li, J.; Hu, Q.; Yang, X.; Wu, X.-Y.; He, H. Eur. J. Org. Chem. 2014, 3607–3613. doi:10.1002/ejoc.201402072 |
| 53. | Differding, E.; Lang, R. W. Helv. Chim. Acta 1989, 72, 1248–1252. doi:10.1002/hlca.19890720609 |
| 52. | Differding, E.; Lang, R. W. Tetrahedron Lett. 1988, 29, 6087–6090. doi:10.1016/s0040-4039(00)82271-6 |
| 106. | Shunatona, H. P.; Früh, N.; Wang, Y.-M.; Rauniyar, V.; Toste, F. D. Angew. Chem., Int. Ed. 2013, 52, 7724–7727. doi:10.1002/anie.201302002 |
| 57. | Davis, F. A.; Han, W. Tetrahedron Lett. 1991, 32, 1631–1634. doi:10.1016/s0040-4039(00)74290-0 |
| 106. | Shunatona, H. P.; Früh, N.; Wang, Y.-M.; Rauniyar, V.; Toste, F. D. Angew. Chem., Int. Ed. 2013, 52, 7724–7727. doi:10.1002/anie.201302002 |
| 55. | Satyamurthy, N.; Bida, G. T.; Phelps, M. E.; Barrio, J. R. J. Org. Chem. 1990, 55, 3373–3374. doi:10.1021/jo00297a071 |
| 28. | Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3 |
| 29. | Umemoto, T.; Kawada, K.; Tomita, K. Tetrahedron Lett. 1986, 27, 4465–4468. doi:10.1016/s0040-4039(00)84980-1 |
| 30. | Umemoto, T.; Tomizawa, G. Bull. Chem. Soc. Jpn. 1986, 59, 3625–3629. doi:10.1246/bcsj.59.3625 |
| 31. | Umemoto, T.; Tomita, K.; Kawada, K. Org. Synth. 1990, 69, 129–140. doi:10.15227/orgsyn.069.0129 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 33. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. Bull. Chem. Soc. Jpn. 1991, 64, 1081–1092. doi:10.1246/bcsj.64.1081 |
| 34. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. J. Fluorine Chem. 1991, 53, 369–377. doi:10.1016/s0022-1139(00)80960-4 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 29. | Umemoto, T.; Kawada, K.; Tomita, K. Tetrahedron Lett. 1986, 27, 4465–4468. doi:10.1016/s0040-4039(00)84980-1 |
| 30. | Umemoto, T.; Tomizawa, G. Bull. Chem. Soc. Jpn. 1986, 59, 3625–3629. doi:10.1246/bcsj.59.3625 |
| 31. | Umemoto, T.; Tomita, K.; Kawada, K. Org. Synth. 1990, 69, 129–140. doi:10.15227/orgsyn.069.0129 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 33. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. Bull. Chem. Soc. Jpn. 1991, 64, 1081–1092. doi:10.1246/bcsj.64.1081 |
| 36. | Umemoto, T.; Tomita, K. N-Fluoropyridinium salt-containing polymer. Jpn. Kokai Tokkyo Koho S63-295610, Dec 2, 1988. |
| 38. | Fukami, S.; Nukui, K.; Kawada, K. Production method of N-fluoropyridinium tetrafluoroborate. Jpn. Kokai Tokkyo Koho H09-255657, Sept 30, 1997. |
| 28. | Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3 |
| 31. | Umemoto, T.; Tomita, K.; Kawada, K. Org. Synth. 1990, 69, 129–140. doi:10.15227/orgsyn.069.0129 |
| 33. | Umemoto, T.; Harasawa, K.; Tomizawa, G.; Kawada, K.; Tomita, K. Bull. Chem. Soc. Jpn. 1991, 64, 1081–1092. doi:10.1246/bcsj.64.1081 |
| 28. | Umemoto, T.; Tomita, K. Tetrahedron Lett. 1986, 27, 3271–3274. doi:10.1016/s0040-4039(00)84772-3 |
| 37. | Nukui, K.; Kamiya, K. Production method of N-fluoropyridinium salts. Jpn. Patent 3038065, May 8, 2000. |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 44. | Banks, R. E.; Du Boisson, R. A.; Morton, W. D.; Tsiliopoulos, E. J. Chem. Soc., Perkin Trans. 1 1988, 2805–2811. doi:10.1039/p19880002805 |
