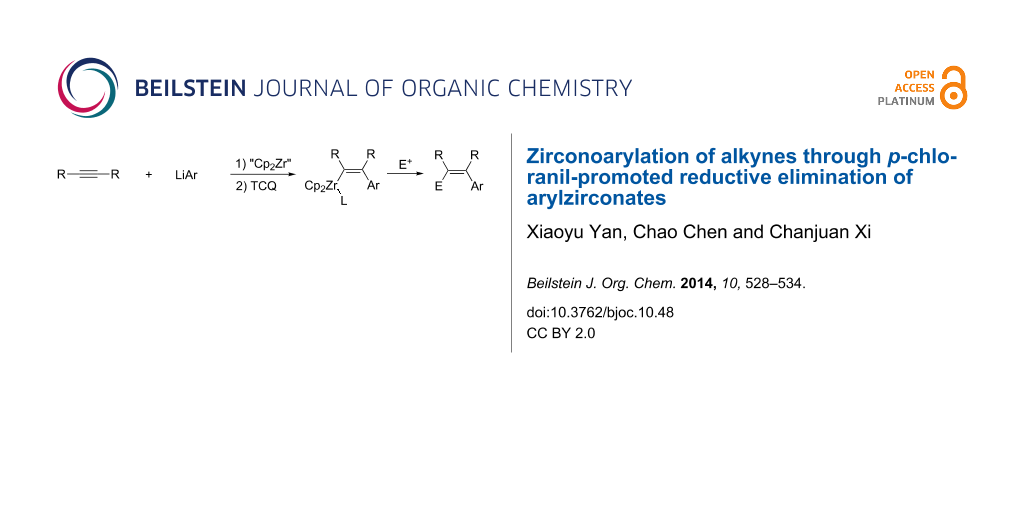
Abstract
A novel method for the zirconoarylation of alkynes was developed. TCQ-promoted reductive elimination of arylzirconate [LiCp2ZrAr(RC≡CR)], which was prepared by the reaction of zirconocene–alkyne complexes with aryllithium compounds, afforded trisubstituted alkenylzirconocenes. This reaction can afford multi-substituted olefins with high stereoselectivity.

Graphical Abstract
Introduction
The controlled synthesis of multi-substituted olefins is one of the most challenging tasks in organic synthesis [1,2]. A series of reactions have been developed for the construction of substituted olefins. Among them, an important route is the addition of various reagents to nonactivated alkynes to form substituted olefins. For example, semihydrogenation of internal alkynes can afford disubstituted olefins (Scheme 1, route a) [3-5]. Hydrocarbonation [6,7] and hydrometalation/functionalization [8-11] of internal alkynes can afford trisubstituted olefins (Scheme 1, routes b and c), respectively. The most exciting progress was the carbometalation of internal alkynes [12-23], which involves simultaneous addition of a metal atom and an organic residue to alkynes. The newly formed carbon–metal bond can be used for further synthetic transformation toward multi-substituted olefins [24-36] (Scheme 1, route d).
![[1860-5397-10-48-i1]](/bjoc/content/inline/1860-5397-10-48-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Transformation of alkynes to olefins.
Scheme 1: Transformation of alkynes to olefins.
A large number of carbometalation reactions of alkynes have been reported. Most of the carbometal reagents, which were used, contained Li, Mg, Cu, Zn, B, or Al [12-16]. In the last several decades also zirconium-mediated or -catalyzed organic reactions have been extensively investigated [37-39]. In addition, a series of organic reactions using zirconocene species have been reported, in particular for the reductive coupling of alkenes or alkynes with other unsaturated compounds [40-43]. On the other hand, carbozirconation of alkynes via reaction of zirconacyclopentenes with alcohols, allyl ethers, homoallyl bromides, vinyl ethers, alkynyl halides, and chloroformates gave ethylzirconation [17], allylzirconation [18,19], cyclopropylmethylzirconation [20], vinylzirconation [21], alkynylzirconation [22], and zirconoestification [23] products of alkynes, respectively. However, to the best of our knowledge, this method failed to fulfill arylzirconation of alkynes (Scheme 2).
![[1860-5397-10-48-i2]](/bjoc/content/inline/1860-5397-10-48-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Carbozirconation of alkynes via zirconacyclopentenes.
Scheme 2: Carbozirconation of alkynes via zirconacyclopentenes.
Recently, we have reported a p-chloranil (TCQ)-promoted reductive elimination reaction of the zirconate complex Li[Cp2Zr(C≡CR)3] toward geminal enediynes [44]. As part of our ongoing project on organozirconate chemistry [45-48], we envisioned that the use of an aryl ligand instead of one of the alkynyl ligands would provide an arylzirconation product of the alkyne. Herein we describe the TCQ-promoted reductive elimination of arylzirconate to afford an arylzirconation product of the alkyne, which can be converted to multi-substituted olefins through coupling with electrophiles (Scheme 3).
![[1860-5397-10-48-i3]](/bjoc/content/inline/1860-5397-10-48-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: TCQ-promoted reductive elimination of arylzirconate.
Scheme 3: TCQ-promoted reductive elimination of arylzirconate.
Results and Discussion
Initially, the reaction of Cp2Zr(PhC≡CPh)(DMAP) (1a), prepared by the reaction of Cp2ZrBu2 [48] with DMAP and diphenylacetylene according to reported literature [49], with phenyllithium produced arylzirconate 2a. To this mixture, 2 equivalents of p-chloranil (TCQ) were added and the reaction mixture was stirred for 12 h at room temperature. After being quenched with HCl solution, the desired triphenylethylene (3a) was isolated in 62% yield. When the reaction mixture was quenched with DCl solution, the deuterated product 3a-D was isolated in 60% yield with >95% of deuterium incorporation (Scheme 4). The deuterium experiment revealed the formation of triphenylvinylzirconocene 4a as intermediate.
![[1860-5397-10-48-i4]](/bjoc/content/inline/1860-5397-10-48-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: TCQ-promoted arylzirconation of diphenylacetylene.
Scheme 4: TCQ-promoted arylzirconation of diphenylacetylene.
