Abstract
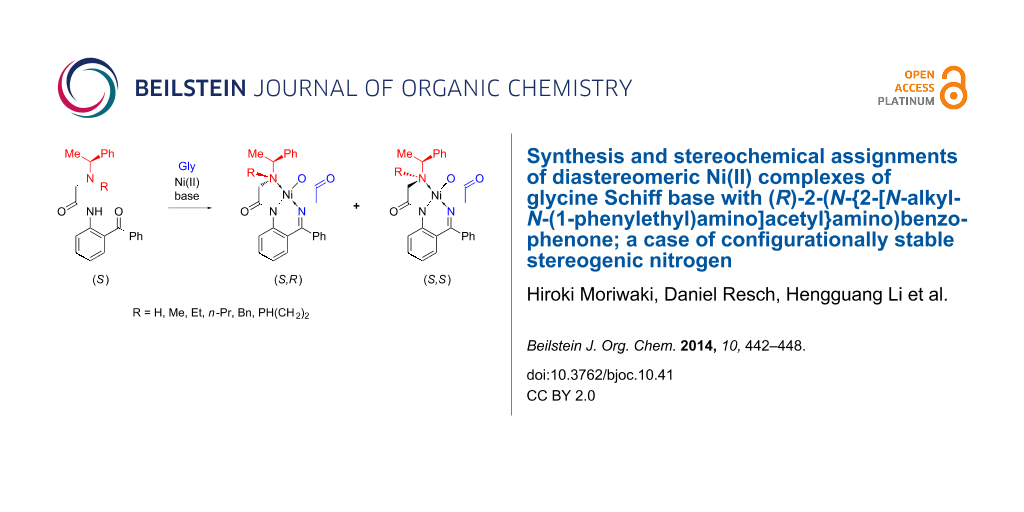
A family of chiral ligands derived from α-phenylethylamine and 2-aminobenzophenone were prepared by alkylation of the nitrogen atom. Upon reaction with glycine and a Ni(II) salt, these ligands were transformed into diastereomeric complexes, as a result of the configurational stability of the stereogenic nitrogen atom. Different diastereomeric ratios were observed depending on the substituent R introduced in the starting ligand, and stereochemical assignments were based on X-ray analysis, along with NMR studies and optical rotation measurements.

Graphical Abstract
Introduction
Since the beginning of organic chemistry, α-amino acids (α-AAs) have been an attractive target for synthetic chemists. The reported synthetic methods for assembling the RCH(NH2)COOH units have been explored in great detail. Structurally complex natural or tailor-made [1] α-AAs can be prepared using the currently available methodologies [2-30]; however, the chemistry of α-AAs continues to evolve [31-38]. Many of the reported synthetic methodologies have shown preference for adjusting stoichiometry or developing catalytic reactions to achieve a specific chemical and stereochemical outcome [2-30]. On the other hand, a measure of the value of a synthetic method, regardless of its stoichiometric or catalytic requirements, is the cost of the final product. In this regard, the synthetic preparation of α-AAs is far behind the biocatalytic methods that dominate their industrial production [39,40]. It was emphasized, in relatively recent reviews [39-41], that currently available, purely chemical methods for preparation of α-AAs are prohibitively expensive. The advantage of biocatalytic processes is that they can be conducted under operationally convenient conditions [42,43] and therefore are more economical. Consequently, the current emphasis in developing synthetic procedures for preparation of α-AAs is focused on simplicity of the experimental procedures and cost of the target α-AAs.
Among various chiral nucleophilic glycine equivalents, the Ni(II) complex of glycine Schiff base 1 (Figure 1) possesses some attractive characteristics that underscore its potential commercial application [44-47]. In particular, homologation of Ni(II) complex 1 via alkyl halide alkylation [48-50], aldol [51-53], Mannich [54,55] and Michael [56-58] addition reactions can be conducted at room temperature and without specially controlled conditions. Achiral derivatives of 1, compounds 2 [59,60] and 3 [61-63], show even greater features of practicality and found application in the convenient synthesis of symmetrically α,α-disubstituted α-AAs [64,65], homologation under asymmetric PTC [66,67] and Michael addition reactions [68-72]. Nevertheless, the commercial application of chiral complex 1 is rather limited. The major shortcomings of compound 1 are: problematic scale up of its synthesis, partial racemization of the N-(benzyl)proline moiety and undesirable stereochemical outcomes. To overcome these deficiencies we initiated a project aiming to design a novel and advanced structural type of Ni(II) complexes using an inexpensive and nonracemizable chiral auxiliary. As the first step in this direction, here we describe the preparation of Ni(II) complexes of glycine Schiff base with α-phenylethylamine-derived ligands. One unusual feature of these new glycine-Ni(II) complexes is that they, along with the chiral amine residue, have a configurationally stable stereogenic nitrogen, leading to formation of two diastereomers. Consequently, stereochemical assignments of the diastereomeric products, based on crystallographic data, and sense of stereochemical preferences, were an important part of this work.
![[1860-5397-10-41-1]](/bjoc/content/figures/1860-5397-10-41-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: A family of chiral and achiral equivalents of nucleophilic glycine.
Figure 1: A family of chiral and achiral equivalents of nucleophilic glycine.
Results and Discussion
Taking advantage of the recently developed new generation of modular ligands/complexes 3 [73,74], we have designed ligands 4a–f (Scheme 1) derived from α-phenylethylamine [75,76], as the source of chirality. As one can see from Scheme 1, the synthesis of ligands 4a–f involves a set of very simple reactions, occurring with virtually quantitative yields [77,78].
![[1860-5397-10-41-i1]](/bjoc/content/inline/1860-5397-10-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of chiral ligands 4a–f.
Scheme 1: Synthesis of chiral ligands 4a–f.
To prepare the target Gly-Ni(II) complexes, ligands 4a–f were heated in MeOH in the presence of glycine, Ni(OAc)2 and K2CO3 (Scheme 2). To compare the results, all reactions were conducted under the same conditions. The reaction mixtures were poured into water and the products were simply filtered off.
![[1860-5397-10-41-i2]](/bjoc/content/inline/1860-5397-10-41-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of diastereomeric Ni(II) complexes 5a–f and 6a–f.
Scheme 2: Preparation of diastereomeric Ni(II) complexes 5a–f and 6a–f.
The chemical and stereochemical outcomes of the reactions, presented in Table 1, were rather unexpected. First of all, one can see a clear tendency that the increase in steric bulk of the substituent R has a significant effect on the reaction rates. In the case of R = H and Me (4a,b; Table 1, entries 1 and 2) the reactions were completed in 1 hour and the corresponding Ni(II) complexes were obtained in high yields. Under the same conditions, the reactions of Et- and n-Pr-containing ligands (4c,d; Table 1, entries 3 and 4) proceeded with noticeably slower rates affording the corresponding products 5 and 6 in lower yields. Still slower reaction rates were observed in the case of Bn (4e) and 2-(phenyl)ethyl (4f) bearing ligands (Table 1, entries 5 and 6). These results revealed that the steric bulk of the amine’s nitrogen plays a significant role and should be minimized to maintain practically sounding reactions rates. On the other hand, the diastereoselectivity in the formation of 5 and 6 was increasing along with the steric bulk of the substituent R, reaching the maximum of ~75/25 in the case of R = n-Pr and Bn groups (Table 1, entries 4 and 5). Another surprising result is that the complex(es) obtained in the reaction of NH (bearing ligand R = H) had unprecedentedly low solubility. During the complex formation reaction, the product(s) precipitated from the reaction mixture and were isolated by filtration. Solvents such as DMF, DMSO, and acetic acid were used in an attempt to dissolve the product without success. The stereochemical outcome in this reaction remains unclear.
