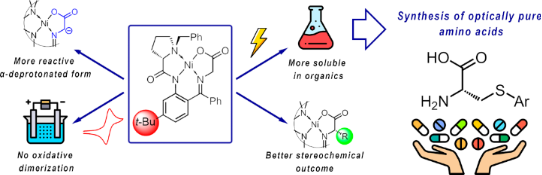
Abstract
A new oxidatively stable (S)-N-benzylproline-derived ligand ((S)-N-(2-benzoyl-5-tert-butylphenyl)-1-benzylpyrrolidine-2-carboxamide) and its Ni(II)–Schiff base complexes formed of glycine, serine, and dehydroalanine are reported. A bulky tert-butyl substituent in the phenylene fragment precludes unwanted oxidative dimerization of the Schiff base complex, making it suitable for targeted electrochemically induced oxidative modification of the amino acid side chain. Experimental and DFT studies showed that the additional tert-butyl group increases the dispersion interactions in the Ni coordination environment making the complexes more conformationally rigid and provides a higher level of thermodynamically controlled stereoselectivity as compared to the parent Belokon complex. Additionally, functionalization with the tert-butyl group significantly enhances the reactivity of the deprotonated glycine complex towards electrophiles as compared to the anionic species formed from the original Belokon complex. Solubility of the t-Bu-containing ligand and its Schiff base complexes is increased, facilitating scaling-up the reaction procedure and isolation of the functionalized amino acid.
Graphical Abstract
Introduction
Asymmetric synthesis of functionalized amino acids is a subject of intense research because these compounds are of great demand for pharmaceutical industry, health care, and food production [1-3]. Various approaches to enantiomerically enriched amino acids have been developed employing chiral auxiliaries [4,5] and asymmetric phase-transfer catalysis [6,7]. The former approach is commonly based on the application of chiral derivatives of glycine containing structurally diverse chiral auxiliaries, both cyclic [8-11] and acyclic [12,13]. Transition-metal complexes derived from glycine Schiff bases containing a source of chirality is the most convenient and widely used template for modification of the glycine fragment under mild conditions. The template can be easily obtained via self-assembly of the starting components in the presence of metal ions (commonly Ni(II)) and includes a chiral auxiliary, an amino acid, and a bifunctional linker capable to arrange the components in the Schiff base complex. Such templates provide a significant C–H acidity at the α-amino acid carbon and a possibility for recycling of the chiral auxiliaries (for reviews see [5,14-18]). In early works, the chiral tridentate ligand based on (S)-N-benzylproline (L1) was used [19,20]. Though the original Belokon complex derived from N-benzylproline and o-aminobenzophenone showed sufficiently high efficiency and is still the most widely used template, considerable efforts on the modification of the chiral auxiliaries as well as of the other fragments of the tridentate ligand have been made to improve stereocontrolling efficiency and to modify physicochemical properties of the template (such as solubility, lipophilicity, etc). Thus, various substituents (4,5-di-CH3 [21], 2-Cl [21], 3,4-di-Cl [21,22], 2/3/4-F [23]) were inserted in the N-benzyl moiety as well as in the aromatic rings of the benzophenone fragment [24-26] (selected examples are given in Scheme 1).
![[1860-5397-19-41-i1]](/bjoc/content/inline/1860-5397-19-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Selected examples of the chiral ligands used for synthesis of the Ni(II)–Schiff base complexes.
Scheme 1: Selected examples of the chiral ligands used for synthesis of the Ni(II)–Schiff base complexes.
Insertion of halogen atoms increased enantioselectivity, e.g., in alkylation reactions [27,28]. Functionalization of the phenyl ring in the benzophenone gives rise to an additional axial chirality (L3), thus improving stereoselectivity observed at the removed stereocenter [24,25]. Replacing N-benzylproline for 2,7-dihydro-1H-azepine (L5) allowed obtaining a new tridentate ligand with chemically stable axial chirality which exhibits no racemization under action of strong bases [29]. The chiral N–H-containing rimantadine-based ligand (L6) is highly lipophilic and it was successfully used for the kinetic resolution of unprotected racemic amino acids [30].
The examples mentioned above show that the metal–Schiff base chiral template is a fairly versatile “tool” that can be adapted to a specific task. A relatively new approach to functionalization of amino acids is a combination of a stereoselective synthesis in a metal-coordination environment with electrochemical activation [31]. It increases the reactivity and the reaction scope, making possible new types of transformations [32-35]. On the other hand, it puts additional requirements on the design of the chiral ligand. Another important structurally tunable parameter comprises the oxidation or reduction potential of the complex. Additionally, as it has been shown in our previous reports [31,36], the protection of the phenylene fragment with substituents is required, to allow oxidative transformations of the amino acid fragment. Otherwise, the oxidative coupling of two phenylene fragments via the para-positions may be the dominant reaction path yielding the bimetallic Ni(II) complex [36]. Thus, L4 in Scheme 1 was shown to be suitable for the oxidative functionalization of the amino acid fragment [37].
A bulky tert-butyl group can be also proposed as a protecting moiety. One could expect that the t-Bu group will prevent fast oxidative dimerization of the Schiff base complex and the radical cation formed under one-electron electrochemical oxidation will be sufficiently stable, opening a route to further oxidative modification of the amino acid side chain under appropriate conditions. Additionally, this bulky group may significantly alter the whole steric construction of a molecule and give rise to additional interactions which would increase the stereocontrolling properties. This idea efficiently works in the enantioselective extraction of the unprotected amino acids [38,39]. In the case of the Ni–Schiff base complexes, the t-Bu group may give rise to additional dispersion interactions with the phenyl ring in the proline auxiliary, making the Schiff base complexes more conformationally rigid, thus increasing the stereochemical outcome of the functionalized amino acids as compared to the parent ligand L1.
In the present paper, the new structurally advantageous (S)-N-benzylproline-derived ligand containing the bulky tert-butyl substituent in the phenylene fragment (L7) is reported. Three Ni(II)–Schiff base derivatives of the new ligand (containing glycine, serine, and dehydroalanine) were obtained. Complexes of this type are most often used as the starting platform for the modification of an amino acid moiety by nucleophilic substitution and addition reactions, including electrochemically induced processes [5,16-18,31]. The electrochemical behavior of the new complexes was investigated to get an experimental support for their oxidative stability. The reactivity of the glycine and dehydroalanine complexes in the nucleophilic functionalization of amino acids was compared to the corresponding derivatives of the parent ligand L1. The results clearly demonstrated the advantages of the new complexes as stereocontrolling templates.