Under similar reaction conditions, a study on the substrate scope was carried out, and the results are summarized in Table 1. When diphenylacetylene was used as starting material, different aryllithium compounds were employed to afford triarylethylene in 38% to 62% isolated yields after being quenched with HCl (Table 1, entries 1–4). Bromination of the reaction mixture by NBS instead hydrolysis afforded bromotriphenylethylene in 37% isolated yield (Table 1, entry 5). When allyl bromide was employed as electrophile in the presence of CuCl, the allyltriarylethylenes were formed in 43% to 45% yields (Table 1, entries 6 and 7). Cross coupling with iodobenzene in the presence of CuCl/Pd(PPh3)4 afforded tetraarylethylene in 31% yield (Table 1, entry 8). When other diarylacetylene was employed in this reaction, the corresponding products were formed in 36% to 59% yields (Table 1, entries 9–12). No desired product was observed when alkylacetylenes were used.
Table 1: Formation of multi-substituted olefins via the reaction of alkynes with [Cp2Zr(1-butene)(DMAP)] and aryllithium in the presence of TCQa.
| entry | alkynes | aryllithium | electrophiles | products | yield (%)b |
|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-48-i7.svg?max-width=637&scale=1.0)
|
PhLi | HCl |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-48-i8.svg?max-width=637&scale=1.0)
3a |
62 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-48-i9.svg?max-width=637&scale=1.0)
|
TolLi | HCl |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-48-i10.svg?max-width=637&scale=1.0)
3b |
58 |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-48-i11.svg?max-width=637&scale=1.0)
|
2-ThLi | HCl |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-48-i12.svg?max-width=637&scale=1.0)
3c |
47 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-48-i13.svg?max-width=637&scale=1.0)
|
NaphLi | HCl |
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-48-i14.svg?max-width=637&scale=1.0)
3d |
38 |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-48-i15.svg?max-width=637&scale=1.0)
|
PhLi | NBS |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-48-i16.svg?max-width=637&scale=1.0)
3e |
37 |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-48-i17.svg?max-width=637&scale=1.0)
|
PhLi | allyl-Br |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-48-i18.svg?max-width=637&scale=1.0)
3f |
45 |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-48-i19.svg?max-width=637&scale=1.0)
|
TolLi | allyl-Br |
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-48-i20.svg?max-width=637&scale=1.0)
3g |
43 |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-10-48-i21.svg?max-width=637&scale=1.0)
|
TolLi | PhI |
![[Graphic 16]](/bjoc/content/inline/1860-5397-10-48-i22.svg?max-width=637&scale=1.0)
3h |
31 |
| 9 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-10-48-i23.svg?max-width=637&scale=1.0)
|
PhLi | HCl |
![[Graphic 18]](/bjoc/content/inline/1860-5397-10-48-i24.svg?max-width=637&scale=1.0)
3i |
57 |
| 10 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-10-48-i25.svg?max-width=637&scale=1.0)
|
PhLi | NBS |
![[Graphic 20]](/bjoc/content/inline/1860-5397-10-48-i26.svg?max-width=637&scale=1.0)
3j |
36 |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-10-48-i27.svg?max-width=637&scale=1.0)
|
PhLi | HCl |
![[Graphic 22]](/bjoc/content/inline/1860-5397-10-48-i28.svg?max-width=637&scale=1.0)
3k |
59 |
| 12 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-10-48-i29.svg?max-width=637&scale=1.0)
|
PhLi | HCl |
![[Graphic 24]](/bjoc/content/inline/1860-5397-10-48-i30.svg?max-width=637&scale=1.0)
3l |
52 |
aReaction conditions: Alkyne (1 mmol), [Cp2Zr(1-butene)(DMAP)] (1 mmol), ArLi (2 mmol), TCQ (2 mmol), electrophile (2 mmol). Tol = p-tolyl, Th = 2-thienyl, Naph = 1-naphthyl. bIsolated yield.
Recently, oxidative dimerization of alkenylcopper was reported to afford conjugated dienes and polyenes [44,50-57]. When triphenylvinylzirconocene 4a was reacted in the presence of CuCl and TCQ, the 1,1,2,3,4,4-hexaphenyl-1,3-butadiene (5) was formed in 43% isolated yield (Scheme 5). It is noteworthy that in this reaction two molecular alkynes and two aryllithium compounds were coupled in one-pot in the presence of Cp2Zr species to afford highly substituted 1,3-butadienes.
![[1860-5397-10-48-i5]](/bjoc/content/inline/1860-5397-10-48-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The pathway of the oxidation of zirconate 2 to vinylzirconocene 4 is not yet clear. A possible mechanism is proposed in Scheme 6. Coordination of TCQ to zirconium results in reductive elimination to afford vinylzirconocene(II) 6. Then intermediate 6 reacts with TCQ to form vinylzirconocene(IV) 4. The intermediate 4 reacts with electrophiles to afford multi-substituted olefin 3.
![[1860-5397-10-48-i6]](/bjoc/content/inline/1860-5397-10-48-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
We have developed a novel method for the zirconoarylation of alkynes through TCQ-promoted reductive elimination of arylzirconate. This reaction can afford multi-substituted olefins with stereoselectivity.
Experimental
General Comments. All manipulations were conducted in Schlenk tubes and under a nitrogen atmosphere with a slightly positive pressure. Unless otherwise noted, all starting materials were commercially available and were used without further purification. Tetrahydrofuran (THF) was refluxed and freshly distilled from dark purple solutions of sodium and benzophenone under nitrogen atmosphere. 1H NMR and 13C NMR spectra were recorded on 300 MHz and 400 MHz NMR spectrometers with TMS as internal standard. GC–MS spectra were recorded on a Hewlett Packard GC–MS system.
Typical procedure for TCQ-promoted arylzirconation of alkynes
To a solution of Cp2ZrCl2 (1.2 mmol, 351 mg) in 5 mL THF, n-BuLi (2.4 mmol, 1.5 mL, 1.6 M in hexane) was added at −78 °C and the mixture was stirred for 1 h at the same temperature. To this solution, 4-dimethylaminopyridine (DMAP, 122 mg, 1.0 mmol) was added. The resulting mixture was warmed to room temperature and stirred for 1 h. Diphenylacetylene (1.0 mmol, 178 mg) was added and the mixture was stirred for 1 h at the same temperature. Subsequently, PhLi (2.0 mmol) was added and the solution was stirred for 12 h at room temperature. Then TCQ (2.0 mmol) was added and stirred for another 12 h to afford alkenylzirconocene 4a. The mixture was quenched with HCl solution to afford product 3a in 62% isolated yield.