Table 1: Preparation of diastereomeric Ni(II) complexes 5a–f and 6a–f.
| Entry | R | 5/6 ratioa | Yield (%) | [α]Db | |
|---|---|---|---|---|---|
| 5 | 6 | ||||
| 1 | H (a) | N/Ac | 87.4 | N/Ac | N/Ac |
| 2 | Me (b) | 57/43 | 96.9 | +88.6 | +1701.8 |
| 3 | Et (c) | 61/39 | 67.7 | −150.8 | +1226.4 |
| 4 | n-Pr (d) | 74/26 | 70.1 | −55.0 | +1033.3 |
| 5 | Bn (e) | 76/24 | 31.5d | +303.6 | +361.5 |
| 6 | Ph(CH2)2 (f) | 67/33 | 12.1d | N/Ae | N/Ae |
aDetermined by 1H NMR analysis on the crude reaction mixtures. bMeasured in dichloromethane, 25 ºC, c = ~1. cThe compound could not be dissolved in any solvent. dThe reaction was incomplete and the starting ligand was also recovered. eThe diastereomers could not be separated.
Since the reactions were conducted in open atmosphere, formation of byproducts (S)-7a–f (5–20%) were observed in all cases. Compounds (S)-7a–f result from the reaction of atmospheric oxygen with the corresponding enolates derived from glycine complexes 5 and 6 and the basic reaction conditions [79,80]. Formation of 4-phenylquinazoline derivatives (S)-7a–f can be prevented by conducting the reactions in oxygen-free atmosphere. Another general trend is apparent when considering the observed optical rotations of the diastereomeric products 5 and 6. In all cases, the major diastereomers 5 had low-magnitude rotations as compared with high-magnitude dextrorotatory rotations of 6. This trend can be used to assign the configurations of products 5 and 6 and related compounds. It is interesting to note that the difference in optical rotation between diastereomers 5e and 6e was the least pronounced in the series. Most likely, some similarity in the structure of N-benzyl and N-α-(methyl)benzyl groups results in low impact on the optical rotation of diastereomers 5e and 6e.
Knowledge of one of the stereocenters in diastereomers 5 and 6 allowed us to use X-ray crystallography to determine the absolute configuration of the major product 5b [81]. The structure presented in Figure 2 shows its (SCRN) absolute configuration. Accordingly, the second product, diastereomer 6b, has (SCSN) configuration. The X-ray structure of 5b revealed the complex is not fully planar as expected. The benzophenone chelate ring system deviates from planarity by 14.4° (0.16 e.s.d.). The torsion angle of Ni2–O5–C10–C29 is −10.9° further indicating a deviation from planarity. One of the rings in the benzophenone chelate is rotated 85.8° (0.36 e.s.d.) out of the plane presumably due to sterics. This rotation minimizes the Ar–H···H–Ar repulsion as well as repulsion with methylene group of the glycine moiety and the Ar–H.
![[1860-5397-10-41-2]](/bjoc/content/figures/1860-5397-10-41-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystallographic structure of (SCRN)-5b.
Figure 2: Crystallographic structure of (SCRN)-5b.
As we have mentioned above, there is a great degree of similarity in the chiroptical properties of complexes 5b–e and 6b–e, sufficient to make the corresponding stereochemical assignments. Moreover, there is also another general trend observed in the 1H NMR spectra of compounds 5 and 6. In particular, as it is evident form Figure 3, the α-phenylethylamine moiety's methyl group is located in relatively close proximity to the Ni(II) atom, causing the methyl protons to shift more downfield [82-85]. Accordingly, in all 1H NMR spectra of compounds 5b–e this methyl group is shifted downfield (2.5–2.9 ppm) as compared with the chemical shift of the same methyl in diastereomers 6b–e (2.2–1.75 ppm).
![[1860-5397-10-41-3]](/bjoc/content/figures/1860-5397-10-41-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Crystallographic structure of (SCRN)-5b showing an exposure of the methyl moiety of α-phenylethylamine to the Ni(II).
Figure 3: Crystallographic structure of (SCRN)-5b showing an exposure of the methyl moiety of α-phenylethylam...
Combining together the crystallographic, chiroptical and 1H NMR data of compounds 5 and 6, we can confidently assign the (SCRN) absolute configuration to all products 5b–e and (SCSN) stereochemistry to the complexes 6b–e.
Conclusion
In conclusion, this exploratory work has revealed that: 1) Application of α-phenylethylamine as the source of stereochemistry information in the novel Ni(II) complexes, results in the formation of diastereomeric glycine Schiff base products due to the configurational stability of the stereogenic nitrogen. 2) The steric bulk of the substituent R on the α-phenylethylamine residue has a negative effect on the reaction rates and a positive influence on the stereochemical outcome. 3) The optimal size of the R group is n-Pr or Bn. 4) Synthesis of this type of Ni(II) complexes should be conducted under oxygen-free conditions to avoid the formation of byproducts, resulting from the oxidation of the corresponding enolates. 5) The (SCRN) stereochemistry of the Ni(II) complexes is generally preferred. Drawing from these results, an improved design of new ligands is currently under investigation in our laboratories.
Experimental
General procedure for the preparation of ligands 4b–f
In analogous manner as previously reported [77], secondary amine 4a [78] (1 equiv), N,N-diisopropylethylamine (1.5 equiv), alkyl halide (1.5–3.5 equiv) and 10 mL of MeCN were placed in a round bottom flask and stirred at room temperature under nitrogen. After completion of the reaction (monitored by TLC), the reaction mixture was evaporated to dryness under vacuum. The residue was dissolved in 5 mL of CH2Cl2 and washed with 5 mL of water. The aqueous layer was washed with 3 × 5 mL fractions of CH2Cl2. The collected organic fractions were dried over MgSO4 and the solvent was removed under vacuum to yield the crude product. Analytically pure samples of 4b–f were obtained by column chromatography (Hex/EtOAc).
(S)-N-(2-benzoylphenyl)-2-(methyl-(1-phenylethyl)amino)acetamide (4b): From 4a (6.14 g, 17.23 mmol) and MeI (4.3 mL, 68.52 mmol), 4.61 g (72.4%) of 4b. White solid. Mp 80–82 °C; [α]D25 +23.7 (c 1.55, CHCl3); 1H NMR (300 MHz, CDCl3) δ 1.54 (d, J = 6.9 Hz, 3H), 2.44 (s, 3H), 3.12 (d, J = 16.8 Hz, 1H), 3.25 (d, J = 16.8 Hz, 1H), 3.82 (q, J = 6.6 Hz, 1H), 6.87–6.92 (m, 1H), 7.15–7.22 (m, 1H), 7.25–7.28 (m, 2H), 7.50–7.72 (m, 7H), 7.73–7.92 (m, 2H), 8.69 (d, J = 8.7 Hz, 1H), 11.67 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 18.4, 40.8, 58.7, 63.5, 121.5, 122.1, 125.1, 127.1, 127.6, 128.2, 130.0, 132.3, 132.5, 133.2, 138.4, 139.0, 142.8, 171.1, 197.9; HRMS: [M + H]+ calcd for C24H25N2O2, 373.1916; found, 373.1923.