Results and Discussion
Synthesis
The synthetic approach to the chiral ligand L7 as well as to its Ni–Schiff base derivatives containing glycine, serine, dehydroalanine, and cysteine is given in Scheme 2. Commercially available (S)-proline was used as the starting material.
![[1860-5397-19-41-i2]](/bjoc/content/inline/1860-5397-19-41-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the chiral ligand L7 and its Ni(II) complexes with glycine, serine, dehydroalanine, and cysteine derivatives.
Scheme 2: Synthesis of the chiral ligand L7 and its Ni(II) complexes with glycine, serine, dehydroalanine, an...
To obtain the starting t-Bu-substituted o-aminobenzophenone, the synthetic procedure reported in [40] was applied. For acylation of the tert-butylacetanilide, PhCCl3 in the presence of AlCl3 was used. In the reported [40] reaction conditions, a complicated mixture of products was obtained. Lowering the temperature to −15 °C (in contrast to rt in [40]) allowed obtaining the targeted compound; however, acylation was accompanied with the migration of the t-Bu group yielding 2-benzoyl-4-tert-butylaniline and the corresponding 5-butylated isomer in a 10:1 ratio. Our attempts to separate the isomers were unsuccessful. However, as it will be shown below, this is not necessary since the isomeric Schiff base template proved efficient in stereoselective modification of amino acids. The products of the oxidative dimerization via the phenylene ring were not detected for both isomeric templates.
The subsequent reaction with (S)-benzylproline lead to (S)-N-(2-benzoyl-5-tert-butylphenyl)-1-benzylpyrrolidine-2-carboxamide (L7) in 73% yield with 96% ee (see Supporting Information File 1). The self-assembly of the three components L7, Ni(NO3)2, and glycine gave the corresponding (GlyNi)L7 complex which was isolated in 66% yield and fully characterized using spectral methods (for HRMS, 1H, 13C NMR, including 2D techniques, see Supporting Information File 1).
To obtain the serine derivative (SerNi)L7, the recently developed electrochemical approach for the stereoselective hydroxyalkylation [32] was used. The reaction protocol is operationally simple and suitable for gram-scale loadings. Galvanostatic electrolysis of the glycine complex (GlyNi)L7 was performed in a one-compartment electrochemical cell in a methanol solution (CH3OH serves as a reactant and a solvent simultaneously) in the presence of KOH. By this route the (SerNi)L7 complex (with ʟ-configuration at the α-stereocenter) was obtained in quantitative yield with high diastereselectivity (83% de). Previously, the same protocol was applied to the parent Belokon complex, affording the corresponding serine derivative in 97% yield with 81% de [32]. For spectral characterization of (SerNi)L7, see Supporting Information File 1.
The serine complex (SerNi)L7 served as the precursor for (ΔAlaNi)L7 which was obtained via dehydration using a commonly used procedure [41]. The new complex (ΔAlaNi)L7 was isolated in 92% yield and fully characterized (HRMS, 1H, 13C NMR, including 2D techniques; for details, see Supporting Information File 1); [α]D20 = 2127 (MeOH).
To test the stereocontrolling efficiency of the new ligand L7 and to obtain new cysteine Ni–Schiff base derivatives (RCysNi)L7 which are of practical interest, complex (ΔAlaNi)L7 was involved in a nucleophilic addition as Michael acceptor. A number of thiols was taken as the model compounds. The reaction was performed under thermodynamic conditions reported in [41], to compare the thermodynamically controlled stereoselectivity of L7 and L1. The use of a 2 molar excess of K2CO3 induces the equilibrium between the isomers (established via the α-protonation/deprotonation in the intermediate α-carbanion in the amino acid moiety) and concomitant epimerization of the final product [41]. The results of the thiolation of complex (ΔAlaNi)L7 are given in Table 1. The corresponding ʟ-cysteine derivatives (RCysNi)L7 were isolated in high yields and with excellent stereoselectivity: only traces of the ᴅ-cysteine derivatives were determined, demonstrating the advantages of the tert-butyl-containing template (for the probable reasons, see below).
Table 1: The yields and the thermodynamically controlled stereoselectivity observed in the thiolation of (ΔAlaNi)L7 complexes with RSH; and comparison with the data for (ΔAlaNi)L1 reported previously [41] given in parentheses.a
| R | Bn | Ph | p-CH3C6H4- | o-BrC6H4- | p-BrC6H4- |
| product | (BnCysNi)L | (PhCysNi)L | (pMeCysNi)L | (oBrCysNi)L | (pBrCysNi)L |
| yield | 85% (97%) | 73% (93%) | 83% | 71% | 78% |
| dr (ʟ:ᴅ)b | >52:1 (19:1) | >44:1 (30:1) | >43:1 | >29:1 | 32:1 |
aConditions: 0.65 М ΔAlaNi, CH3CN/DMF, 2 equiv K2CO3, 1.05 equiv RSH, 50–55 °C. bdr was determined as the ratio of the isolated diastereomers. For accuracy, less than 1 mg amounts of the minor isomer were rounded up to 1 mg.
The relative ʟ-configuration of the amino acid α-stereocenter in the major diastereomers of (RCysNi)L7 formed in the Michael addition reaction was confirmed using the NOESY spectrum. It is illustrated in Figure 1 for (oBrCysNi)L7 complex as the representative example. A correlation between the signal of the H-2 proton at the α-amino acid stereocenter with the signal of the ortho-protons of the benzyl fragment was observed. This indicated that H-2 is located on the same side of the nickel coordination plane as the benzyl substituent at the proline nitrogen atom, leading to the ʟ-configuration of the α-amino acid stereocenter. Notably, the major stereoisomer of all thiolated compounds (RCysNi)L7 has the same configuration (ʟ), which was additionally confirmed by the similarity of the H-8 proton chemical shifts for all these complexes (the value falls within the 8.27–8.30 ppm range). It should be emphasized that the H-8 chemical shift is very sensitive to the configuration of the α-stereocenter [32]; a significant downfield shift of this signal (up to 0.5 ppm) could be expected for the diastereomer with the ᴅ-α-stereocenter with respect to the ʟ-isomer.