1,1,2-Triphenylethene (3a) [58]: 1H NMR (300 MHz, CDCl3, Me4Si) δ 6.93 (s, 1H), 6.99–7.30 (m, 15H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 126.6, 126.8, 127.5, 127.6, 127.7, 128.1, 128.3, 128.7, 129.7, 130.5, 137.5, 140.5, 142.7, 143.5; GC–MS m/z: 256.
2-Deuterium-1,1,2-triphenylethene (3a-D) [59]: The reaction mixture containing 4a was quenched with DCl, and 3a-D was isolated in 60% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 7.00–7.51 (m, 15H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 127.0, 127.6, 127.7, 127.7 (t, JDC = 7.2 Hz), 127.8, 128.2, 128.4, 128.8, 129.7, 130.6, 137.5, 140.5, 142.7, 143.6; GC–MS m/z: 257.
1,2-Diphenyl-1-(p-tolyl)ethene (3b) [58]: The reaction was using p-tolyllithium instead of phenyllithium, and 3b was isolated in 58% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 2.45 (s, 3H), 7.06 (s 1H), 7.20–7.42 (m, 14H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 21.4, 126.8, 127.6, 127.7, 128.2, 128.8, 129.1, 129.7, 130.6, 137.5, 137.7, 140.7, 140.9, 142.8; GC–MS m/z: 270.
(E)-1,2-Diphenyl-1-(2-thienyl)ethene (3c): The reaction was performed using 2-thienyllithium instead of phenyllithium, and 3c was isolated in 47% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 6.69–6.71 (dd, JHH = 3.8, 1.1 Hz, 1H), 6.87–6.90 (m, 1H), 6.93–6.96 (m, 2H), 7.04–7.09 (m, 4H), 7.13–7.15 (dd, JHH = 4.8, 2.9 Hz, 1H), 7.26–7.36 (m, 5H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 124.8, 126.2, 126.4, 126.9, 127.5, 127.9, 128.1, 128.9, 129.5, 130.0, 136.3, 136.7, 139.5, 148.0; GC–MS m/z: 262; HRMS: calcd for C18H14S, 262.0816; found, 262.0813.
(E)-1-(1-Naphthyl)-1,2-diphenylethene (3d) [59]: The reaction was performed using 1-naphthyllithium instead of phenyllithium, and 3d was isolated in 38% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 6.85 (s, 1H), 7.23–8.17 (m, 17H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 125.4, 125.8, 126.1, 126.3, 127.1, 127.4, 127.5, 128.0, 128.2, 128.4, 128.5, 129.6, 129.9, 131.8, 132.0, 134.1, 137.5, 141.1, 141.4, 142.3; GC–MS m/z: 306.
1-Bromo-1,2,2-triphenylethene (3e) [60]: The reaction mixture containing 4a was further treated with NBS (2 mmol) for 4 h at room temperature and 3e was isolated in 37% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 6.93–6.97 (m, 3H), 7.02–7.05 (m, 3H), 7.11–7.17 (m, 3H), 7.27–7.33 (m, 3H), 7.35–7.37 (m, 3H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 122.3, 127.1, 127.7, 128.0, 128.1, 128.2, 128.4, 129.7, 130.4, 130.4, 141.1, 141.2, 143.7, 143.9; GC–MS m/z: 334, 336.
1,1,2-Triphenylpenta-1,4-diene (3f) [27]: To the reaction mixture containing 4a, CuCl (1 mmol) and allyl bromide (2 mmol) were added. The reaction mixture was stirred for 12 h at room temperature and 3f was isolated in 45% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 3.28–3.30 (d, JHH = 6.1 Hz, 2H), 4.97–5.05 (m, 2H), 5.73–5.79 (m, 1H), 6.91–7.40 (m, 15H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 40.6, 116.1, 126.2, 126.5, 127.1, 127.6, 127.7, 128.0, 128.3, 129.8, 130.0, 131.0, 136.5, 138.0, 140.9, 142.4, 143.1, 143.4; GC–MS m/z: 296.
(E)-1,2-Diphenyl-1-(p-tolyl)penta-1,4-diene (3g): The reaction was performed using p-tolyllithium instead of phenyllithium. After treatment with TCQ for 12 h, CuCl (1 mmol) and allyl bromide (2 mmol) was added. The reaction mixture was stirred for 12 h at room temperature and 3g was isolated in 43% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 2.44 (s, 3H), 3.36–3.38 (d, JHH = 6.1 Hz, 2H), 5.80–5.89 (m, 2H), 7.01–7.25 (m, 14H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 21.4, 40.6, 116.0, 126.1, 126.3, 127.6, 128.0, 128.9, 129.7, 130.0, 131.0, 136.6, 136.6, 137.7, 140.5, 140.8, 142.5, 143.3; GC–MS m/z: 310; HRMS: calcd for C24H22, 310.1722; found, 310.1724.
1,2,2-Triphenyl-1-(p-tolyl)ethene (3h) [61]: The reaction was performed using p-tolyllithium instead of phenyllithium. To the reaction mixture, CuCl (1 mmol), Pd(PPh3)4 (0.05 mmol), and iodobenzene (2 mmol) were added. The reaction mixture was stirred for 12 h at room temperature and 3h was isolated in 31% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 2.28 (s, 3H), 3.94–7.12 (m, 19H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 21.3, 126.4, 126.4, 127.7, 127.8, 128.5, 131.3, 131.4, 131.5, 136.1 ,140.6, 140.9 ,141.0, 144.1; GC–MS m/z: 346.
(Z)-1-Phenyl-1,2-di(p-tolyl)ethene (3i) [58]: The reaction was performed using ditolylacetylene instead of diphenylacetylene, and 3i was isolated in 57% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 2.31 (s, 3H), 2.43 (s, 3H), 6.96 (s, 1H), 7.00–7.36 (m, 13H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 21.3, 21.5, 127.4, 127.7, 128.0, 128.3, 128.8, 129.5, 129.6, 130.4, 134.8, 136.6, 137.1, 137.7, 141.8, 144.0; GC–MS m/z: 284.
