Abstract
A series of novel (1-aminoalkyl)(trifluoromethyl)- and -(difluoromethyl)phosphinic acids – analogues of proteinogenic and nonproteinogenic α-amino acids were prepared. The synthetic methodology was based on nucleophilic addition of (trifluoromethyl)phosphinic acid or (difluoromethyl)phosphinic acid or its ethyl ester to substrates with C=N or activated C=C double bonds. Analogues of glycine, phenylglycine, alanine, valine, proline, aminomalonic and aspartic acids were thus prepared. Three-component one-pot reactions of (trifluoromethyl)phosphinic acid and dibenzylamine with aldehydes were also tested to prepare the title compounds.

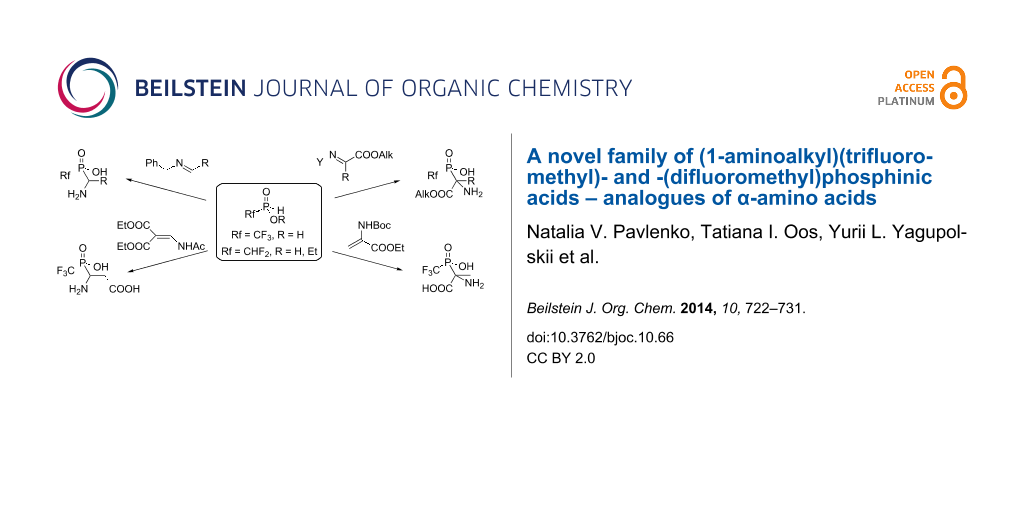
Graphical Abstract
Introduction
For a long time aminophosphonic and aminophosphinic acids as isosters of aminocarboxylic acids have attracted a particular interest for the preparation of analogues of numerous natural products. Among the literature concerning various aspects of the chemistry and biological activity of aminophosphonic and aminophosphinic acids, several monographs and reviews have appeared over the last decade [1-6]. The chemistry of fluorinated aminophosphonic and -phosphinic acids is a relatively new area of research. Incorporation of fluorine or fluorinated moieties can be used for the alteration of physiological properties of many biologically significant substances. The changes of their biological properties caused by this fluorination are influenced by complex factors, however. The similarity of the diameters of fluorine and hydrogen atoms in organic compounds makes fluorine an obvious choice as a substituent for biologically active substances, frequently without disrupting the shape and geometry of the substituted molecules. Nevertheless fluorine influences the electronic properties of a compound drastically because of its strong electronegativity. This enables modulation of the lipophilicity profile, of electrostatic interactions with the target structure and inhibition of some metabolic pathways [7-9]. Data concerning the biological activity and synthetic approaches toward fluorinated aminophosphonates, bearing side chain C–F linkages are well documented in a review [10].