![[1860-5397-19-41-1]](/bjoc/content/figures/1860-5397-19-41-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Fragment of the NOESY spectrum of the ʟ-(oBrCysNi)L7 complex indicating the correlation between the H-2 and H-17,21 protons.
Figure 1: Fragment of the NOESY spectrum of the ʟ-(oBrCysNi)L7 complex indicating the correlation between the...
To prove the recyclability of the t-Bu-containing (S)-N-benzylproline-derived ligand L7, the ʟ-(pMeCysNi)L7 complex was decomposed by heating of its MeOH/HCl solution at 60 °C under conditions similar to the ones previously reported for (RCysNi)L4 [42] (see Supporting Information File 1 for details). The corresponding amino acid was obtained, along with the L7 ligand which was recovered in ca. 70% yield and then reused.
DFT study
The level of stereoselectivity induction is important for the evaluation of the chiral templates efficiency. The estimation of the thermodynamically and kinetically controlled stereoselectivity is based on different approaches. The diastereomeric ratio of the products formed under kinetic control is related to the transition-state energy. However, the problem is that the transition state is rather specific and dependent on the particular reaction type. Thus, it hardly can be generalized. In contrast, the thermodynamically controlled stereoselectivity refers to the difference in the thermodynamic stability of the (S)- and (R)-diastereomers which is related to the conformational flexibility of the template. The rigidity of the chiral Ni–Schiff base complexes is mainly determined by the noncovalent interactions in the Ni coordination environment. Among them, the most important is the π-stacking between the o-phenylene fragment and the benzyl moiety in the proline. The stronger the π-stacking interactions are, the more rigid are the complexes and the higher is the difference in the relative energies of the amino acid derivatives with different configuration of the α-stereocenter. As it has been shown previously [31,37], this difference determines the stereochemical outcome of the reactions performed under thermodynamic control.
To determine the conformational changes in the Ni(II) coordination environment induced by the additional tert-butyl group in the o-phenylene moiety, DFT calculations were performed for the model diastereomeric α-ʟ- and α-ᴅ-alanine complexes derived from new ligand L7; the approach for visualization of the noncovalent interactions suggested in [43] was applied.
The images obtained for the ʟ- and ᴅ-(AlaNi)L7 complexes are given in Figure 2. The types of interactions are presented in different colors: the hydrogen bonding are labeled in blue color of the reduced density gradient isosurface; green color corresponds to the dispersion interactions (van der Waals interactions, the π-stacking); red color represents steric clashes. The interplay of these through-space interactions gives rise to the difference in the relative energies of the ʟ- and ᴅ-isomers. The most important are the π-stacking between the benzyl group in the proline fragment and the o-phenylene moiety which bring these two fragments closer to each other. These interactions are much stronger in the α-ʟ isomer (see Figure 2). The presence of the tert-butyl group induces an additional dispersion interaction between this group and the benzyl fragment in the ʟ-(AlaNi)L7 complex, bringing the benzyl group and the o-phenylene moiety even closer to each other (the distance between the C20 and C8 atoms is 3.45 Å in ʟ-(AlaNi)L7 and 3.49 Å in ʟ-(AlaNi)L1; for numbering of the atoms, see Figure 1). This makes the former complex more conformationally rigid. Indeed, the calculated (PBE-D4 /def2-SVP) energy difference between the ᴅ- and ʟ-diastereomers is more significant in (AlaNi)L7 than in (AlaNi)L1 (3.6 kcal/mol vs 3.3 kcal/mol, see Table 2) giving rise to a higher level of thermodynamically controlled stereoselectivity in the ligand L7-derived complexes.
![[1860-5397-19-41-2]](/bjoc/content/figures/1860-5397-19-41-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Low-gradient isosurfaces with low densities (blue color of the isosurface corresponds to the hydrogen bonding; the dispersion interactions (van der Waals interactions, the π-stacking) are marked in green color; red color indicates steric clashes) obtained for the ʟ- (left image) and ᴅ-alanine (right image) Ni–Schiff base derivatives with ligand L7.
Figure 2: Low-gradient isosurfaces with low densities (blue color of the isosurface corresponds to the hydrog...
Table 2: The calculated (PBE-D4 /def2-SVP) energy differences between the ᴅ- and ʟ-(AlaNi).
| Complex | (AlaNi)L7 | (AlaNi)L1 | (AlaNi)L4 [37] |
| ΔE, kcal/mol | 3.6 | 3.3 | 3.7 |
However, the energy difference between the ᴅ- and ʟ-diastereomeric (AlaNi)L7 is somewhat lower than that for (AlaNi)L4 (3.6 kcal/mol vs 3.7 kcal/mol, see Table 2); consequently, a stereoinduction efficiency expected for ligand L7 may be lower than that of L4. However, taking into account the simplicity of our procedure and the atom-economy principle, the ligand L7 can be considered as a cheaper alternative to L4, still providing a significant stereoselectivity level.
Additionally, Schiff base derivatives of L7 have a considerably higher solubility in acetonitrile as compared to the L4-based complexes (see Table 3 and Supporting Information File 1); this makes it easier to scale-up the synthesis (e.g., for the SN2 alkylations or nucleophilic addition including the thiolation discussed above). Insertion of the t-Bu group also makes the (GlyNi)L7 complex more soluble in diethyl ether as compared to the parent (GlyNi)L1 complex (21.3 mg/1 mL vs <1 mg/1 mL in Et2O; see Figure 3). This is beneficial for electrosynthesis, simplifying the separation of the complexes from the supporting electrolyte (which is usually insoluble in Et2O).
![[1860-5397-19-41-3]](/bjoc/content/figures/1860-5397-19-41-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Saturated solutions of (GlyNi)L1 (left) and (GlyNi)L7 (right) in diethyl ether.
Figure 3: Saturated solutions of (GlyNi)L1 (left) and (GlyNi)L7 (right) in diethyl ether.
Electrophilicity of the deprotonated (GlyNi)L7 complex
The α-deprotonated Ni–Schiff base derivatives of glycine (which can be considered as a chiral nucleophilic equivalent of glycine) can be involved in various types of chemical transformations. The oxidation potential value of the deprotonated glycine complex correlates with the reactivity of the α-carbanionic species towards electrophiles [37]. Therefore, the electrochemical approach is very convenient and informative for the estimation of the relative reactivity of the complexes derived from various chiral ligands. The oxidation potential can be determined from the voltammetry curve measured for the quantitatively deprotonated complex. The electrochemical deprotonation using an electro-generated base is the most convenient approach [37,44]. Comparison with the Eox values determined previously for the parent Belokon complex (GlyNi)L1 (−0.32 V vs Ag/AgCl/KCl(sat.) [37]) and (GlyNi)L4 (−0.22 V [37]) showed that the nucleophilic equivalent of (GlyNi)L7 (Eox = −0.41 V, see Table 4) should be much more reactive towards electrophiles than the deprotonated (GlyNi)L1 and (GlyNi)L4 complexes.