(E)-1-Bromo-2-phenyl-1,2-di(p-tolyl)ethene (3j): The reaction was performed using ditolylacetylene instead of diphenylacetylene. After bromination by NBS (2 mmol), 3j was isolated in 36% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 2.18 (s, 3H), 2.25 (s, 3H), 6.80–7.38 (m, 13H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 21.3, 21.4, 122.1, 127.5, 128.2, 128.7, 128.8, 129.6, 130.3, 130.3, 136.7, 137.8, 138.4, 138.5, 143.0, 144.3; GC-MS: 362, 364; HRMS: calcd for C22H19Br, 362.0670; found, 362.0674.
(Z)-1-Phenyl-1,2-di(p-fluorophenyl)ethene (3k) [59]: The reaction was performed using 4,4′-difluorodiphenylacetylene instead of diphenylacetylene, and 3k was formed in 59% yield. 1H NMR (400 MHz, CDCl3, Me4Si) δ 6.87–6.78 (m, 2H), 6.90 (s, 1H), 7.05–6.95 (m, 4H), 7.18–7.10 (m, 2H), 7.25–7.35 (m, 5H); 13C NMR (100 MHz, CDCl3, Me4Si) δ115.2 (d, J = 21.4 Hz), 115.9 (d, J = 21.5 Hz), 127.4, 127.7, 127.9, 128.4, 131.2 (d, J = 7.8 Hz), 132.2 (d, J = 7.8 Hz), 133.4, 136.1, 141.5, 143.2, 161.6 (d, J = 247.4 Hz), 162.3 (d, J = 246.7.8 Hz); GC–MS m/z: 292.
(Z)-1-Phenyl-1,2-di(p-methoxylphenyl)ethene (3l) [59]: The reaction was performed using 4,4’-dimethoxyldiphenylacetylene instead of diphenylacetylene, and 3l was formed in 52% yield. 1H NMR (400 MHz, CDCl3, Me4Si) δ 3.76 (s, 3H), 3.85 (s, 3H), 6.71 (d, J = 8.8 Hz, 2H), 6.88 (s, 1H), 6.90 (d, J = 8.8 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 7.15 (d, J = 8.7 Hz, 2H), 7.37–7.24 (m, 5H); 13C NMR (100 MHz, CDCl3, Me4Si) δ 55.2, 55.3, 113.5, 114.2, 127.3, 127.5, 127.6, 128.2, 130.4, 130.9, 131.7, 132.9, 140.4, 144.1, 158.4, 158.9.
1,1,2,3,4,4-Hexaphenyl-1,3-butadiene (5) [62]: To the reaction mixture containing 4a, CuCl (1 mmol) and TCQ (2 mmol) were added. The reaction mixture was stirred for 12 h at room temperature and compound 5 was isolated in 43% yield. 1H NMR (300 MHz, CDCl3, Me4Si) δ 6.96–7.40 (m, 30H); 13C NMR (75 MHz, CDCl3, Me4Si) δ 127.1, 127.6, 128.0, 128.0, 128.1, 128.2, 139.5, 140.4, 141.1, 142.0.
References
-
Flynn, A. B.; Ogilvie, W. W. Chem. Rev. 2007, 107, 4698–4745. doi:10.1021/cr050051k
Return to citation in text: [1] -
Negishi, E.; Huang, Z.; Wang, G.; Mohan, S.; Wang, C.; Hattori, H. Acc. Chem. Res. 2008, 41, 1474–1485. doi:10.1021/ar800038e
Return to citation in text: [1] -
Lindlar, H. Helv. Chim. Acta 1952, 35, 446–450. doi:10.1002/hlca.19520350205
Return to citation in text: [1] -
Rajaram, J.; Narula, A. P. S.; Chawla, H. P. S.; Dev, S. Tetrahedron 1983, 39, 2315–2322. doi:10.1016/S0040-4020(01)91960-X
Return to citation in text: [1] -
Hauwert, P.; Maestri, G.; Sprengers, J. W.; Catellani, M.; Elsevier, C. J. Angew. Chem., Int. Ed. 2008, 47, 3223–3226. doi:10.1002/anie.200705638
Return to citation in text: [1] -
Kitamura, T. Eur. J. Org. Chem. 2009, 1111–1125. doi:10.1002/ejoc.200801054
Return to citation in text: [1] -
Nevado, C.; Echavarren, A. M. Synthesis 2005, 167–182. doi:10.1055/s-2005-861781
Return to citation in text: [1] -
Hart, D. W.; Schwartz, J. J. Am. Chem. Soc. 1974, 96, 8115–8116. doi:10.1021/ja00833a048
Return to citation in text: [1] -
Ilies, L.; Yoshida, T.; Nakamura, E. J. Am. Chem. Soc. 2012, 134, 16951–16954. doi:10.1021/ja307631v
Return to citation in text: [1] -
Greenhalgh, M. D.; Thomas, S. P. Synlett 2013, 24, 531–534. doi:10.1055/s-0032-1318075
Return to citation in text: [1] -
Shen, R.; Chen, T.; Zhao, Y.; Qiu, R.; Zhou, Y.; Yin, S.; Wang, X.; Goto, M.; Han, L.-B. J. Am. Chem. Soc. 2011, 133, 17037–17044. doi:10.1021/ja2069246
Return to citation in text: [1] -
Knochel, P. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 865–911.