General procedure for the preparation of Ni(II) complexes 5b–f and 6b–f
To a flask containing a methanol solution of ligand 4b–f (1 equiv), Ni(OAc)2·4H2O (2 equiv) and glycine (5.0 equiv), was added K2CO3 (9 equiv), and the reaction mixture was stirred at 60–70 °C. The progress of the reaction was monitored by TLC and upon completion (consumption of the reagent 4), the reaction mixture was poured into ice water. The target product was extracted three times with CH2Cl2. The combined organic layer was dried over anhydrous MgSO4 and evaporated under vacuum. After evaporation of the solvents, the target complexes 5b–f and 6b–f were obtained in diastereomerically pure form by separation on silica-gel column chromatography.
5b and 6b: From 4b (512 mg, 1.375 mmol), 647.3 mg (96.9 %) of a 57:43 mixture of 5b and 6b. Data of 5b: Red solid. Mp 265–267 °C; [α]D25 +88.6 (c 1.0, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ 2.49 (d, J = 6.9 Hz, 3H), 2.57 (d, J = 16.8 Hz, 1H), 2.83 (s, 3H), 3.65–3.85 (m, 2H), 3.90–4.00 (m, 1H), 3.97 (d, J =16.8 Hz, 1H), 6.77–7.05 (m, 4H), 7.26–7.40 (m, 6H), 7.51–7.53 (m, 3H), 8.63 (d, J = 9.0 Hz, 1H). 13C NMR (75 MHz, CDCl3): 19.7, 47.5, 58.9, 61.3, 64.6, 121.1, 124.2, 125.1, 125.8, 126.0, 128.5, 129.0, 129.5, 129.6, 130.1, 132.7, 133.5, 134.1, 134.6, 142.5, 172.0, 177.2, 178.5. HRMS: [M + H]+ calcd for C26H26N3O3Ni, 486.1328; found, 486.1335. Data of 6b: Red solid. Mp 269–272 °C. [α]D25 +1701.8 (c 1.0, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ 2.27 (s, 3H), 2.34 (d, J = 6.9 Hz, 3H), 3.35 (d, J = 16.5 Hz, 1H), 3.60–3.72 (m, 3H), 4.32 (q, J = 6.9 Hz, 1H), 6.75–6.93 (m, 3H), 7.13–7.18 (m, 1H), 7.30–7.51 (m, 9H), 8.48 (d, J = 8.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 17.6, 42.2, 61.1, 62.2, 66.2, 121.3, 124.3, 125.5, 125.6, 126.2, 128.7, 129.1, 129.3, 129.7, 129.9, 130.2, 132.5, 133.4, 134.5, 134.8, 142.3, 171.9, 176.0, 177.2. HRMS: [M + H]+ calcd for C26H26N3O3Ni, 486.1328; found, 486.1337.
Supporting Information
| Supporting Information File 1: Experimental data for compounds 4c–f, 5c–e and 6c–e. | ||
| Format: PDF | Size: 217.7 KB | Download |
Acknowledgements
We thank IKERBASQUE, Basque Foundation for Science; the Basque Government (SAIOTEK S-PE12UN044), Spanish Ministry of Science and Innovation (CTQ2010-19974) and Hamari Chemicals (Osaka, Japan) for generous financial support. The Stony Brook University single-crystal diffractometer was obtained through the support of the National Science Foundation (NSF) grant CHE-0840483. JLA and VAS are very grateful to Professor Dieter Seebach (ETH-Zürich) for fruitful and inspirational discussions.
References
-
Soloshonok, V. A.; Cai, C.; Hruby, V. J.; Van Meervelt, L.; Mischenko, N. Tetrahedron 1999, 55, 12031–12044. doi:10.1016/S0040-4020(99)00711-5
Return to citation in text: [1] -
Schöllkopf, U.; Groth, U.; Deng, C. Angew. Chem., Int. Ed. Engl. 1981, 20, 798–799. doi:10.1002/anie.198107981
Return to citation in text: [1] [2] -
Williams, R. M.; Sinclair, P. J.; Zhai, W. J. Am. Chem. Soc. 1988, 110, 482–483. doi:10.1021/ja00210a027
Return to citation in text: [1] [2] -
Fitzi, R.; Seebach, D. Tetrahedron 1988, 44, 5277–5292. doi:10.1016/S0040-4020(01)86036-1
Return to citation in text: [1] [2] -
O’Donnell, M. J.; Eckrich, T. M. Tetrahedron Lett. 1978, 19, 4625–4628. doi:10.1016/S0040-4039(01)85688-4
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Hayashi, T.; Ishikawa, K.; Nagashima, N. Tetrahedron Lett. 1994, 35, 1055–1058. doi:10.1016/S0040-4039(00)79964-3
Return to citation in text: [1] [2] -
Lygo, B.; Wainwright, P. G. Tetrahedron Lett. 1997, 38, 8595–8598. doi:10.1016/S0040-4039(97)10293-3
Return to citation in text: [1] [2] -
Corey, E. J.; Xu, F.; Noe, M. C. J. Am. Chem. Soc. 1997, 119, 12414–12415. doi:10.1021/ja973174y
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Kacharov, A. D.; Avilov, D. V.; Ishikawa, K.; Nagashima, N.; Hayashi, T. J. Org. Chem. 1997, 62, 3470–3479. doi:10.1021/jo9623402
Return to citation in text: [1] [2] -
Ooi, T.; Kameda, M.; Maruoka, K. J. Am. Chem. Soc. 1999, 121, 6519–6520. doi:10.1021/ja991062w
Return to citation in text: [1] [2] -
Chinchilla, R.; Mazón, P.; Nájera, C. Tetrahedron: Asymmetry 2000, 11, 3277–3281. doi:10.1016/S0957-4166(00)00317-7
Return to citation in text: [1] [2] -
Park, H.-g.; Jeong, B.-S.; Yoo, M.-S.; Lee, J.-H.; Park, M.-k.; Lee, Y.-J.; Kim, M.-J.; Jew, S.-s. Angew. Chem., Int. Ed. 2002, 41, 3036–3038. doi:10.1002/1521-3773(20020816)41:16<3036::AID-ANIE3036>3.0.CO;2-3
Return to citation in text: [1] [2] -
Shibuguchi, T.; Fukuta, Y.; Akachi, Y.; Sekine, A.; Ohshima, T.; Shibasaki, M. Tetrahedron Lett. 2002, 43, 9539–9543. doi:10.1016/S0040-4039(02)02416-4
Return to citation in text: [1] [2] -
Solladié-Cavallo, A.; Sedy, O.; Salisova, M.; Schmitt, M. Eur. J. Org. Chem. 2002, 3042–3049. doi:10.1002/1099-0690(200209)2002:17<3042::AID-EJOC3042>3.0.CO;2-S
Return to citation in text: [1] [2] -
Park, H.; Kim, K. M.; Lee, A.; Ham, S.; Nam, W.; Chin, J. J. Am. Chem. Soc. 2007, 129, 1518–1519. doi:10.1021/ja067724g
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ellis, T. K.; Ueki, H.; Ono, T. J. Am. Chem. Soc. 2009, 131, 7208–7209. doi:10.1021/ja9026055
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Ueki, H.; Aceña, J. L.; Ellis, T. K.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 152, 114–118. doi:10.1016/j.jfluchem.2013.02.022