| 24. | Purrington, S. T.; Jones, W. A. J. Org. Chem. 1983, 48, 761–762. doi:10.1021/jo00153a037 |
| 25. | Purrington, S. T.; Jones, W. A. J. Fluorine Chem. 1984, 26, 43–46. doi:10.1016/s0022-1139(00)85117-9 |
| 42. | Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595 |
| 43. | Banks, R. E.; Du Boisson, R. A.; Tsiliopoulos, E. J. Fluorine Chem. 1986, 32, 461–466. doi:10.1016/s0022-1139(00)81953-3 |
| 40. | Naruta, Y.; Tani, F.; Maruyama, K. Tetrahedron Lett. 1992, 33, 1069–1072. doi:10.1016/s0040-4039(00)91863-x |
| 41. | Poss, A. J.; Van der Puy, M.; Nalewajek, D.; Shia, G. A.; Wagner, W. J.; Frenette, R. L. J. Org. Chem. 1991, 56, 5962–5964. doi:10.1021/jo00020a051 |
| 30. | Umemoto, T.; Tomizawa, G. Bull. Chem. Soc. Jpn. 1986, 59, 3625–3629. doi:10.1246/bcsj.59.3625 |
| 32. | Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. J. Am. Chem. Soc. 1990, 112, 8563–8575. doi:10.1021/ja00179a047 |
| 39. | Kawada, K. N-Fluoropyridinium Salt Fluorination for Preparing Aryl Fluorides. In Fluorination; Hu, J.; Umemoto, T., Eds.; Springer: Singapore, 2020. doi:10.1007/978-981-10-1855-8_40-2 |
| 13. | Rozen, S.; Lerman, O. J. Am. Chem. Soc. 1979, 101, 2782–2784. doi:10.1021/ja00504a073 |
| 14. | Rozen, S.; Lerman, O.; Kol, M.; Hebel, D. J. Org. Chem. 1985, 50, 4753–4758. doi:10.1021/jo00224a019 |
| 11. | Barton, D. H. R. Pure Appl. Chem. 1977, 49, 1241–1249. doi:10.1351/pac197749091241 |
| 12. | Appelman, E. H.; Basile, L. J.; Thompson, R. C. J. Am. Chem. Soc. 1979, 101, 3384–3385. doi:10.1021/ja00506a046 |
| 11. | Barton, D. H. R. Pure Appl. Chem. 1977, 49, 1241–1249. doi:10.1351/pac197749091241 |
| 20. | Polishchuk, V. R.; German, L. S. Tetrahedron Lett. 1972, 13, 5169–5172. doi:10.1016/s0040-4039(01)85198-4 |
| 17. | Banks, R. E.; Cheng, W. M.; Haszeldine, R. N. J. Chem. Soc. 1962, 3407–3416. doi:10.1039/jr9620003407 |
| 18. | Banks, R. E.; Ginsberg, A. E.; Haszeldine, R. N. J. Chem. Soc. 1961, 1740–1743. doi:10.1039/jr9610001740 |
| 19. | Banks, R. E.; Haszeldine, R. N.; Hatton, R. Tetrahedron Lett. 1967, 8, 3993–3996. doi:10.1016/s0040-4039(01)89722-7 |
| 75. | Davis, F. A.; Zhou, P.; Murphy, C. K. Tetrahedron Lett. 1993, 34, 3971–3974. doi:10.1016/s0040-4039(00)60592-0 |
| 42. | Banks, R. E.; Mohialdin-Khaffaf, S. N.; Lal, G. S.; Sharif, I.; Syvret, R. G. J. Chem. Soc., Chem. Commun. 1992, 595–596. doi:10.1039/c39920000595 |
| 61. | Banks, R. E.; Lawrence, N. J.; Popplewell, A. L. J. Chem. Soc., Chem. Commun. 1994, 343–344. doi:10.1039/c39940000343 |
| 62. | Abdul-Ghani, M.; Banks, E. R.; Besheesh, M. K.; Sharif, I.; Syvret, R. G. J. Fluorine Chem. 1995, 73, 255–257. doi:10.1016/0022-1139(94)03225-o |
| 63. | Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Chem. Soc., Perkin Trans. 1 1996, 2069–2076. doi:10.1039/p19960002069 |
| 64. | Banks, R. E.; Besheesh, M. K.; Mohialdin-Khaffaf, S. N.; Sharif, I. J. Fluorine Chem. 1996, 78, 43–50. doi:10.1016/0022-1139(96)03395-7 |