Return to citation in text: [1] [2] -
Normant, J. F.; Alexakis, A. Synthesis 1981, 841–870. doi:10.1055/s-1981-29622
Return to citation in text: [1] [2] -
Negishi, E.; Wang, G.; Rao, H.; Xu, Z. J. Org. Chem. 2010, 75, 3151–3182. doi:10.1021/jo1003218
Return to citation in text: [1] [2] -
Shirakawa, E.; Ikeda, D.; Masui, S.; Yoshida, M.; Hayashi, T. J. Am. Chem. Soc. 2012, 134, 272–279. doi:10.1021/ja206745w
Return to citation in text: [1] [2] -
Murakami, K.; Yorimitsu, H. Beilstein J. Org. Chem. 2013, 9, 278–302. doi:10.3762/bjoc.9.34
Return to citation in text: [1] [2] -
Takahashi, T.; Aoyagi, K.; Hara, R.; Suzuki, N. J. Chem. Soc., Chem. Commun. 1993, 1042–1044. doi:10.1039/c39930001042
Return to citation in text: [1] [2] -
Takahashi, T.; Suzuki, N.; Kageyama, M.; Kondakov, D. Y.; Hara, R. Tetrahedron Lett. 1993, 34, 4811–4814. doi:10.1016/S0040-4039(00)74095-0
Return to citation in text: [1] [2] -
Suzuki, N.; Kondakov, D. Y.; Kageyama, M.; Kotora, M.; Hara, R.; Takahashi, T. Tetrahedron 1995, 51, 4519–4540. doi:10.1016/0040-4020(94)01138-P
Return to citation in text: [1] [2] -
Takahashi, T.; Kondakov, D. Y.; Suzuki, N. Tetrahedron Lett. 1993, 34, 6571–6574. doi:10.1016/0040-4039(93)88107-T
Return to citation in text: [1] [2] -
Takahashi, T.; Kondakov, D. Y.; Xi, Z.; Suzuki, N. J. Am. Chem. Soc. 1995, 117, 5871–5872. doi:10.1021/ja00126a035
Return to citation in text: [1] [2] -
Liu, Y.; Zhong, Z.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2002, 67, 7451–7456. doi:10.1021/jo026037e
Return to citation in text: [1] [2] -
Takahashi, T.; Xi, C.; Ura, Y.; Nakajima, K. J. Am. Chem. Soc. 2000, 122, 3228–3229. doi:10.1021/ja994234y
Return to citation in text: [1] [2] -
Negishi, E.; Liu, F. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F.; Stang, P. J., Eds.; Wiley-VCH: New York, 1998; Chapter 1.
Return to citation in text: [1] -
Negishi, E. Overview of the Negishi Protocol with Zn, Al, Zr, and Related Metals. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; Wiley-Interscience: New York, 2002; pp 229–247. doi:10.1002/0471212466.ch15
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Hara, R.; Nishihara, Y.; Landré, P. D.; Takahashi, T. Tetrahedron Lett. 1997, 38, 447–450. doi:10.1016/S0040-4039(96)02322-2
Return to citation in text: [1] [2] -
Hara, R.; Liu, Y.; Sun, W.-H.; Takahashi, T. Tetrahedron Lett. 1997, 38, 4103–4106. doi:10.1016/S0040-4039(97)00838-1
Return to citation in text: [1] -
Miyaji, T.; Xi, Z.; Nakajima, K.; Takahashi, T. Organometallics 2001, 20, 2859–2863. doi:10.1021/om000592u
Return to citation in text: [1] -
Miyaji, T.; Xi, Z.; Ogasawara, M.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2007, 72, 8737–8740. doi:10.1021/jo071089v
Return to citation in text: [1] -
Nishihara, Y.; Miyasaka, M.; Okamoto, M.; Takahashi, H.; Inoue, E.; Tanemura, K.; Takagi, K. J. Am. Chem. Soc. 2007, 129, 12634–12635. doi:10.1021/ja075234y
Return to citation in text: [1] -
Nishihara, Y.; Saito, D.; Tanemura, K.; Noyori, S.; Takagi, K. Org. Lett. 2009, 11, 3546–3549. doi:10.1021/ol901236s
Return to citation in text: [1] -
Nishihara, Y.; Okada, Y.; Jiao, J.; Suetsugu, M.; Lan, M.-T.; Kinoshita, M.; Iwasaki, M.; Takagi, K. Angew. Chem., Int. Ed. 2011, 50, 8660–8664. doi:10.1002/anie.201103601
Return to citation in text: [1] -
Yan, X.; Chen, C.; Zhou, Y.; Xi, C. Org. Lett. 2012, 14, 4750–4753. doi:10.1021/ol302004t
Return to citation in text: [1] -
Liu, H.; Yan, X.; Chen, C.; Liu, Q.; Xi, C. Chem. Commun. 2013, 49, 5513–5515. doi:10.1039/c3cc41574k
Return to citation in text: [1] -
Liu, H.; Zhou, Y.; Yan, X.; Chen, C.; Liu, Q.; Xi, C. Org. Lett. 2013, 15, 5174–5177. doi:10.1021/ol402212g
Return to citation in text: [1] -
Alt, H. G.; Köppl, A. Chem. Rev. 2000, 100, 1205–1222. doi:10.1021/cr9804700
Return to citation in text: [1] -
Erker, G. Acc. Chem. Res. 2001, 34, 309–317. doi:10.1021/ar9800183
Return to citation in text: [1] -
Marek, I., Ed. Titanium and Zirconium in Organic Synthesis; Wiley-VCH: Weinheim, 2002. doi:10.1002/3527600671
Return to citation in text: [1] -
Takahashi, T.; Kageyama, M.; Denisov, V.; Hara, R.; Negishi, E. Tetrahedron Lett. 1993, 34, 687–690. doi:10.1016/S0040-4039(00)61653-2
Return to citation in text: [1] -
Xi, Z.; Hara, R.; Takahashi, T. J. Org. Chem. 1995, 60, 4444–4448. doi:10.1021/jo00119a022