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Ueki, H.; Aceña, J. L.; Ellis, T. K.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Org. Biomol. Chem. 2013, 11, 4503–4507. doi:10.1039/c3ob40541a
Return to citation in text: [1] [2] -
Duthaler, R. O. Tetrahedron 1994, 50, 1539–1650. doi:10.1016/S0040-4020(01)80840-1
Return to citation in text: [1] [2] -
Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e
Return to citation in text: [1] [2] -
Ma, J.-A. Angew. Chem., Int. Ed. 2003, 42, 4290–4299. doi:10.1002/anie.200301600
Return to citation in text: [1] [2] -
Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o
Return to citation in text: [1] [2] -
Soloshonok, V. A. Curr. Org. Chem. 2002, 6, 341–364. doi:10.2174/1385272024605014
Return to citation in text: [1] [2] -
Kukhar, V. P.; Sorochinsky, A. E.; Soloshonok, V. A. Future Med. Chem. 2009, 1, 793–819. doi:10.4155/fmc.09.70
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Soloshonok, V. A. J. Fluorine Chem. 2010, 131, 127–139. doi:10.1016/j.jfluchem.2009.09.015
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Sorochinsky, A. E. Synthesis 2010, 2319–2344. doi:10.1055/s-0029-1220013
Return to citation in text: [1] [2] -
Aceña, J. L.; Sorochinsky, A. E.; Soloshonok, V. A. Synthesis 2012, 44, 1591–1602. doi:10.1055/s-0031-1289756
Return to citation in text: [1] [2] -
Aceña, J. L.; Sorochinsky, A. E.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 155, 21–38. doi:10.1016/j.jfluchem.2013.06.004
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 691–718. doi:10.1007/s00726-013-1539-4
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 1017–1033. doi:10.1007/s00726-013-1580-3
Return to citation in text: [1] [2] -
Wang, H.; Jiang, T.; Xu, M.-H. J. Am. Chem. Soc. 2013, 135, 971–974. doi:10.1021/ja3110818
Return to citation in text: [1] -
Pericas, À.; Shafir, A.; Vallribera, A. Org. Lett. 2013, 15, 1448–1451. doi:10.1021/ol400136y
Return to citation in text: [1] -
Yadav, S.; Taylor, C. M. J. Org. Chem. 2013, 78, 5401–5409. doi:10.1021/jo400558t
Return to citation in text: [1] -
Hugelshofer, C. L.; Mellem, K. T.; Myers, A. G. Org. Lett. 2013, 15, 3134–3137. doi:10.1021/ol401337p
Return to citation in text: [1] -
Sathe, A. A.; Hartline, D. R.; Radosevich, A. T. Chem. Commun. 2013, 49, 5040–5042. doi:10.1039/c3cc42057d
Return to citation in text: [1] -
Nash, A.; Soheili, A.; Tambar, U. K. Org. Lett. 2013, 15, 4770–4773. doi:10.1021/ol402129h
Return to citation in text: [1] -
Chen, K.; Hu, F.; Zhang, S.-Q.; Shi, B.-F. Chem. Sci. 2013, 4, 3906–3911. doi:10.1039/c3sc51747k
Return to citation in text: [1] -
Noisier, A. F. M.; Harris, C. S.; Brimble, M. A. Chem. Commun. 2013, 49, 7744–7746. doi:10.1039/c3cc44717k
Return to citation in text: [1] -
Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keßeler, M.; Stürmer, R.; Zelinski, T. Angew. Chem., Int. Ed. 2004, 43, 788–824. doi:10.1002/anie.200300599
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Izawa, K., Eds. Asymmetric Synthesis and Application of α-Amino Acids; ACS Symposium Series, Vol. 1009; American Chemical Society: Washington DC, 2009.
Return to citation in text: [1] [2] -
Fogassy, E.; Nógrádi, M.; Pálovics, E.; Schindler, J. Synthesis 2005, 1555–1568. doi:10.1055/s-2005-869903
Return to citation in text: [1] -
Ellis, T. K.; Martin, C. H.; Tsai, G. M.; Ueki, H.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 6208–6214. doi:10.1021/jo030075w
Return to citation in text: [1] -
Soloshonok, V. A.; Ohkura, H.; Yasumoto, M. J. Fluorine Chem. 2006, 127, 924–929. doi:10.1016/j.jfluchem.2006.04.003
Return to citation in text: [1] -
Belokon, Yu. N.; Zel'tser, E.; Bakhmutov, V. I.; Saporovskaya, M. B.; Ryzhov, M. G.; Yanovskii, A. I.; Struchkov, Yu. T.; Belikov, V. M. J. Am. Chem. Soc. 1983, 105, 2010–2017. doi:10.1021/ja00345a057
Return to citation in text: [1] -
Belokon, Yu. N.; Bulychev, A. G.; Vitt, S. V.; Struchkov, Yu. T.; Batsanov, A. S.; Timofeeva, T. V.; Tsyryapkin, V. A.; Ryzhov, M. G.; Lysova, L. A.; Bakhmutov, V. I.; Belikov, V. M. J. Am. Chem. Soc. 1985, 107, 4252–4259. doi:10.1021/ja00300a030
Return to citation in text: [1] -
Belokon, Y. N.; Tararov, V. I.; Maleev, V. I.; Savel’eva, T. F.; Ryzhov, M. G. Tetrahedron: Asymmetry 1998, 9, 4249–4252. doi:10.1016/S0957-4166(98)00449-2
Return to citation in text: [1] -
Ueki, H.; Ellis, T. K.; Martin, C. H.; Boettiger, T. U.; Bolene, S. B.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 7104–7107. doi:10.1021/jo0301494
Return to citation in text: [1] -
Tang, X.; Soloshonok, V. A.; Hruby, V. J. Tetrahedron: Asymmetry 2000, 11, 2917–2925. doi:10.1016/S0957-4166(00)00250-0
Return to citation in text: [1] -
Qiu, W.; Soloshonok, V. A.; Cai, C.; Tang, X.; Hruby, V. J. Tetrahedron 2000, 56, 2577–2582. doi:10.1016/S0040-4020(00)00176-9
Return to citation in text: [1] -
Soloshonok, V. A.; Tang, X.; Hruby, V. J. Tetrahedron 2001, 57, 6375–6382. doi:10.1016/S0040-4020(01)00504-X
Return to citation in text: [1] -
Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Tararov, V. I.; Savel'eva, T. F.; Churkina, T. D.; Ikonnikov, N. S.; Kochetkov, K. A.; Orlova, S. A.; Pysarevsky, A. P.; Struchkov, Y. T.; Raevsky, N. I.; Belokon, Y. N. Tetrahedron: Asymmetry 1995, 6, 1741–1756. doi:10.1016/0957-4166(95)00220-J
Return to citation in text: [1] -
Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P. Tetrahedron 1996, 52, 12433–12442. doi:10.1016/0040-4020(96)00741-7
Return to citation in text: [1] -