| 65. | Lal, G. S. J. Org. Chem. 1993, 58, 2791–2796. doi:10.1021/jo00062a023 |
| 66. | Stavber, S.; Sotler, T.; Zupan, M. Tetrahedron Lett. 1994, 35, 1105–1108. doi:10.1016/s0040-4039(00)79977-1 |
| 67. | Lal, G. S. Synth. Commun. 1995, 25, 725–737. doi:10.1080/00397919508011410 |
| 68. | Brown, D. S.; Marples, B. A.; Smith, P.; Walton, L. Tetrahedron 1995, 51, 3587–3606. doi:10.1016/0040-4020(95)00075-j |
| 69. | Lal, G. S.; Pastore, W.; Pesaresi, R. J. Org. Chem. 1995, 60, 7340–7342. doi:10.1021/jo00127a046 |
| 70. | Zupan, M.; Iskra, J.; Stavber, S. J. Org. Chem. 1995, 60, 259–260. doi:10.1021/jo00106a045 |
| 71. | Brunavs, M.; Dell, C. P.; Owton, W. M. J. Fluorine Chem. 1994, 68, 201–203. doi:10.1016/0022-1139(93)03042-k |
| 72. | Matthews, D. P.; Miller, S. C.; Jarvi, E. T.; Sabol, J. S.; McCarthy, J. R. Tetrahedron Lett. 1993, 34, 3057–3060. doi:10.1016/s0040-4039(00)93378-1 |
| 73. | Hodson, H. F.; Madge, D. J.; Slawin, A. N. Z.; Widdowson, D. A.; Williams, D. J. Tetrahedron 1994, 50, 1899–1906. doi:10.1016/s0040-4020(01)80862-0 |
| 74. | McClinton, M. A.; Sik, V. J. Chem. Soc., Perkin Trans. 1 1992, 1891–1895. doi:10.1039/p19920001891 |
| 78. | Poss, A. J.; Shia, G. 1-Substituted-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts and their application as fluorinating agents. U.S. Patent US5,459,267, Oct 17, 1995. |
| 79. | Stavber, S.; Zupan, M. Tetrahedron Lett. 1996, 37, 3591–3594. doi:10.1016/0040-4039(96)00630-2 |
| 80. | Poss, A. J.; Shia, G. A. Tetrahedron Lett. 1999, 40, 2673–2676. doi:10.1016/s0040-4039(99)00303-2 |
| 76. | Umemoto, T.; Tomizawa, G. J. Org. Chem. 1995, 60, 6563–6570. doi:10.1021/jo00125a049 |
| 57. | Davis, F. A.; Han, W. Tetrahedron Lett. 1991, 32, 1631–1634. doi:10.1016/s0040-4039(00)74290-0 |
| 58. | Davis, F. A.; Han, W.; Murphy, C. K. J. Org. Chem. 1995, 60, 4730–4737. doi:10.1021/jo00120a014 |
| 55. | Satyamurthy, N.; Bida, G. T.; Phelps, M. E.; Barrio, J. R. J. Org. Chem. 1990, 55, 3373–3374. doi:10.1021/jo00297a071 |
| 56. | Grakauskas, V.; Baum, K. J. Org. Chem. 1970, 35, 1545–1549. doi:10.1021/jo00830a061 |
| 60. | Gakh, A. A.; Romaniko, S. V.; Ugrak, B. I.; Fainzilberg, A. A. Tetrahedron 1991, 47, 7447–7458. doi:10.1016/s0040-4020(01)89746-5 |
| 26. | Barnette, W. E. J. Am. Chem. Soc. 1984, 106, 452–454. doi:10.1021/ja00314a050 |
| 27. | Lee, S. H.; Schwartz, J. J. Am. Chem. Soc. 1986, 108, 2445–2447. doi:10.1021/ja00269a052 |
| 25. | Purrington, S. T.; Jones, W. A. J. Fluorine Chem. 1984, 26, 43–46. doi:10.1016/s0022-1139(00)85117-9 |
| 24. | Purrington, S. T.; Jones, W. A. J. Org. Chem. 1983, 48, 761–762. doi:10.1021/jo00153a037 |
| 24. | Purrington, S. T.; Jones, W. A. J. Org. Chem. 1983, 48, 761–762. doi:10.1021/jo00153a037 |
| 21. | Banks, R. E.; Tsiliopoulos, E. J. Fluorine Chem. 1986, 34, 281–285. doi:10.1016/s0022-1139(00)85081-2 |
| 22. | Banks, R. E.; Murtagh, V.; Tsiliopoulos, E. J. Fluorine Chem. 1991, 52, 389–401. doi:10.1016/s0022-1139(00)80353-x |
© 2021 Umemoto et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)