Table 4: Oxidation and reduction potential values for (GlyNi)L7 and (ΔAlaNi)L7 and comparison with previously reported data for (GlyNi)L1, (ΔAlaNi)L1, (GlyNi)L4 and (ΔAlaNi)L4 [36,37] (Pt, CH3CN, 0.1 M Bu4NBF4, vs Ag/AgCl/KCl(sat.), 100 mV/s).
| Complex | Ligand L7 | Ligand L1 [36,37] | Ligand L4 [36,37] | |||
| Oxidation, V | Reduction, V | Oxidation, V | Reduction, V | Oxidation, V | Reduction, V | |
| ΔAlaNi |
Еp = 1.32
Е1/2 = 1.285 |
Еp = −1.34
Е1/2 = −1.31 |
Еp = 1.34
Е1/2 = 1.255 |
Еp = −1.30 |
Еp = 1.4
Е1/2 = 1.37 |
Еp = −1.32 |
| GlyNi |
Еp = 1.31
Е1/2 = 1.27 |
Еp = −1.59
Е1/2 = −1.53 |
Еp = 1.32
Е1/2 = 1.24 |
Еp = −1.57
Е1/2 = −1.51 |
Еp = 1.4
Е1/2 = 1.36 |
Еp = −1.53
Е1/2 = −1.43 |
| Gly−HNi (deprotonated) | Еp = −0.41 | – | Еp = −0.32 | – | Еp = −0.22 | – |
Electrochemical properties
For further applications of the (GlyNi)L7 and (ΔAlaNi)L7 complexes in electrochemically activated functionalization of amino acid fragments, a more detailed electrochemical study is an important precondition. Electrochemical properties were investigated using cyclic voltammetry (CV) in acetonitrile with 0.1 M Bu4NBF4 as a supporting electrolyte. A platinum disk was used as a working electrode. Formal potential values are given in Table 4. As could be expected, the potential values are rather close to that for the corresponding derivatives of the original ligand L1 [37].
Meanwhile, an important difference is the reversibility of the oxidation observed at low potential scan rates (100 mV/s) for the complexes (GlyNi)L7 and (ΔAlaNi)L7 (see Figure 4), in contrast to their t-Bu-free analogs. The similarity of the potential values for the complexes derived from L1 and L7 allows assuming that the HOMO/LUMO localization is not significantly influenced by the tert-butyl group. Indeed, the HOMO is a combination of the π-orbitals of the substituted phenylene fragment, the p-orbital of the amide nitrogen and the Ni d-orbitals (see Supporting Information File 1). Although the phenylene fragment in the oxidized (GlyNi)L7 and (ΔAlaNi)L7 complexes bears a certain spin density (as it has been previously shown for the t-Bu-free analogs [31]), the bulky t-Bu substituent does prevent dimerization of the radical cations formed in the electron-transfer step increasing their kinetic stability. This result is important; it indicates the possibility for further oxidative functionalization of the amino acid fragment using the Ni–Schiff base templates derived from the new ligand L7.
![[1860-5397-19-41-4]](/bjoc/content/figures/1860-5397-19-41-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The CV curves observed for (GlyNi)L7 and (ΔAlaNi)L7 in the anodic and cathodic regions (Pt, CH3CN, 0.1 M Bu4NBF4, 100 mV/s, vs Ag/AgCl/KCl(sat.)). Oxidation of (GlyNi)L1 is given for comparison.
Figure 4: The CV curves observed for (GlyNi)L7 and (ΔAlaNi)L7 in the anodic and cathodic regions (Pt, CH3CN, ...
One-electron reduction of the complex (GlyNi)L7 is mainly metal centered (with some impact of the π* orbital of the C=N bond), diffusion controlled (the slope of the ln ipc vs ln v dependence is 0.48), and reversible; the direct and reverse peak separation value was 57 mV at 100 mV/s, and Ia/Ic peak current ratio was close to unity. This means that the radical anion of (GlyNi)L7 is stable, at least in the cyclic voltammetry time scale. The spin density in the radical anion is mainly localized on the Ni d-orbitals and on the N and O atoms of its coordination environment (see Supporting Information File 1).
The reduction of the complex (ΔAlaNi)L7 is irreversible at scan rates lower 2000 mV/s. Full reversibility can be reached at scan rates above 20 V/s allowing the E1/2 estimation. Similarly to previously studied complex (ΔAlaNi)L1 [36], electron transfer to (ΔAlaNi)L7 is mainly ligand centered. The DFT-estimated LUMO is formed as an overlap of the π antibonding orbitals of the dehydroalanine and imine fragments with much smaller impacts of the Ni orbitals and π orbitals of the aromatic moiety. The radical anion formed behaves like a nucleophilic С-radical, reacting with the C–C double bond of the starting ΔAlaNi complex yielding the dimeric binuclear complex.
Conclusion
The new structurally advantageous (S)-N-benzylproline-derived ligand (S)-N-(2-benzoyl-5-tert-butylphenyl)-1-benzylpyrrolidine-2-carboxamide and its Ni(II)–Schiff base complexes formed of glycine, serine, and dehydroalanine are reported. The bulky tert-butyl substituent inserted in the phenylene fragment of the ligand allowed to solve the following problems. First of all, radical cations formed under a one-electron electrochemical oxidation of the glycine and dehydroalanine complexes become sufficiently stable. The fast side reaction of the oxidative dimerization of the Schiff base complex via the phenylene fragments (inherent to the original Belokon complexes) is prevented, thus opening a route to the targeted electrochemically induced oxidative modification of the amino acids side chain. Second, as follows from the comparison of the oxidation potential values, the reactivity of the deprotonated glycine complex towards electrophiles is significantly enhanced as compared to the anionic species formed from the original Belokon complex. Third, the dehydroalanine complex showed the superior performance in the stereoselective arylthiolation of the double bond yielding the cysteine derivatives as compared to the original complex derived from N-benzylproline. The quantum chemical study showed that the t-Bu group increases the dispersion interactions between the benzyl group in the proline fragment and the o-phenylene moiety making the Ni(II) complex more conformationally rigid and providing a higher level of thermodynamically controlled stereoselectivity. Finally, the solubility of the t-Bu-containing ligand and its Schiff base complexes is increased, facilitating scaling-up the reaction procedure and isolation of the functionalized amino acid.