Soloshonok, V. A.; Kukhar, V. P.; Galushko, S. V.; Svistunova, N. Yu.; Avilov, D. V.; Kuz'mina, N. A.; Raevski, N. I.; Struchkov, Y. T.; Pysarevsky, A. P.; Belokon, Y. N. J. Chem. Soc., Perkin Trans. 1 1993, 3143–3155. doi:10.1039/p19930003143
Return to citation in text: [1] -
Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Van Meervelt, L.; Mischenko, N. Tetrahedron Lett. 1997, 38, 4671–4674. doi:10.1016/S0040-4039(97)00963-5
Return to citation in text: [1] -
Wang, J.; Shi, T.; Deng, G.; Jiang, H.; Liu, H. J. Org. Chem. 2008, 73, 8563–8570. doi:10.1021/jo8019169
Return to citation in text: [1] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron: Asymmetry 1999, 10, 4265–4269. doi:10.1016/S0957-4166(99)00483-8
Return to citation in text: [1] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 135–139. doi:10.1016/S0040-4039(99)02018-3
Return to citation in text: [1] -
Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561
Return to citation in text: [1] -
Ueki, H.; Ellis, T. K.; Martin, C. H.; Soloshonok, V. A. Eur. J. Org. Chem. 2003, 1954–1957. doi:10.1002/ejoc.200200688
Return to citation in text: [1] -
Deng, G.; Wang, J.; Zhou, Y.; Jiang, H.; Liu, H. J. Org. Chem. 2007, 72, 8932–8934. doi:10.1021/jo071011e
Return to citation in text: [1] -
Ellis, T. K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V. A. J. Org. Chem. 2006, 71, 8572–8578. doi:10.1021/jo0616198
Return to citation in text: [1] -
Ellis, T. K.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2005, 46, 941–944. doi:10.1016/j.tetlet.2004.12.050
Return to citation in text: [1] -
Soloshonok, V. A.; Ueki, H.; Ellis, T. K.; Yamada, T.; Ohfune, Y. Tetrahedron Lett. 2005, 46, 1107–1110. doi:10.1016/j.tetlet.2004.12.093
Return to citation in text: [1] -
Ellis, T. K.; Hochla, V. M.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 4973–4976. doi:10.1021/jo030065v
Return to citation in text: [1] -
Ellis, T. K.; Martin, C. H.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2003, 44, 1063–1066. doi:10.1016/S0040-4039(02)02719-3
Return to citation in text: [1] -
Belokon, Y. N.; Kochetkov, K. A.; Churkina, T. D.; Ikonnikov, N. S.; Larionov, O. V.; Harutyunyan, S. R.; Vyskočil, Š.; North, M.; Kagan, H. B. Angew. Chem., Int. Ed. 2001, 40, 1948–1951. doi:10.1002/1521-3773(20010518)40:10<1948::AID-ANIE1948>3.3.CO;2-F
Return to citation in text: [1] -
Belokon, Y. N.; Bespalova, N. B.; Churkina, T. D.; Císařová, I.; Ezernitskaya, M. G.; Harutyunyan, S. R.; Hrdina, R.; Kagan, H. B.; Kočovský, P.; Kochetkov, K. A.; Larionov, O. V.; Lyssenko, K. A.; North, M.; Polášek, M.; Peregudov, A. S.; Prisyazhnyuk, V. V.; Vyskočil, Š. J. Am. Chem. Soc. 2003, 125, 12860–12871. doi:10.1021/ja035465e
Return to citation in text: [1] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 9645–9649. doi:10.1016/S0040-4039(00)01737-8
Return to citation in text: [1] -
Soloshonok, V. A.; Ueki, H.; Tiwari, R.; Cai, C.; Hruby, V. J. J. Org. Chem. 2004, 69, 4984–4990. doi:10.1021/jo0495438
Return to citation in text: [1] -
Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V. A. Org. Lett. 2006, 8, 5625–5628. doi:10.1021/ol0623668
Return to citation in text: [1] -
Yamada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y.; Soloshonok, V. A. Tetrahedron: Asymmetry 2008, 19, 2789–2795. doi:10.1016/j.tetasy.2008.11.036
Return to citation in text: [1] -
Ellis, T. K.; Soloshonok, V. A. Synlett 2006, 533–538. doi:10.1055/s-2006-926252
Return to citation in text: [1] -
Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Synlett 2009, 704–715. doi:10.1055/s-0028-1087929
Return to citation in text: [1] -
Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Chim. Oggi 2008, 26, 51–54.
Return to citation in text: [1] -
Juaristi, E.; Escalante, J.; León-Romo, J. L.; Reyes, A. Tetrahedron: Asymmetry 1998, 9, 715–740. doi:10.1016/S0957-4166(98)00058-5
Return to citation in text: [1] -
Juaristi, E.; León-Romo, J. L.; Reyes, A.; Escalante, J. Tetrahedron: Asymmetry 1999, 10, 2441–2495. doi:10.1016/S0957-4166(99)00242-6
Return to citation in text: [1] -
Moore, J. L.; Taylor, S. M.; Soloshonok, V. A. ARKIVOC 2005, No. vi, 287–292. doi:10.3998/ark.5550190.0006.624
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090
Return to citation in text: [1] [2] -
Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215
Return to citation in text: [1] -
Soloshonok, V. A.; Aceña, J. L.; Ueki, H.; Han, J. Beilstein J. Org. Chem. 2012, 8, 1920–1928. doi:10.3762/bjoc.8.223
Return to citation in text: [1] -
CCDC 943686 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Soloshonok, V. A.; Cai, C.; Hruby, V. J.; Van Meervelt, L. Tetrahedron 1999, 55, 12045–12058. doi:10.1016/S0040-4020(99)00710-3
Return to citation in text: [1] -
Belokon, Y. N.; Bulychev, A. G.; Ryzhov, M. G.; Vitt, S. V.; Batsanov, A. S.; Struchkov, Y. T.; Bakhmutov, V. I.; Belikov, V. M. J. Chem. Soc., Perkin Trans. 1 1986, 1865–1872. doi:10.1039/p19860001865
Return to citation in text: [1] -
Belokon, Y. N.; Bulychev, A. G.; Pavlov, V. A.; Fedorova, E. B.; Tsyryapkin, V. A.; Bakhmutov, V. I.; Belikov, V. M. J. Chem. Soc., Perkin Trans. 1 1988, 2075–2083. doi:10.1039/P19880002075
Return to citation in text: [1] -
Belokon, Y. N.; Maleyev, V. I.; Vitt, S. V.; Ryzhov, M. G.; Kondrashov, Y. D.; Golubev, S. N.; Vauchskii, Y. P.; Kazika, A. I.; Novikova, M. I.; Krasutskii, P. A.; Yurchenko, A. G.; Dubchak, I. L.; Shklover, V. E.; Struchkov, Y. T.; Bakhmutov, V. I.; Belikov, V. M. J. Chem. Soc., Dalton Trans. 1985, 17–26. doi:10.1039/dt9850000017