Return to citation in text: [1] -
Takahashi, T.; Xi, C.; Xi, Z.; Kageyama, M.; Fischer, R.; Nakajima, K.; Negishi, E. J. Org. Chem. 1998, 63, 6802–6806. doi:10.1021/jo980022s
Return to citation in text: [1] -
Takahashi, T.; Xi, Z.; Yamazaki, A.; Liu, Y.; Nakajima, K.; Kotora, M. J. Am. Chem. Soc. 1998, 120, 1672–1680. doi:10.1021/ja970869q
Return to citation in text: [1] -
Xi, C.; Liu, Y.; Yan, X.; Chen, C. J. Organomet. Chem. 2007, 692, 4612–4617. doi:10.1016/j.jorganchem.2007.04.028
Return to citation in text: [1] [2] -
Xi, C.; Yan, X.; You, W.; Takahashi, T. Angew. Chem., Int. Ed. 2009, 48, 8120–8123. doi:10.1002/anie.200904255
Return to citation in text: [1] -
Yan, X.; Lai, C.; Xi, C. Chem. Commun. 2009, 6026–6028. doi:10.1039/b912175g
Return to citation in text: [1] -
Yan, X.; Zhou, Y.; Xi, C. Chem. Commun. 2010, 46, 7801–7803. doi:10.1039/c0cc02997a
Return to citation in text: [1] -
Yan, X.; Zhou, Y.; Xi, C. Organometallics 2013, 32, 869–873. doi:10.1021/om301176e
Return to citation in text: [1] [2] -
Negishi, E.; Cederbaum, F. E.; Takahashi, T. Tetrahedron Lett. 1986, 27, 2829–2832. doi:10.1016/S0040-4039(00)84653-5
Return to citation in text: [1] -
Wagenen, B. C. V.; Livinghouse, T. Tetrahedron Lett. 1989, 30, 3495–3498. doi:10.1016/S0040-4039(00)99422-X
Return to citation in text: [1] -
Banks, R. B.; Walborsky, H. M. J. Am. Chem. Soc. 1976, 98, 3732–3734. doi:10.1021/ja00428a071
Return to citation in text: [1] -
Piers, E.; Romero, M. A. J. Am. Chem. Soc. 1996, 118, 1215–1216. doi:10.1021/ja953648y
Return to citation in text: [1] -
Chen, C.; Xi, C.; Lai, C.; Wang, R.; Hong, X. Eur. J. Org. Chem. 2004, 647–650. doi:10.1002/ejoc.200300485
Return to citation in text: [1] -
Chen, C.; Xi, C.; Liu, Y.; Hong, X. J. Org. Chem. 2006, 71, 5373–5376. doi:10.1021/jo060334s
Return to citation in text: [1] -
Wang, C.; Yuan, J.; Li, G.; Wang, Z.; Zhang, S.; Xi, Z. J. Am. Chem. Soc. 2006, 128, 4564–4565. doi:10.1021/ja0579208
Return to citation in text: [1] -
Luo, Q.; Wang, C.; Zhang, W.-X.; Xi, Z. Chem. Commun. 2008, 1593–1595. doi:10.1039/b719007g
Return to citation in text: [1] -
Wei, J.; Wang, Z.; Zhang, W.-X.; Xi, Z. Org. Lett. 2013, 15, 1222–1225. doi:10.1021/ol400140n
Return to citation in text: [1] -
Zhang, W.; Liu, M.; Wu, H.; Ding, J.; Cheng, J. Tetrahedron Lett. 2008, 49, 5214–5216. doi:10.1016/j.tetlet.2008.05.140
Return to citation in text: [1] [2] [3] -
Xu, X.; Chen, J.; Gao, W.; Wu, H.; Ding, J.; Su, W. Tetrahedron 2010, 66, 2433–2438. doi:10.1016/j.tet.2010.01.086
Return to citation in text: [1] [2] [3] [4] -
Kahveci, M. U.; Uygun, M.; Tasdelen, M. A.; Schnabel, W.; Cook, W. D.; Yagci, Y. Macromolecules 2009, 42, 4443–4448. doi:10.1021/ma900359c
Return to citation in text: [1] -
Banerjee, M.; Emond, S. J.; Lindeman, S. V.; Rathore, R. J. Org. Chem. 2007, 72, 8054–8061. doi:10.1021/jo701474y
Return to citation in text: [1] -
Horiguchi, H.; Tsurugi, H.; Satoh, T.; Miura, M. Adv. Synth. Catal. 2008, 350, 509–514. doi:10.1002/adsc.200700533
Return to citation in text: [1]
| 58. | Zhang, W.; Liu, M.; Wu, H.; Ding, J.; Cheng, J. Tetrahedron Lett. 2008, 49, 5214–5216. doi:10.1016/j.tetlet.2008.05.140 |
| 59. | Xu, X.; Chen, J.; Gao, W.; Wu, H.; Ding, J.; Su, W. Tetrahedron 2010, 66, 2433–2438. doi:10.1016/j.tet.2010.01.086 |
| 59. | Xu, X.; Chen, J.; Gao, W.; Wu, H.; Ding, J.; Su, W. Tetrahedron 2010, 66, 2433–2438. doi:10.1016/j.tet.2010.01.086 |
| 1. | Flynn, A. B.; Ogilvie, W. W. Chem. Rev. 2007, 107, 4698–4745. doi:10.1021/cr050051k |
| 2. | Negishi, E.; Huang, Z.; Wang, G.; Mohan, S.; Wang, C.; Hattori, H. Acc. Chem. Res. 2008, 41, 1474–1485. doi:10.1021/ar800038e |
| 12. | Knochel, P. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 865–911. |
| 13. | Normant, J. F.; Alexakis, A. Synthesis 1981, 841–870. doi:10.1055/s-1981-29622 |
| 14. | Negishi, E.; Wang, G.; Rao, H.; Xu, Z. J. Org. Chem. 2010, 75, 3151–3182. doi:10.1021/jo1003218 |
| 15. | Shirakawa, E.; Ikeda, D.; Masui, S.; Yoshida, M.; Hayashi, T. J. Am. Chem. Soc. 2012, 134, 272–279. doi:10.1021/ja206745w |
| 16. | Murakami, K.; Yorimitsu, H. Beilstein J. Org. Chem. 2013, 9, 278–302. doi:10.3762/bjoc.9.34 |
| 17. | Takahashi, T.; Aoyagi, K.; Hara, R.; Suzuki, N. J. Chem. Soc., Chem. Commun. 1993, 1042–1044. doi:10.1039/c39930001042 |