We hope that the chiral templates derived from the easily available and inexpensive new ligand and amino acids (glycine and dehydroalanine) will find their rightful place in the stereoselective synthesis of tailor-made amino acids (including oxidative transformations).
Supporting Information
| Supporting Information File 1: Experimental details, characterization, and copies of spectra. | ||
| Format: PDF | Size: 8.3 MB | Download |
References
-
Trabocchi, A.; Guarna, F.; Guarna, A. Curr. Org. Chem. 2005, 9, 1127–1153. doi:10.2174/1385272054553631
Return to citation in text: [1] -
Adhikari, A.; Bhattarai, B. R.; Aryal, A.; Thapa, N.; KC, P.; Adhikari, A.; Maharjan, S.; Chanda, P. B.; Regmi, B. P.; Parajuli, N. RSC Adv. 2021, 11, 38126–38145. doi:10.1039/d1ra07028b
Return to citation in text: [1] -
Han, J.; Lyutenko, N. V.; Sorochinsky, A. E.; Okawara, A.; Konno, H.; White, S.; Soloshonok, V. A. Chem. – Eur. J. 2021, 27, 17510–17528. doi:10.1002/chem.202102485
Return to citation in text: [1] -
Nagaoka, K.; Nakano, A.; Han, J.; Sakamoto, T.; Konno, H.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V. A. Chirality 2021, 33, 685–702. doi:10.1002/chir.23350
Return to citation in text: [1] -
Zou, Y.; Han, J.; Saghyan, A. S.; Mkrtchyan, A. F.; Konno, H.; Moriwaki, H.; Izawa, K.; Soloshonok, V. A. Molecules 2020, 25, 2739. doi:10.3390/molecules25122739
Return to citation in text: [1] [2] [3] -
O'Donnell, M. J. Tetrahedron 2019, 75, 3667–3696. doi:10.1016/j.tet.2019.03.029
Return to citation in text: [1] -
Maruoka, K. Chem. Rec. 2010, 10, 254–259. doi:10.1002/tcr.201000019
Return to citation in text: [1] -
Undheim, K. Amino Acids 2008, 34, 357–402. doi:10.1007/s00726-007-0512-5
Return to citation in text: [1] -
Schollkopf, U. Pure Appl. Chem. 1983, 55, 1799–1806. doi:10.1351/pac198355111799
Return to citation in text: [1] -
Sebahar, P. R.; Williams, R. M. J. Am. Chem. Soc. 2000, 122, 5666–5667. doi:10.1021/ja001133n
Return to citation in text: [1] -
Williams, R. M.; Im, M. N. J. Am. Chem. Soc. 1991, 113, 9276–9286. doi:10.1021/ja00024a038
Return to citation in text: [1] -
Myers, A. G.; Gleason, J. L.; Yoon, T.; Kung, D. W. Asymmetric Synthesis: Construction of Chiral Molecules Using Amino Acids; Pergamon Press, 1992; Vol. 114.
Return to citation in text: [1] -
Evans, D. A.; Weber, A. E. J. Am. Chem. Soc. 1986, 108, 6757–6761. doi:10.1021/ja00281a049
Return to citation in text: [1] -
Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V. A.; Liu, H. Amino Acids 2017, 49, 1487–1520. doi:10.1007/s00726-017-2458-6
Return to citation in text: [1] -
Soloshonok, V.; Ueki, H.; Ellis, T. Synlett 2009, 704–715. doi:10.1055/s-0028-1087929
Return to citation in text: [1] -
Aceña, J. L.; Sorochinsky, A. E.; Soloshonok, V. Amino Acids 2014, 46, 2047–2073. doi:10.1007/s00726-014-1764-5
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. Amino Acids 2013, 45, 1017–1033. doi:10.1007/s00726-013-1580-3
Return to citation in text: [1] [2] -
Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 691–718. doi:10.1007/s00726-013-1539-4
Return to citation in text: [1] [2] -
Belokon, Y. N.; Bulychev, A. G.; Vitt, S. V.; Struchkov, Y. T.; Batsanov, A. S.; Timofeeva, T. V.; Tsyryapkin, V. A.; Ryzhov, M. G.; Lysova, L. A. J. Am. Chem. Soc. 1985, 107, 4252–4259. doi:10.1021/ja00300a030
Return to citation in text: [1] -
Belokon, Y. N.; Zel'tser, I. E.; Bakhmutov, V. I.; Saporovskaya, M. B.; Ryzhov, M. G.; Yanovskii, A. I.; Struchkov, Y. T.; Belikov, V. M. J. Am. Chem. Soc. 1983, 105, 2010–2017. doi:10.1021/ja00345a057
Return to citation in text: [1] -
Saghiyan, A. S.; Dadayan, S. A.; Petrosyan, S. G.; Manasyan, L. L.; Geolchanyan, A. V.; Djamgaryan, S. M.; Andreasyan, S. A.; Maleev, V. I.; Khrustalev, V. N. Tetrahedron: Asymmetry 2006, 17, 455–467. doi:10.1016/j.tetasy.2006.01.026
Return to citation in text: [1] [2] [3] -
Belokon, Y. N.; Maleev, V. I.; Petrosyan, A. A.; Savel"eva, T. F.; Ikonnikov, N. S.; Peregudov, A. S.; Khrustalev, V. N.; Saghiyan, A. S. Russ. Chem. Bull. 2002, 51, 1593–1599. doi:10.1023/a:1020952115556
Return to citation in text: [1] -
Saghyan, A. S.; Dadayan, A. S.; Dadayan, S. A.; Mkrtchyan, A. F.; Geolchanyan, A. V.; Manasyan, L. L.; Ajvazyan, H. R.; Khrustalev, V. N.; Hambardzumyan, H. H.; Maleev, V. I. Tetrahedron: Asymmetry 2010, 21, 2956–2965. doi:10.1016/j.tetasy.2010.11.024
Return to citation in text: [1] -
Jörres, M.; Chen, X.; Aceña, J. L.; Merkens, C.; Bolm, C.; Liu, H.; Soloshonok, V. A. Adv. Synth. Catal. 2014, 356, 2203–2208. doi:10.1002/adsc.201400405