The isolation of phosphinothricin, a naturally occurring phosphorus analogue of glutamic acid and the discovery of its antibiotic, fungicidal and herbicidal properties [11] has led to an increased activity in the study of methylphosphinic acid analogues of the protein amino acids [12] and those of glycine [13], alanine [14], valine [14], leucine [15], proline [16], aspartic [17] and glutamic [11] acids and GABA [18] have been described. But almost nothing is known about phosphorus isosters of aminocarboxylic acids bearing a (trifluoromethyl)- or (difluoromethyl)phosphonyl moiety instead of the carboxylate function. To the best of our knowledge there is only one report on the application of ethyl (difluoromethyl)phosphinate CHF2(H)P(O)(OEt) in the synthesis of a (difluoromethyl)phosphinic acid analogue of GABA, as a potent agonist of the GABAB receptor [18].
In light of the above and in connection with our interest in the chemistry of fluorinated compounds of phosphorus we report here the preparation of a series of novel (1-aminoalkyl)phosphinic acids bearing CF3 or CHF2 groups at phosphorus.
Results and Discussion
Research efforts have established H-phosphinates R(H)P(O)(OR′) or appropriate P(III) acids R(H)P(O)(OH) as appropriate starting materials for the preparation of aminophosphinic acids. The most typical route involves the three-component reaction of an aldehyde, an amine and a P–H substrate in a one-pot Mannich type protocol [19-21]. An alternative to this approach involves the simple addition of alkyl H-phosphinates or H-phosphinic acids to Schiff bases [5,22]. In this paper we exploit both routes to prepare (1-aminoalkyl)(trifluoromethyl)- and -(difluoromethyl)phosphinic acids using P–H compounds bearing CF3 and CHF2 groups attached to phosphorus.
P–H Substrates
(Trifluoromethyl)phosphinic acid CF3P(O)H(OH) (1) was first prepared in 1954 [23], but since then little chemistry has been reported involving 1. Also monoesters of 1 such as CF3P(O)H(OAlk) [24,25] have not been widely applied. These compounds, contain a labile P–H bond and synthetic problems underly their preparation. Emeléus and Haszeldine were the first [26] to prepare CF3P(III) compounds via the interaction of red phosphorus and CF3I in an autoclave. This gave mixtures of CF3-containing phosphanes and phosphane iodides but in poor yields. More recently, Ruppert described the reaction between CF3Br, P(NEt2)3 and PCl3, which gave CF3P(NEt2)2 in a good yield [27]. We applied this procedure to prepare CF3PCl2 [27] by interaction of diamide CF3P(NEt2)2 with gaseous HCl and then the chlorine was replaced by neutral hydrolysis to give (trifluoromethyl)phosphinic acid (1) [23] or by alcoholysis with ethanol or isopropanol to reach the appropriate esters 2–4 [24,25] (Scheme 1).
![[1860-5397-10-66-i1]](/bjoc/content/inline/1860-5397-10-66-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of (trifluoromethyl)phosphinic acid (1) and ethyl and isopropyl esters 2–4. Reagents and conditions: i) 1.8 equiv H2O per 1 equiv CF3PCl2, hexane, −10 °C → 0 °C, 3 h, then rt overnight, argon atmosphere, 84%. ii) 2 equiv anhydrous iPrOH per 1 equiv CF3PCl2, −40 °C → 0 °C, 3 h, then rt overnight, argon atmosphere, 68%. iii) 2 equiv anhydrous EtOH per 1 equiv CF3PCl2, −40 °C → 0 °C, 3 h, then rt overnight, argon atmosphere, mixture 3:4 ~ 10:1, ~70%.
Scheme 1: Synthesis of (trifluoromethyl)phosphinic acid (1) and ethyl and isopropyl esters 2–4. Reagents and ...
In our hands CF3PCl2 was hydrolyzed by two equivalents of water in hexane over the temperature range −10 °C → 0 °C, to give water-free (trifluoromethyl)phosphinic acid (1). Acid 1 proved easy to handle as a distillable liquid when prepared in this way and is stable under storage for months in contrast to that prepared by Emeléus and Haszeldine [23]. Phosphinate 2 was prepared with anhydrous isopropanol. When stored under anhydrous conditions at room temperature, ester 2 was partially converted to acid 1 as determined by 31P NMR. Alcoholysis of CF3PCl2 with ethanol under the same conditions produced diethyl phosphinate 3 admixed with monoester 4 (~10%), as previously described [25]. 31P NMR of these products indicated that they were converted to esters 3 and 4 on storage in a ratio of ~ 3:2 with acid 1 as an impurity. The low stability of all three esters 2–4 can be attributed to the lability of the O–C ester bond, resulting from the electron-withdrawing effect of the CF3 group attached to phosphorus, therefore esters 2, 3 and 4 were used in the syntheses only when freshly prepared and distilled.