| 18. | Takahashi, T.; Suzuki, N.; Kageyama, M.; Kondakov, D. Y.; Hara, R. Tetrahedron Lett. 1993, 34, 4811–4814. doi:10.1016/S0040-4039(00)74095-0 |
| 19. | Suzuki, N.; Kondakov, D. Y.; Kageyama, M.; Kotora, M.; Hara, R.; Takahashi, T. Tetrahedron 1995, 51, 4519–4540. doi:10.1016/0040-4020(94)01138-P |
| 20. | Takahashi, T.; Kondakov, D. Y.; Suzuki, N. Tetrahedron Lett. 1993, 34, 6571–6574. doi:10.1016/0040-4039(93)88107-T |
| 21. | Takahashi, T.; Kondakov, D. Y.; Xi, Z.; Suzuki, N. J. Am. Chem. Soc. 1995, 117, 5871–5872. doi:10.1021/ja00126a035 |
| 22. | Liu, Y.; Zhong, Z.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2002, 67, 7451–7456. doi:10.1021/jo026037e |
| 23. | Takahashi, T.; Xi, C.; Ura, Y.; Nakajima, K. J. Am. Chem. Soc. 2000, 122, 3228–3229. doi:10.1021/ja994234y |
| 23. | Takahashi, T.; Xi, C.; Ura, Y.; Nakajima, K. J. Am. Chem. Soc. 2000, 122, 3228–3229. doi:10.1021/ja994234y |
| 8. | Hart, D. W.; Schwartz, J. J. Am. Chem. Soc. 1974, 96, 8115–8116. doi:10.1021/ja00833a048 |
| 9. | Ilies, L.; Yoshida, T.; Nakamura, E. J. Am. Chem. Soc. 2012, 134, 16951–16954. doi:10.1021/ja307631v |
| 10. | Greenhalgh, M. D.; Thomas, S. P. Synlett 2013, 24, 531–534. doi:10.1055/s-0032-1318075 |
| 11. | Shen, R.; Chen, T.; Zhao, Y.; Qiu, R.; Zhou, Y.; Yin, S.; Wang, X.; Goto, M.; Han, L.-B. J. Am. Chem. Soc. 2011, 133, 17037–17044. doi:10.1021/ja2069246 |
| 44. | Xi, C.; Liu, Y.; Yan, X.; Chen, C. J. Organomet. Chem. 2007, 692, 4612–4617. doi:10.1016/j.jorganchem.2007.04.028 |
| 6. | Kitamura, T. Eur. J. Org. Chem. 2009, 1111–1125. doi:10.1002/ejoc.200801054 |
| 7. | Nevado, C.; Echavarren, A. M. Synthesis 2005, 167–182. doi:10.1055/s-2005-861781 |
| 21. | Takahashi, T.; Kondakov, D. Y.; Xi, Z.; Suzuki, N. J. Am. Chem. Soc. 1995, 117, 5871–5872. doi:10.1021/ja00126a035 |
| 3. | Lindlar, H. Helv. Chim. Acta 1952, 35, 446–450. doi:10.1002/hlca.19520350205 |
| 4. | Rajaram, J.; Narula, A. P. S.; Chawla, H. P. S.; Dev, S. Tetrahedron 1983, 39, 2315–2322. doi:10.1016/S0040-4020(01)91960-X |
| 5. | Hauwert, P.; Maestri, G.; Sprengers, J. W.; Catellani, M.; Elsevier, C. J. Angew. Chem., Int. Ed. 2008, 47, 3223–3226. doi:10.1002/anie.200705638 |
| 22. | Liu, Y.; Zhong, Z.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2002, 67, 7451–7456. doi:10.1021/jo026037e |
| 40. | Takahashi, T.; Kageyama, M.; Denisov, V.; Hara, R.; Negishi, E. Tetrahedron Lett. 1993, 34, 687–690. doi:10.1016/S0040-4039(00)61653-2 |
| 41. | Xi, Z.; Hara, R.; Takahashi, T. J. Org. Chem. 1995, 60, 4444–4448. doi:10.1021/jo00119a022 |
| 42. | Takahashi, T.; Xi, C.; Xi, Z.; Kageyama, M.; Fischer, R.; Nakajima, K.; Negishi, E. J. Org. Chem. 1998, 63, 6802–6806. doi:10.1021/jo980022s |
| 43. | Takahashi, T.; Xi, Z.; Yamazaki, A.; Liu, Y.; Nakajima, K.; Kotora, M. J. Am. Chem. Soc. 1998, 120, 1672–1680. doi:10.1021/ja970869q |
| 18. | Takahashi, T.; Suzuki, N.; Kageyama, M.; Kondakov, D. Y.; Hara, R. Tetrahedron Lett. 1993, 34, 4811–4814. doi:10.1016/S0040-4039(00)74095-0 |
| 19. | Suzuki, N.; Kondakov, D. Y.; Kageyama, M.; Kotora, M.; Hara, R.; Takahashi, T. Tetrahedron 1995, 51, 4519–4540. doi:10.1016/0040-4020(94)01138-P |
| 37. | Alt, H. G.; Köppl, A. Chem. Rev. 2000, 100, 1205–1222. doi:10.1021/cr9804700 |
| 38. | Erker, G. Acc. Chem. Res. 2001, 34, 309–317. doi:10.1021/ar9800183 |
| 39. | Marek, I., Ed. Titanium and Zirconium in Organic Synthesis; Wiley-VCH: Weinheim, 2002. doi:10.1002/3527600671 |
| 20. | Takahashi, T.; Kondakov, D. Y.; Suzuki, N. Tetrahedron Lett. 1993, 34, 6571–6574. doi:10.1016/0040-4039(93)88107-T |
| 12. | Knochel, P. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 4, pp 865–911. |
| 13. | Normant, J. F.; Alexakis, A. Synthesis 1981, 841–870. doi:10.1055/s-1981-29622 |
| 14. | Negishi, E.; Wang, G.; Rao, H.; Xu, Z. J. Org. Chem. 2010, 75, 3151–3182. doi:10.1021/jo1003218 |
| 15. | Shirakawa, E.; Ikeda, D.; Masui, S.; Yoshida, M.; Hayashi, T. J. Am. Chem. Soc. 2012, 134, 272–279. doi:10.1021/ja206745w |
| 16. | Murakami, K.; Yorimitsu, H. Beilstein J. Org. Chem. 2013, 9, 278–302. doi:10.3762/bjoc.9.34 |
| 62. | Horiguchi, H.; Tsurugi, H.; Satoh, T.; Miura, M. Adv. Synth. Catal. 2008, 350, 509–514. doi:10.1002/adsc.200700533 |
| 24. | Negishi, E.; Liu, F. In Metal-Catalyzed Cross-Coupling Reactions; Diederich, F.; Stang, P. J., Eds.; Wiley-VCH: New York, 1998; Chapter 1. |