Return to citation in text: [1]
| 1. | Soloshonok, V. A.; Cai, C.; Hruby, V. J.; Van Meervelt, L.; Mischenko, N. Tetrahedron 1999, 55, 12031–12044. doi:10.1016/S0040-4020(99)00711-5 |
| 39. | Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keßeler, M.; Stürmer, R.; Zelinski, T. Angew. Chem., Int. Ed. 2004, 43, 788–824. doi:10.1002/anie.200300599 |
| 40. | Soloshonok, V. A.; Izawa, K., Eds. Asymmetric Synthesis and Application of α-Amino Acids; ACS Symposium Series, Vol. 1009; American Chemical Society: Washington DC, 2009. |
| 64. | Ellis, T. K.; Hochla, V. M.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 4973–4976. doi:10.1021/jo030065v |
| 65. | Ellis, T. K.; Martin, C. H.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2003, 44, 1063–1066. doi:10.1016/S0040-4039(02)02719-3 |
| 2. | Schöllkopf, U.; Groth, U.; Deng, C. Angew. Chem., Int. Ed. Engl. 1981, 20, 798–799. doi:10.1002/anie.198107981 |
| 3. | Williams, R. M.; Sinclair, P. J.; Zhai, W. J. Am. Chem. Soc. 1988, 110, 482–483. doi:10.1021/ja00210a027 |
| 4. | Fitzi, R.; Seebach, D. Tetrahedron 1988, 44, 5277–5292. doi:10.1016/S0040-4020(01)86036-1 |
| 5. | O’Donnell, M. J.; Eckrich, T. M. Tetrahedron Lett. 1978, 19, 4625–4628. doi:10.1016/S0040-4039(01)85688-4 |
| 6. | Soloshonok, V. A.; Hayashi, T.; Ishikawa, K.; Nagashima, N. Tetrahedron Lett. 1994, 35, 1055–1058. doi:10.1016/S0040-4039(00)79964-3 |
| 7. | Lygo, B.; Wainwright, P. G. Tetrahedron Lett. 1997, 38, 8595–8598. doi:10.1016/S0040-4039(97)10293-3 |
| 8. | Corey, E. J.; Xu, F.; Noe, M. C. J. Am. Chem. Soc. 1997, 119, 12414–12415. doi:10.1021/ja973174y |
| 9. | Soloshonok, V. A.; Kacharov, A. D.; Avilov, D. V.; Ishikawa, K.; Nagashima, N.; Hayashi, T. J. Org. Chem. 1997, 62, 3470–3479. doi:10.1021/jo9623402 |
| 10. | Ooi, T.; Kameda, M.; Maruoka, K. J. Am. Chem. Soc. 1999, 121, 6519–6520. doi:10.1021/ja991062w |
| 11. | Chinchilla, R.; Mazón, P.; Nájera, C. Tetrahedron: Asymmetry 2000, 11, 3277–3281. doi:10.1016/S0957-4166(00)00317-7 |
| 12. | Park, H.-g.; Jeong, B.-S.; Yoo, M.-S.; Lee, J.-H.; Park, M.-k.; Lee, Y.-J.; Kim, M.-J.; Jew, S.-s. Angew. Chem., Int. Ed. 2002, 41, 3036–3038. doi:10.1002/1521-3773(20020816)41:16<3036::AID-ANIE3036>3.0.CO;2-3 |
| 13. | Shibuguchi, T.; Fukuta, Y.; Akachi, Y.; Sekine, A.; Ohshima, T.; Shibasaki, M. Tetrahedron Lett. 2002, 43, 9539–9543. doi:10.1016/S0040-4039(02)02416-4 |
| 14. | Solladié-Cavallo, A.; Sedy, O.; Salisova, M.; Schmitt, M. Eur. J. Org. Chem. 2002, 3042–3049. doi:10.1002/1099-0690(200209)2002:17<3042::AID-EJOC3042>3.0.CO;2-S |
| 15. | Park, H.; Kim, K. M.; Lee, A.; Ham, S.; Nam, W.; Chin, J. J. Am. Chem. Soc. 2007, 129, 1518–1519. doi:10.1021/ja067724g |
| 16. | Soloshonok, V. A.; Ellis, T. K.; Ueki, H.; Ono, T. J. Am. Chem. Soc. 2009, 131, 7208–7209. doi:10.1021/ja9026055 |
| 17. | Sorochinsky, A. E.; Ueki, H.; Aceña, J. L.; Ellis, T. K.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 152, 114–118. doi:10.1016/j.jfluchem.2013.02.022 |
| 18. | Sorochinsky, A. E.; Ueki, H.; Aceña, J. L.; Ellis, T. K.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Org. Biomol. Chem. 2013, 11, 4503–4507. doi:10.1039/c3ob40541a |
| 19. | Duthaler, R. O. Tetrahedron 1994, 50, 1539–1650. doi:10.1016/S0040-4020(01)80840-1 |
| 20. | Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e |
| 21. | Ma, J.-A. Angew. Chem., Int. Ed. 2003, 42, 4290–4299. doi:10.1002/anie.200301600 |
| 22. | Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o |
| 23. | Soloshonok, V. A. Curr. Org. Chem. 2002, 6, 341–364. doi:10.2174/1385272024605014 |
| 24. | Kukhar, V. P.; Sorochinsky, A. E.; Soloshonok, V. A. Future Med. Chem. 2009, 1, 793–819. doi:10.4155/fmc.09.70 |
| 25. | Sorochinsky, A. E.; Soloshonok, V. A. J. Fluorine Chem. 2010, 131, 127–139. doi:10.1016/j.jfluchem.2009.09.015 |
| 26. | Soloshonok, V. A.; Sorochinsky, A. E. Synthesis 2010, 2319–2344. doi:10.1055/s-0029-1220013 |
| 27. | Aceña, J. L.; Sorochinsky, A. E.; Soloshonok, V. A. Synthesis 2012, 44, 1591–1602. doi:10.1055/s-0031-1289756 |
| 28. | Aceña, J. L.; Sorochinsky, A. E.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 155, 21–38. doi:10.1016/j.jfluchem.2013.06.004 |
| 29. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 691–718. doi:10.1007/s00726-013-1539-4 |
| 30. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 1017–1033. doi:10.1007/s00726-013-1580-3 |
| 66. | Belokon, Y. N.; Kochetkov, K. A.; Churkina, T. D.; Ikonnikov, N. S.; Larionov, O. V.; Harutyunyan, S. R.; Vyskočil, Š.; North, M.; Kagan, H. B. Angew. Chem., Int. Ed. 2001, 40, 1948–1951. doi:10.1002/1521-3773(20010518)40:10<1948::AID-ANIE1948>3.3.CO;2-F |
| 67. | Belokon, Y. N.; Bespalova, N. B.; Churkina, T. D.; Císařová, I.; Ezernitskaya, M. G.; Harutyunyan, S. R.; Hrdina, R.; Kagan, H. B.; Kočovský, P.; Kochetkov, K. A.; Larionov, O. V.; Lyssenko, K. A.; North, M.; Polášek, M.; Peregudov, A. S.; Prisyazhnyuk, V. V.; Vyskočil, Š. J. Am. Chem. Soc. 2003, 125, 12860–12871. doi:10.1021/ja035465e |
| 31. | Wang, H.; Jiang, T.; Xu, M.-H. J. Am. Chem. Soc. 2013, 135, 971–974. doi:10.1021/ja3110818 |
| 32. | Pericas, À.; Shafir, A.; Vallribera, A. Org. Lett. 2013, 15, 1448–1451. doi:10.1021/ol400136y |
| 33. | Yadav, S.; Taylor, C. M. J. Org. Chem. 2013, 78, 5401–5409. doi:10.1021/jo400558t |