We next explored the CHF2 group attached to phosphorus. Ethyl (difluoromethyl)phosphinate CHF2P(O)H(OEt) (5) was prepared as previously described [18]. The appropriate (difluoromethyl)phosphinic acid CHF2P(O)H(OH) (6) was obtained from 5 by ester deprotection with NaHCO3, as a viscous undistillable liquid, which was stable for weeks on storage.
Three-component reactions
At the outset of our work three-component reactions of formaldehyde, dibenzylamine and the esters 2 or 5 were explored as model transformations to evaluate the feasibility of the Kabachnik–Fields procedure [19,20] to the synthesis of fluorinated (1-aminoalkyl)phosphinate 7 (Scheme 2).
![[1860-5397-10-66-i2]](/bjoc/content/inline/1860-5397-10-66-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Three-component Kabachnik–Fields reaction of CF3(H)P(O)(OiPr) (2) with formaldehyde and dibenzylamine. Reagents and conditions: i) an equimolar mixture of reagents, H2O, 80 °C, 3 h, argon atmosphere, yield 10 ~80% or an equimolar mixture of reagents, dioxane, 100 °C, 3 h, argon atmosphere, yield 10 ~90%.
Scheme 2: Three-component Kabachnik–Fields reaction of CF3(H)P(O)(OiPr) (2) with formaldehyde and dibenzylami...
It turned out that this method is unsuitable for the synthesis of phosphinate 7. Formalin was added to an equimolar mixture of dibenzylamine and ester 2 at 80 °C under an oxygen-free atmosphere to give reaction mixtures with a low content of P–C products (31P NMR). A similar outcome was obtained when the reaction was run at room temperature or in dioxane with simultaneous water azeotropic distillation. Such an result might be explained by high reactivity of the starting ester, which readily reacted with formaldehyde, forming (α-hydroxymethyl)phosphinate 8. Its further irreversible rearrangement [28] to the corresponding phosphonate 9 was accompanied with hydrolysis of the ester function and formation of (trifluoromethyl)phosphonic acid (10) [29] as the main product. Analogous results were obtained, when CHF2 containing ester 5 was introduced into the reaction with formaline and dibenzylamine to give CHF2P(O)(OH)2 (11) as the major product [30].
Such results prompted us to explore the Mannich-type procedure of Moedritzer and Irani [21] for the syntheses of the desired aminophosphinic acids starting from acid 1. This resulted in the preparation of the analogues of glycine 14a and phenylglycine 14b (Scheme 3).
![[1860-5397-10-66-i3]](/bjoc/content/inline/1860-5397-10-66-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Three-component synthesis of CF3 containing α-aminophosphinic acids 14a,b. Reagents and conditions: i) An equimolar mixture of acid 1, dibenzylamine, HCl and two fold excess of aldehyde, H2O, 80 °C, 3 h; isolated yields: 13a (52%), 13b (28%); yields, determined by 31P and 19F NMR: 13c (<10%). ii) H2, ethanol, catalysis 10% Pd/C, rt, normal pressure, yields: 14a (95%), 14b (90%).
Scheme 3: Three-component synthesis of CF3 containing α-aminophosphinic acids 14a,b. Reagents and conditions:...