| 25. | Negishi, E. Overview of the Negishi Protocol with Zn, Al, Zr, and Related Metals. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; Wiley-Interscience: New York, 2002; pp 229–247. doi:10.1002/0471212466.ch15 |
| 26. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 27. | Hara, R.; Nishihara, Y.; Landré, P. D.; Takahashi, T. Tetrahedron Lett. 1997, 38, 447–450. doi:10.1016/S0040-4039(96)02322-2 |
| 28. | Hara, R.; Liu, Y.; Sun, W.-H.; Takahashi, T. Tetrahedron Lett. 1997, 38, 4103–4106. doi:10.1016/S0040-4039(97)00838-1 |
| 29. | Miyaji, T.; Xi, Z.; Nakajima, K.; Takahashi, T. Organometallics 2001, 20, 2859–2863. doi:10.1021/om000592u |
| 30. | Miyaji, T.; Xi, Z.; Ogasawara, M.; Nakajima, K.; Takahashi, T. J. Org. Chem. 2007, 72, 8737–8740. doi:10.1021/jo071089v |
| 31. | Nishihara, Y.; Miyasaka, M.; Okamoto, M.; Takahashi, H.; Inoue, E.; Tanemura, K.; Takagi, K. J. Am. Chem. Soc. 2007, 129, 12634–12635. doi:10.1021/ja075234y |
| 32. | Nishihara, Y.; Saito, D.; Tanemura, K.; Noyori, S.; Takagi, K. Org. Lett. 2009, 11, 3546–3549. doi:10.1021/ol901236s |
| 33. | Nishihara, Y.; Okada, Y.; Jiao, J.; Suetsugu, M.; Lan, M.-T.; Kinoshita, M.; Iwasaki, M.; Takagi, K. Angew. Chem., Int. Ed. 2011, 50, 8660–8664. doi:10.1002/anie.201103601 |
| 34. | Yan, X.; Chen, C.; Zhou, Y.; Xi, C. Org. Lett. 2012, 14, 4750–4753. doi:10.1021/ol302004t |
| 35. | Liu, H.; Yan, X.; Chen, C.; Liu, Q.; Xi, C. Chem. Commun. 2013, 49, 5513–5515. doi:10.1039/c3cc41574k |
| 36. | Liu, H.; Zhou, Y.; Yan, X.; Chen, C.; Liu, Q.; Xi, C. Org. Lett. 2013, 15, 5174–5177. doi:10.1021/ol402212g |
| 17. | Takahashi, T.; Aoyagi, K.; Hara, R.; Suzuki, N. J. Chem. Soc., Chem. Commun. 1993, 1042–1044. doi:10.1039/c39930001042 |
| 49. | Negishi, E.; Cederbaum, F. E.; Takahashi, T. Tetrahedron Lett. 1986, 27, 2829–2832. doi:10.1016/S0040-4039(00)84653-5 |
| 45. | Xi, C.; Yan, X.; You, W.; Takahashi, T. Angew. Chem., Int. Ed. 2009, 48, 8120–8123. doi:10.1002/anie.200904255 |
| 46. | Yan, X.; Lai, C.; Xi, C. Chem. Commun. 2009, 6026–6028. doi:10.1039/b912175g |
| 47. | Yan, X.; Zhou, Y.; Xi, C. Chem. Commun. 2010, 46, 7801–7803. doi:10.1039/c0cc02997a |
| 48. | Yan, X.; Zhou, Y.; Xi, C. Organometallics 2013, 32, 869–873. doi:10.1021/om301176e |
| 48. | Yan, X.; Zhou, Y.; Xi, C. Organometallics 2013, 32, 869–873. doi:10.1021/om301176e |
| 27. | Hara, R.; Nishihara, Y.; Landré, P. D.; Takahashi, T. Tetrahedron Lett. 1997, 38, 447–450. doi:10.1016/S0040-4039(96)02322-2 |
| 61. | Banerjee, M.; Emond, S. J.; Lindeman, S. V.; Rathore, R. J. Org. Chem. 2007, 72, 8054–8061. doi:10.1021/jo701474y |
| 59. | Xu, X.; Chen, J.; Gao, W.; Wu, H.; Ding, J.; Su, W. Tetrahedron 2010, 66, 2433–2438. doi:10.1016/j.tet.2010.01.086 |
| 60. | Kahveci, M. U.; Uygun, M.; Tasdelen, M. A.; Schnabel, W.; Cook, W. D.; Yagci, Y. Macromolecules 2009, 42, 4443–4448. doi:10.1021/ma900359c |
| 59. | Xu, X.; Chen, J.; Gao, W.; Wu, H.; Ding, J.; Su, W. Tetrahedron 2010, 66, 2433–2438. doi:10.1016/j.tet.2010.01.086 |
| 58. | Zhang, W.; Liu, M.; Wu, H.; Ding, J.; Cheng, J. Tetrahedron Lett. 2008, 49, 5214–5216. doi:10.1016/j.tetlet.2008.05.140 |
| 44. | Xi, C.; Liu, Y.; Yan, X.; Chen, C. J. Organomet. Chem. 2007, 692, 4612–4617. doi:10.1016/j.jorganchem.2007.04.028 |
| 50. | Wagenen, B. C. V.; Livinghouse, T. Tetrahedron Lett. 1989, 30, 3495–3498. doi:10.1016/S0040-4039(00)99422-X |
| 51. | Banks, R. B.; Walborsky, H. M. J. Am. Chem. Soc. 1976, 98, 3732–3734. doi:10.1021/ja00428a071 |
| 52. | Piers, E.; Romero, M. A. J. Am. Chem. Soc. 1996, 118, 1215–1216. doi:10.1021/ja953648y |
| 53. | Chen, C.; Xi, C.; Lai, C.; Wang, R.; Hong, X. Eur. J. Org. Chem. 2004, 647–650. doi:10.1002/ejoc.200300485 |
| 54. | Chen, C.; Xi, C.; Liu, Y.; Hong, X. J. Org. Chem. 2006, 71, 5373–5376. doi:10.1021/jo060334s |
| 55. | Wang, C.; Yuan, J.; Li, G.; Wang, Z.; Zhang, S.; Xi, Z. J. Am. Chem. Soc. 2006, 128, 4564–4565. doi:10.1021/ja0579208 |
| 56. | Luo, Q.; Wang, C.; Zhang, W.-X.; Xi, Z. Chem. Commun. 2008, 1593–1595. doi:10.1039/b719007g |
| 57. | Wei, J.; Wang, Z.; Zhang, W.-X.; Xi, Z. Org. Lett. 2013, 15, 1222–1225. doi:10.1021/ol400140n |
| 58. | Zhang, W.; Liu, M.; Wu, H.; Ding, J.; Cheng, J. Tetrahedron Lett. 2008, 49, 5214–5216. doi:10.1016/j.tetlet.2008.05.140 |
© 2014 Yan et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)