| 34. | Hugelshofer, C. L.; Mellem, K. T.; Myers, A. G. Org. Lett. 2013, 15, 3134–3137. doi:10.1021/ol401337p |
| 35. | Sathe, A. A.; Hartline, D. R.; Radosevich, A. T. Chem. Commun. 2013, 49, 5040–5042. doi:10.1039/c3cc42057d |
| 36. | Nash, A.; Soheili, A.; Tambar, U. K. Org. Lett. 2013, 15, 4770–4773. doi:10.1021/ol402129h |
| 37. | Chen, K.; Hu, F.; Zhang, S.-Q.; Shi, B.-F. Chem. Sci. 2013, 4, 3906–3911. doi:10.1039/c3sc51747k |
| 38. | Noisier, A. F. M.; Harris, C. S.; Brimble, M. A. Chem. Commun. 2013, 49, 7744–7746. doi:10.1039/c3cc44717k |
| 59. | Ueki, H.; Ellis, T. K.; Martin, C. H.; Soloshonok, V. A. Eur. J. Org. Chem. 2003, 1954–1957. doi:10.1002/ejoc.200200688 |
| 60. | Deng, G.; Wang, J.; Zhou, Y.; Jiang, H.; Liu, H. J. Org. Chem. 2007, 72, 8932–8934. doi:10.1021/jo071011e |
| 2. | Schöllkopf, U.; Groth, U.; Deng, C. Angew. Chem., Int. Ed. Engl. 1981, 20, 798–799. doi:10.1002/anie.198107981 |
| 3. | Williams, R. M.; Sinclair, P. J.; Zhai, W. J. Am. Chem. Soc. 1988, 110, 482–483. doi:10.1021/ja00210a027 |
| 4. | Fitzi, R.; Seebach, D. Tetrahedron 1988, 44, 5277–5292. doi:10.1016/S0040-4020(01)86036-1 |
| 5. | O’Donnell, M. J.; Eckrich, T. M. Tetrahedron Lett. 1978, 19, 4625–4628. doi:10.1016/S0040-4039(01)85688-4 |
| 6. | Soloshonok, V. A.; Hayashi, T.; Ishikawa, K.; Nagashima, N. Tetrahedron Lett. 1994, 35, 1055–1058. doi:10.1016/S0040-4039(00)79964-3 |
| 7. | Lygo, B.; Wainwright, P. G. Tetrahedron Lett. 1997, 38, 8595–8598. doi:10.1016/S0040-4039(97)10293-3 |
| 8. | Corey, E. J.; Xu, F.; Noe, M. C. J. Am. Chem. Soc. 1997, 119, 12414–12415. doi:10.1021/ja973174y |
| 9. | Soloshonok, V. A.; Kacharov, A. D.; Avilov, D. V.; Ishikawa, K.; Nagashima, N.; Hayashi, T. J. Org. Chem. 1997, 62, 3470–3479. doi:10.1021/jo9623402 |
| 10. | Ooi, T.; Kameda, M.; Maruoka, K. J. Am. Chem. Soc. 1999, 121, 6519–6520. doi:10.1021/ja991062w |
| 11. | Chinchilla, R.; Mazón, P.; Nájera, C. Tetrahedron: Asymmetry 2000, 11, 3277–3281. doi:10.1016/S0957-4166(00)00317-7 |
| 12. | Park, H.-g.; Jeong, B.-S.; Yoo, M.-S.; Lee, J.-H.; Park, M.-k.; Lee, Y.-J.; Kim, M.-J.; Jew, S.-s. Angew. Chem., Int. Ed. 2002, 41, 3036–3038. doi:10.1002/1521-3773(20020816)41:16<3036::AID-ANIE3036>3.0.CO;2-3 |
| 13. | Shibuguchi, T.; Fukuta, Y.; Akachi, Y.; Sekine, A.; Ohshima, T.; Shibasaki, M. Tetrahedron Lett. 2002, 43, 9539–9543. doi:10.1016/S0040-4039(02)02416-4 |
| 14. | Solladié-Cavallo, A.; Sedy, O.; Salisova, M.; Schmitt, M. Eur. J. Org. Chem. 2002, 3042–3049. doi:10.1002/1099-0690(200209)2002:17<3042::AID-EJOC3042>3.0.CO;2-S |
| 15. | Park, H.; Kim, K. M.; Lee, A.; Ham, S.; Nam, W.; Chin, J. J. Am. Chem. Soc. 2007, 129, 1518–1519. doi:10.1021/ja067724g |
| 16. | Soloshonok, V. A.; Ellis, T. K.; Ueki, H.; Ono, T. J. Am. Chem. Soc. 2009, 131, 7208–7209. doi:10.1021/ja9026055 |
| 17. | Sorochinsky, A. E.; Ueki, H.; Aceña, J. L.; Ellis, T. K.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 152, 114–118. doi:10.1016/j.jfluchem.2013.02.022 |
| 18. | Sorochinsky, A. E.; Ueki, H.; Aceña, J. L.; Ellis, T. K.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Org. Biomol. Chem. 2013, 11, 4503–4507. doi:10.1039/c3ob40541a |
| 19. | Duthaler, R. O. Tetrahedron 1994, 50, 1539–1650. doi:10.1016/S0040-4020(01)80840-1 |
| 20. | Maruoka, K.; Ooi, T. Chem. Rev. 2003, 103, 3013–3028. doi:10.1021/cr020020e |
| 21. | Ma, J.-A. Angew. Chem., Int. Ed. 2003, 42, 4290–4299. doi:10.1002/anie.200301600 |
| 22. | Nájera, C.; Sansano, J. M. Chem. Rev. 2007, 107, 4584–4671. doi:10.1021/cr050580o |
| 23. | Soloshonok, V. A. Curr. Org. Chem. 2002, 6, 341–364. doi:10.2174/1385272024605014 |
| 24. | Kukhar, V. P.; Sorochinsky, A. E.; Soloshonok, V. A. Future Med. Chem. 2009, 1, 793–819. doi:10.4155/fmc.09.70 |
| 25. | Sorochinsky, A. E.; Soloshonok, V. A. J. Fluorine Chem. 2010, 131, 127–139. doi:10.1016/j.jfluchem.2009.09.015 |
| 26. | Soloshonok, V. A.; Sorochinsky, A. E. Synthesis 2010, 2319–2344. doi:10.1055/s-0029-1220013 |
| 27. | Aceña, J. L.; Sorochinsky, A. E.; Soloshonok, V. A. Synthesis 2012, 44, 1591–1602. doi:10.1055/s-0031-1289756 |
| 28. | Aceña, J. L.; Sorochinsky, A. E.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. J. Fluorine Chem. 2013, 155, 21–38. doi:10.1016/j.jfluchem.2013.06.004 |
| 29. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 691–718. doi:10.1007/s00726-013-1539-4 |
| 30. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 1017–1033. doi:10.1007/s00726-013-1580-3 |
| 61. | Ellis, T. K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V. A. J. Org. Chem. 2006, 71, 8572–8578. doi:10.1021/jo0616198 |
| 62. | Ellis, T. K.; Ueki, H.; Soloshonok, V. A. Tetrahedron Lett. 2005, 46, 941–944. doi:10.1016/j.tetlet.2004.12.050 |
| 63. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K.; Yamada, T.; Ohfune, Y. Tetrahedron Lett. 2005, 46, 1107–1110. doi:10.1016/j.tetlet.2004.12.093 |
| 48. | Tang, X.; Soloshonok, V. A.; Hruby, V. J. Tetrahedron: Asymmetry 2000, 11, 2917–2925. doi:10.1016/S0957-4166(00)00250-0 |
| 49. | Qiu, W.; Soloshonok, V. A.; Cai, C.; Tang, X.; Hruby, V. J. Tetrahedron 2000, 56, 2577–2582. doi:10.1016/S0040-4020(00)00176-9 |
| 50. | Soloshonok, V. A.; Tang, X.; Hruby, V. J. Tetrahedron 2001, 57, 6375–6382. doi:10.1016/S0040-4020(01)00504-X |
| 54. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Van Meervelt, L.; Mischenko, N. Tetrahedron Lett. 1997, 38, 4671–4674. doi:10.1016/S0040-4039(97)00963-5 |