Return to citation in text: [1] [2] -
Jörres, M.; Aceña, J. L.; Soloshonok, V. A.; Bolm, C. ChemCatChem 2015, 7, 1265–1269. doi:10.1002/cctc.201500102
Return to citation in text: [1] [2] -
Nian, Y.; Wang, J.; Zhou, S.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V. A.; Liu, H. Angew. Chem. 2015, 127, 13110–13114. doi:10.1002/ange.201507273
Return to citation in text: [1] -
Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V. A. Symmetry 2019, 11, 578. doi:10.3390/sym11040578
Return to citation in text: [1] -
Mei, H.; Hiramatsu, T.; Takeda, R.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V. A. Org. Process Res. Dev. 2019, 23, 629–634. doi:10.1021/acs.oprd.8b00404
Return to citation in text: [1] -
Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J. L.; Soloshonok, V. A. Angew. Chem., Int. Ed. 2014, 53, 12214–12217. doi:10.1002/anie.201407944
Return to citation in text: [1] -
Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Abe, H.; Soloshonok, V. A. Org. Biomol. Chem. 2018, 16, 4968–4972. doi:10.1039/c8ob00963e
Return to citation in text: [1] -
Magdesieva, T. V. Chem. Rec. 2021, 21, 2178–2192. doi:10.1002/tcr.202100019
Return to citation in text: [1] [2] [3] [4] [5] -
Levitskiy, O. A.; Grishin, Y. K.; Magdesieva, T. V. Eur. J. Org. Chem. 2019, 3174–3182. doi:10.1002/ejoc.201900466
Return to citation in text: [1] [2] [3] [4] -
Levitskiy, O. A.; Aglamazova, O. I.; Dmitrieva, A. V.; Soloshonok, V. A.; Moriwaki, H.; Grishin, Y. K.; Magdesieva, T. V. Mendeleev Commun. 2021, 31, 337–340. doi:10.1016/j.mencom.2021.04.018
Return to citation in text: [1] -
Levitskiy, O. A.; Aglamazova, O. I.; Grishin, Y. K.; Paseshnichenko, K. A.; Magdesieva, T. V. ChemElectroChem 2020, 7, 3361–3367. doi:10.1002/celc.202000970
Return to citation in text: [1] -
Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Kiskin, M. A.; Churakov, A. V.; Babievsky, K. K.; Kochetkov, K. A. Organometallics 2014, 33, 4629–4638. doi:10.1021/om500070n
Return to citation in text: [1] -
Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Jin, Y.; Chen, Q.; Zhang, Y.; Lee, Y.; Kang, D.; Kim, Y.; Kim, S.-J.; Su, Z.; Huang, H.; Kim, K. M. Chirality 2022, 34, 999–1007. doi:10.1002/chir.23449
Return to citation in text: [1] -
Huang, H.; Jin, Y.; Shirbhate, M. E.; Kang, D.; Choi, M.; Chen, Q.; Kim, Y.; Kim, S.-J.; Byun, I.-S.; Wang, M.; Bouffard, J.; Kim, S. K.; Kim, K. M. Nat. Commun. 2021, 12, 125. doi:10.1038/s41467-020-20402-x
Return to citation in text: [1] -
Ayyangar, N. R.; Lahoti, R. J.; Srinivasan, K. V.; Daniel, T. Synthesis 1991, 322–324. doi:10.1055/s-1991-26457
Return to citation in text: [1] [2] [3] -
Belokon', Y. N.; Sagyan, A. S.; Djamgaryan, S. M.; Bakhmutov, V. I.; Belikov, V. M. Tetrahedron 1988, 44, 5507–5514. doi:10.1016/s0040-4020(01)86056-7
Return to citation in text: [1] [2] [3] [4] -
Yamamoto, J.; Kawashima, A.; Kawamura, A.; Abe, H.; Moriwaki, H.; Shibata, N.; Soloshonok, V. A. Eur. J. Org. Chem. 2017, 1931–1939. doi:10.1002/ejoc.201700018
Return to citation in text: [1] -
Johnson, E. R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A. J.; Yang, W. J. Am. Chem. Soc. 2010, 132, 6498–6506. doi:10.1021/ja100936w
Return to citation in text: [1] -
Cheng, S.; Hawley, M. D. J. Org. Chem. 1985, 50, 3388–3392. doi:10.1021/jo00218a028
Return to citation in text: [1]
| 32. | Levitskiy, O. A.; Grishin, Y. K.; Magdesieva, T. V. Eur. J. Org. Chem. 2019, 3174–3182. doi:10.1002/ejoc.201900466 |
| 41. | Belokon', Y. N.; Sagyan, A. S.; Djamgaryan, S. M.; Bakhmutov, V. I.; Belikov, V. M. Tetrahedron 1988, 44, 5507–5514. doi:10.1016/s0040-4020(01)86056-7 |
| 41. | Belokon', Y. N.; Sagyan, A. S.; Djamgaryan, S. M.; Bakhmutov, V. I.; Belikov, V. M. Tetrahedron 1988, 44, 5507–5514. doi:10.1016/s0040-4020(01)86056-7 |
| 1. | Trabocchi, A.; Guarna, F.; Guarna, A. Curr. Org. Chem. 2005, 9, 1127–1153. doi:10.2174/1385272054553631 |
| 2. | Adhikari, A.; Bhattarai, B. R.; Aryal, A.; Thapa, N.; KC, P.; Adhikari, A.; Maharjan, S.; Chanda, P. B.; Regmi, B. P.; Parajuli, N. RSC Adv. 2021, 11, 38126–38145. doi:10.1039/d1ra07028b |
| 3. | Han, J.; Lyutenko, N. V.; Sorochinsky, A. E.; Okawara, A.; Konno, H.; White, S.; Soloshonok, V. A. Chem. – Eur. J. 2021, 27, 17510–17528. doi:10.1002/chem.202102485 |
| 12. | Myers, A. G.; Gleason, J. L.; Yoon, T.; Kung, D. W. Asymmetric Synthesis: Construction of Chiral Molecules Using Amino Acids; Pergamon Press, 1992; Vol. 114. |