| 55. | Wang, J.; Shi, T.; Deng, G.; Jiang, H.; Liu, H. J. Org. Chem. 2008, 73, 8563–8570. doi:10.1021/jo8019169 |
| 44. | Belokon, Yu. N.; Zel'tser, E.; Bakhmutov, V. I.; Saporovskaya, M. B.; Ryzhov, M. G.; Yanovskii, A. I.; Struchkov, Yu. T.; Belikov, V. M. J. Am. Chem. Soc. 1983, 105, 2010–2017. doi:10.1021/ja00345a057 |
| 45. | Belokon, Yu. N.; Bulychev, A. G.; Vitt, S. V.; Struchkov, Yu. T.; Batsanov, A. S.; Timofeeva, T. V.; Tsyryapkin, V. A.; Ryzhov, M. G.; Lysova, L. A.; Bakhmutov, V. I.; Belikov, V. M. J. Am. Chem. Soc. 1985, 107, 4252–4259. doi:10.1021/ja00300a030 |
| 46. | Belokon, Y. N.; Tararov, V. I.; Maleev, V. I.; Savel’eva, T. F.; Ryzhov, M. G. Tetrahedron: Asymmetry 1998, 9, 4249–4252. doi:10.1016/S0957-4166(98)00449-2 |
| 47. | Ueki, H.; Ellis, T. K.; Martin, C. H.; Boettiger, T. U.; Bolene, S. B.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 7104–7107. doi:10.1021/jo0301494 |
| 56. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron: Asymmetry 1999, 10, 4265–4269. doi:10.1016/S0957-4166(99)00483-8 |
| 57. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 135–139. doi:10.1016/S0040-4039(99)02018-3 |
| 58. | Soloshonok, V. A.; Cai, C.; Yamada, T.; Ueki, H.; Ohfune, Y.; Hruby, V. J. J. Am. Chem. Soc. 2005, 127, 15296–15303. doi:10.1021/ja0535561 |
| 42. | Ellis, T. K.; Martin, C. H.; Tsai, G. M.; Ueki, H.; Soloshonok, V. A. J. Org. Chem. 2003, 68, 6208–6214. doi:10.1021/jo030075w |
| 43. | Soloshonok, V. A.; Ohkura, H.; Yasumoto, M. J. Fluorine Chem. 2006, 127, 924–929. doi:10.1016/j.jfluchem.2006.04.003 |
| 39. | Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keßeler, M.; Stürmer, R.; Zelinski, T. Angew. Chem., Int. Ed. 2004, 43, 788–824. doi:10.1002/anie.200300599 |
| 40. | Soloshonok, V. A.; Izawa, K., Eds. Asymmetric Synthesis and Application of α-Amino Acids; ACS Symposium Series, Vol. 1009; American Chemical Society: Washington DC, 2009. |
| 41. | Fogassy, E.; Nógrádi, M.; Pálovics, E.; Schindler, J. Synthesis 2005, 1555–1568. doi:10.1055/s-2005-869903 |
| 51. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P.; Tararov, V. I.; Savel'eva, T. F.; Churkina, T. D.; Ikonnikov, N. S.; Kochetkov, K. A.; Orlova, S. A.; Pysarevsky, A. P.; Struchkov, Y. T.; Raevsky, N. I.; Belokon, Y. N. Tetrahedron: Asymmetry 1995, 6, 1741–1756. doi:10.1016/0957-4166(95)00220-J |
| 52. | Soloshonok, V. A.; Avilov, D. V.; Kukhar, V. P. Tetrahedron 1996, 52, 12433–12442. doi:10.1016/0040-4020(96)00741-7 |
| 53. | Soloshonok, V. A.; Kukhar, V. P.; Galushko, S. V.; Svistunova, N. Yu.; Avilov, D. V.; Kuz'mina, N. A.; Raevski, N. I.; Struchkov, Y. T.; Pysarevsky, A. P.; Belokon, Y. N. J. Chem. Soc., Perkin Trans. 1 1993, 3143–3155. doi:10.1039/p19930003143 |
| 75. | Juaristi, E.; Escalante, J.; León-Romo, J. L.; Reyes, A. Tetrahedron: Asymmetry 1998, 9, 715–740. doi:10.1016/S0957-4166(98)00058-5 |
| 76. | Juaristi, E.; León-Romo, J. L.; Reyes, A.; Escalante, J. Tetrahedron: Asymmetry 1999, 10, 2441–2495. doi:10.1016/S0957-4166(99)00242-6 |
| 68. | Soloshonok, V. A.; Cai, C.; Hruby, V. J. Tetrahedron Lett. 2000, 41, 9645–9649. doi:10.1016/S0040-4039(00)01737-8 |
| 69. | Soloshonok, V. A.; Ueki, H.; Tiwari, R.; Cai, C.; Hruby, V. J. J. Org. Chem. 2004, 69, 4984–4990. doi:10.1021/jo0495438 |
| 70. | Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V. A. Org. Lett. 2006, 8, 5625–5628. doi:10.1021/ol0623668 |
| 71. | Yamada, T.; Sakaguchi, K.; Shinada, T.; Ohfune, Y.; Soloshonok, V. A. Tetrahedron: Asymmetry 2008, 19, 2789–2795. doi:10.1016/j.tetasy.2008.11.036 |
| 72. | Ellis, T. K.; Soloshonok, V. A. Synlett 2006, 533–538. doi:10.1055/s-2006-926252 |
| 73. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Synlett 2009, 704–715. doi:10.1055/s-0028-1087929 |
| 74. | Soloshonok, V. A.; Ueki, H.; Ellis, T. K. Chim. Oggi 2008, 26, 51–54. |
| 77. | Moore, J. L.; Taylor, S. M.; Soloshonok, V. A. ARKIVOC 2005, No. vi, 287–292. doi:10.3998/ark.5550190.0006.624 |
| 78. | Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090 |
| 81. | CCDC 943686 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 82. | Soloshonok, V. A.; Cai, C.; Hruby, V. J.; Van Meervelt, L. Tetrahedron 1999, 55, 12045–12058. doi:10.1016/S0040-4020(99)00710-3 |
| 83. | Belokon, Y. N.; Bulychev, A. G.; Ryzhov, M. G.; Vitt, S. V.; Batsanov, A. S.; Struchkov, Y. T.; Bakhmutov, V. I.; Belikov, V. M. J. Chem. Soc., Perkin Trans. 1 1986, 1865–1872. doi:10.1039/p19860001865 |
| 84. | Belokon, Y. N.; Bulychev, A. G.; Pavlov, V. A.; Fedorova, E. B.; Tsyryapkin, V. A.; Bakhmutov, V. I.; Belikov, V. M. J. Chem. Soc., Perkin Trans. 1 1988, 2075–2083. doi:10.1039/P19880002075 |
| 85. | Belokon, Y. N.; Maleyev, V. I.; Vitt, S. V.; Ryzhov, M. G.; Kondrashov, Y. D.; Golubev, S. N.; Vauchskii, Y. P.; Kazika, A. I.; Novikova, M. I.; Krasutskii, P. A.; Yurchenko, A. G.; Dubchak, I. L.; Shklover, V. E.; Struchkov, Y. T.; Bakhmutov, V. I.; Belikov, V. M. J. Chem. Soc., Dalton Trans. 1985, 17–26. doi:10.1039/dt9850000017 |
| 77. | Moore, J. L.; Taylor, S. M.; Soloshonok, V. A. ARKIVOC 2005, No. vi, 287–292. doi:10.3998/ark.5550190.0006.624 |
| 78. | Soloshonok, V. A.; Ueki, H. Synthesis 2010, 49–56. doi:10.1055/s-0029-1217090 |
| 79. | Soloshonok, V. A.; Ueki, H. J. Am. Chem. Soc. 2007, 129, 2426–2427. doi:10.1021/ja0671215 |
| 80. | Soloshonok, V. A.; Aceña, J. L.; Ueki, H.; Han, J. Beilstein J. Org. Chem. 2012, 8, 1920–1928. doi:10.3762/bjoc.8.223 |
© 2014 Moriwaki et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)