| 13. | Evans, D. A.; Weber, A. E. J. Am. Chem. Soc. 1986, 108, 6757–6761. doi:10.1021/ja00281a049 |
| 29. | Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J. L.; Soloshonok, V. A. Angew. Chem., Int. Ed. 2014, 53, 12214–12217. doi:10.1002/anie.201407944 |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 8. | Undheim, K. Amino Acids 2008, 34, 357–402. doi:10.1007/s00726-007-0512-5 |
| 9. | Schollkopf, U. Pure Appl. Chem. 1983, 55, 1799–1806. doi:10.1351/pac198355111799 |
| 10. | Sebahar, P. R.; Williams, R. M. J. Am. Chem. Soc. 2000, 122, 5666–5667. doi:10.1021/ja001133n |
| 11. | Williams, R. M.; Im, M. N. J. Am. Chem. Soc. 1991, 113, 9276–9286. doi:10.1021/ja00024a038 |
| 30. | Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Abe, H.; Soloshonok, V. A. Org. Biomol. Chem. 2018, 16, 4968–4972. doi:10.1039/c8ob00963e |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 6. | O'Donnell, M. J. Tetrahedron 2019, 75, 3667–3696. doi:10.1016/j.tet.2019.03.029 |
| 7. | Maruoka, K. Chem. Rec. 2010, 10, 254–259. doi:10.1002/tcr.201000019 |
| 27. | Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V. A. Symmetry 2019, 11, 578. doi:10.3390/sym11040578 |
| 28. | Mei, H.; Hiramatsu, T.; Takeda, R.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V. A. Org. Process Res. Dev. 2019, 23, 629–634. doi:10.1021/acs.oprd.8b00404 |
| 31. | Magdesieva, T. V. Chem. Rec. 2021, 21, 2178–2192. doi:10.1002/tcr.202100019 |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 4. | Nagaoka, K.; Nakano, A.; Han, J.; Sakamoto, T.; Konno, H.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V. A. Chirality 2021, 33, 685–702. doi:10.1002/chir.23350 |
| 5. | Zou, Y.; Han, J.; Saghyan, A. S.; Mkrtchyan, A. F.; Konno, H.; Moriwaki, H.; Izawa, K.; Soloshonok, V. A. Molecules 2020, 25, 2739. doi:10.3390/molecules25122739 |
| 24. | Jörres, M.; Chen, X.; Aceña, J. L.; Merkens, C.; Bolm, C.; Liu, H.; Soloshonok, V. A. Adv. Synth. Catal. 2014, 356, 2203–2208. doi:10.1002/adsc.201400405 |
| 25. | Jörres, M.; Aceña, J. L.; Soloshonok, V. A.; Bolm, C. ChemCatChem 2015, 7, 1265–1269. doi:10.1002/cctc.201500102 |
| 43. | Johnson, E. R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A. J.; Yang, W. J. Am. Chem. Soc. 2010, 132, 6498–6506. doi:10.1021/ja100936w |
| 21. | Saghiyan, A. S.; Dadayan, S. A.; Petrosyan, S. G.; Manasyan, L. L.; Geolchanyan, A. V.; Djamgaryan, S. M.; Andreasyan, S. A.; Maleev, V. I.; Khrustalev, V. N. Tetrahedron: Asymmetry 2006, 17, 455–467. doi:10.1016/j.tetasy.2006.01.026 |
| 23. | Saghyan, A. S.; Dadayan, A. S.; Dadayan, S. A.; Mkrtchyan, A. F.; Geolchanyan, A. V.; Manasyan, L. L.; Ajvazyan, H. R.; Khrustalev, V. N.; Hambardzumyan, H. H.; Maleev, V. I. Tetrahedron: Asymmetry 2010, 21, 2956–2965. doi:10.1016/j.tetasy.2010.11.024 |
| 32. | Levitskiy, O. A.; Grishin, Y. K.; Magdesieva, T. V. Eur. J. Org. Chem. 2019, 3174–3182. doi:10.1002/ejoc.201900466 |
| 21. | Saghiyan, A. S.; Dadayan, S. A.; Petrosyan, S. G.; Manasyan, L. L.; Geolchanyan, A. V.; Djamgaryan, S. M.; Andreasyan, S. A.; Maleev, V. I.; Khrustalev, V. N. Tetrahedron: Asymmetry 2006, 17, 455–467. doi:10.1016/j.tetasy.2006.01.026 |
| 24. | Jörres, M.; Chen, X.; Aceña, J. L.; Merkens, C.; Bolm, C.; Liu, H.; Soloshonok, V. A. Adv. Synth. Catal. 2014, 356, 2203–2208. doi:10.1002/adsc.201400405 |
| 25. | Jörres, M.; Aceña, J. L.; Soloshonok, V. A.; Bolm, C. ChemCatChem 2015, 7, 1265–1269. doi:10.1002/cctc.201500102 |
| 26. | Nian, Y.; Wang, J.; Zhou, S.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V. A.; Liu, H. Angew. Chem. 2015, 127, 13110–13114. doi:10.1002/ange.201507273 |
| 42. | Yamamoto, J.; Kawashima, A.; Kawamura, A.; Abe, H.; Moriwaki, H.; Shibata, N.; Soloshonok, V. A. Eur. J. Org. Chem. 2017, 1931–1939. doi:10.1002/ejoc.201700018 |
| 19. | Belokon, Y. N.; Bulychev, A. G.; Vitt, S. V.; Struchkov, Y. T.; Batsanov, A. S.; Timofeeva, T. V.; Tsyryapkin, V. A.; Ryzhov, M. G.; Lysova, L. A. J. Am. Chem. Soc. 1985, 107, 4252–4259. doi:10.1021/ja00300a030 |
| 20. | Belokon, Y. N.; Zel'tser, I. E.; Bakhmutov, V. I.; Saporovskaya, M. B.; Ryzhov, M. G.; Yanovskii, A. I.; Struchkov, Y. T.; Belikov, V. M. J. Am. Chem. Soc. 1983, 105, 2010–2017. doi:10.1021/ja00345a057 |
| 41. | Belokon', Y. N.; Sagyan, A. S.; Djamgaryan, S. M.; Bakhmutov, V. I.; Belikov, V. M. Tetrahedron 1988, 44, 5507–5514. doi:10.1016/s0040-4020(01)86056-7 |
| 5. | Zou, Y.; Han, J.; Saghyan, A. S.; Mkrtchyan, A. F.; Konno, H.; Moriwaki, H.; Izawa, K.; Soloshonok, V. A. Molecules 2020, 25, 2739. doi:10.3390/molecules25122739 |
| 14. | Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V. A.; Liu, H. Amino Acids 2017, 49, 1487–1520. doi:10.1007/s00726-017-2458-6 |
| 15. | Soloshonok, V.; Ueki, H.; Ellis, T. Synlett 2009, 704–715. doi:10.1055/s-0028-1087929 |
| 16. | Aceña, J. L.; Sorochinsky, A. E.; Soloshonok, V. Amino Acids 2014, 46, 2047–2073. doi:10.1007/s00726-014-1764-5 |
| 17. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. Amino Acids 2013, 45, 1017–1033. doi:10.1007/s00726-013-1580-3 |
| 18. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 691–718. doi:10.1007/s00726-013-1539-4 |
| 21. | Saghiyan, A. S.; Dadayan, S. A.; Petrosyan, S. G.; Manasyan, L. L.; Geolchanyan, A. V.; Djamgaryan, S. M.; Andreasyan, S. A.; Maleev, V. I.; Khrustalev, V. N. Tetrahedron: Asymmetry 2006, 17, 455–467. doi:10.1016/j.tetasy.2006.01.026 |
| 22. | Belokon, Y. N.; Maleev, V. I.; Petrosyan, A. A.; Savel"eva, T. F.; Ikonnikov, N. S.; Peregudov, A. S.; Khrustalev, V. N.; Saghiyan, A. S. Russ. Chem. Bull. 2002, 51, 1593–1599. doi:10.1023/a:1020952115556 |
| 41. | Belokon', Y. N.; Sagyan, A. S.; Djamgaryan, S. M.; Bakhmutov, V. I.; Belikov, V. M. Tetrahedron 1988, 44, 5507–5514. doi:10.1016/s0040-4020(01)86056-7 |
| 31. | Magdesieva, T. V. Chem. Rec. 2021, 21, 2178–2192. doi:10.1002/tcr.202100019 |
| 36. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 44. | Cheng, S.; Hawley, M. D. J. Org. Chem. 1985, 50, 3388–3392. doi:10.1021/jo00218a028 |
| 32. | Levitskiy, O. A.; Grishin, Y. K.; Magdesieva, T. V. Eur. J. Org. Chem. 2019, 3174–3182. doi:10.1002/ejoc.201900466 |
| 33. | Levitskiy, O. A.; Aglamazova, O. I.; Dmitrieva, A. V.; Soloshonok, V. A.; Moriwaki, H.; Grishin, Y. K.; Magdesieva, T. V. Mendeleev Commun. 2021, 31, 337–340. doi:10.1016/j.mencom.2021.04.018 |
| 34. | Levitskiy, O. A.; Aglamazova, O. I.; Grishin, Y. K.; Paseshnichenko, K. A.; Magdesieva, T. V. ChemElectroChem 2020, 7, 3361–3367. doi:10.1002/celc.202000970 |
| 35. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Kiskin, M. A.; Churakov, A. V.; Babievsky, K. K.; Kochetkov, K. A. Organometallics 2014, 33, 4629–4638. doi:10.1021/om500070n |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 40. | Ayyangar, N. R.; Lahoti, R. J.; Srinivasan, K. V.; Daniel, T. Synthesis 1991, 322–324. doi:10.1055/s-1991-26457 |
| 32. | Levitskiy, O. A.; Grishin, Y. K.; Magdesieva, T. V. Eur. J. Org. Chem. 2019, 3174–3182. doi:10.1002/ejoc.201900466 |
| 40. | Ayyangar, N. R.; Lahoti, R. J.; Srinivasan, K. V.; Daniel, T. Synthesis 1991, 322–324. doi:10.1055/s-1991-26457 |
| 40. | Ayyangar, N. R.; Lahoti, R. J.; Srinivasan, K. V.; Daniel, T. Synthesis 1991, 322–324. doi:10.1055/s-1991-26457 |
| 36. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x |
| 38. | Jin, Y.; Chen, Q.; Zhang, Y.; Lee, Y.; Kang, D.; Kim, Y.; Kim, S.-J.; Su, Z.; Huang, H.; Kim, K. M. Chirality 2022, 34, 999–1007. doi:10.1002/chir.23449 |
| 39. | Huang, H.; Jin, Y.; Shirbhate, M. E.; Kang, D.; Choi, M.; Chen, Q.; Kim, Y.; Kim, S.-J.; Byun, I.-S.; Wang, M.; Bouffard, J.; Kim, S. K.; Kim, K. M. Nat. Commun. 2021, 12, 125. doi:10.1038/s41467-020-20402-x |
| 36. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 5. | Zou, Y.; Han, J.; Saghyan, A. S.; Mkrtchyan, A. F.; Konno, H.; Moriwaki, H.; Izawa, K.; Soloshonok, V. A. Molecules 2020, 25, 2739. doi:10.3390/molecules25122739 |
| 16. | Aceña, J. L.; Sorochinsky, A. E.; Soloshonok, V. Amino Acids 2014, 46, 2047–2073. doi:10.1007/s00726-014-1764-5 |
| 17. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. Amino Acids 2013, 45, 1017–1033. doi:10.1007/s00726-013-1580-3 |
| 18. | Sorochinsky, A. E.; Aceña, J. L.; Moriwaki, H.; Sato, T.; Soloshonok, V. A. Amino Acids 2013, 45, 691–718. doi:10.1007/s00726-013-1539-4 |
| 31. | Magdesieva, T. V. Chem. Rec. 2021, 21, 2178–2192. doi:10.1002/tcr.202100019 |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 36. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x |
| 36. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
| 36. | Magdesieva, T. V.; Levitskiy, O. A.; Grishin, Y. K.; Ambartsumyan, A. A.; Paseshnichenko, K. A.; Kolotyrkina, N. G.; Kochetkov, K. A. Organometallics 2014, 33, 4639–4654. doi:10.1021/om500034x |
| 37. | Levitskiy, O. A.; Aglamazova, O. I.; Soloshonok, V. A.; Moriwaki, H.; Magdesieva, T. V. Chem. – Eur. J. 2020, 26, 7074–7082. doi:10.1002/chem.201905708 |
© 2023 Dmitrieva et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.