The three-component reaction with formaldehyde gave the best results and N-protected aminophosphinic acid 13a was isolated in a moderate yield, alongside phosphonic acid 10. The analogous reaction with benzaldehyde provided acid 13b in 28% yield and the main product of this reaction was an adduct of acid 1 with benzaldehyde which was isolated from the reaction mixture as ammonium salt 15 in 60% yield. Reaction with acetaldehyde was less successful and generated aminophosphinic acid 13c in low yield (<10%). Attempts to improve conversions products 13a–c by increasing the reaction temperature or varying the amino component (MeC(O)NH2, BnOC(O)NH2 or NH4OAc instead of Bn2NH) and molar equivalent of HCl were unsuccessful. It should be noted, that in contrast to the non-fluorinated counterparts the adducts 13a,b did not form hydrochlorides under this procedure consistent with the strongly acidic nature of the CF3 phosphinic acid group. Catalytic hydrogenation of intermediates 13a,b with Pd/C removed the benzyl groups and produced the corresponding acids 14a,b in high yields.
The hydrophosphinylation of azomethines
The addition of the P–H functionality to C=N double bonds is a very general procedure for the formation of P–C–N systems. Based on our experience of these three-component reactions (Scheme 2 and Scheme 3) we investigated the scope and limitations of the addition of (trifluoromethyl)phosphinic acid (1) to a series of N-benzylimines 16a–e in order to obtain fluorinated phosphorus analogues of glycine 14a, phenylglycine 14b, alanine 14c, valine 14d and proline 14e (Table 1).
Table 1: The interaction of (trifluoromethyl)phosphinic acid (1) with Shiff bases.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-10-66-i11.svg?max-width=637&scale=1.0)
|
||||
| Entry | Shiff base | R | 17 (yield, %)b | 14 (yield, %)b |
|---|---|---|---|---|
| 1c |
![[Graphic 2]](/bjoc/content/inline/1860-5397-10-66-i12.svg?max-width=637&scale=1.0)
|
H | 17a (83) | 14a (95) |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-10-66-i13.svg?max-width=637&scale=1.0)
|
Ph | 17b (79) | 14b (96) |
| 3d |
![[Graphic 4]](/bjoc/content/inline/1860-5397-10-66-i14.svg?max-width=637&scale=1.0)
|
Me | 17c (59) | 14c (96) |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-10-66-i15.svg?max-width=637&scale=1.0)
|
iPr | 17d (92) | 14d (98) |
| 5c |
![[Graphic 6]](/bjoc/content/inline/1860-5397-10-66-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-10-66-i17.svg?max-width=637&scale=1.0)
|
– | 14e (66) |
aReagents and conditions: i) an equimolar mixture of acid 1 and Shiff base, DME, rt, 31P NMR control; ii) H2, ethanol, catalysis 10% Pd/C, rt, normal pressure. bIsolated yields. cSymmetrical cyclic triazinanes (masked imines) were used to generate unstable imines. dThe best yield was obtained with 2 mol equivalents of imine.
The transformations were mildly exothermic and were monitored by 31P NMR. Acid 1 undergoes the typical P–C bond forming reactions with Shiff bases to give adducts 17 in satisfactory yields and these were successfully transformed into the appropriate free acids 14.
The same series of Shiff bases was used to explore the reactivity of ethyl (difluoromethyl)phosphinate (5) in reactions with C=N double bonds and the desired (α-aminoalkyl)phosphinic acids 20a–e were accordingly prepared (Table 2).
Table 2: The interaction of ethyl (difluoromethyl)phosphinate (5) with Shiff bases.a
![[Graphic 8]](/bjoc/content/inline/1860-5397-10-66-i18.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Shiff base | R | 18 (yield, %)b | 19 (yield, %)b | 20 (yield, %)b |
|---|---|---|---|---|---|
| 1c |
![[Graphic 9]](/bjoc/content/inline/1860-5397-10-66-i19.svg?max-width=637&scale=1.0)
|
H | 18a (78)d | 19a (68) | 20a (91) |
| 2 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-10-66-i20.svg?max-width=637&scale=1.0)
|
Ph | 18b (58)e | 19b (86)f | 20b (96) |
| 3g |
![[Graphic 11]](/bjoc/content/inline/1860-5397-10-66-i21.svg?max-width=637&scale=1.0)
|
Me | 18c (56)e | 19c (80)f | 20c (95) |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-10-66-i22.svg?max-width=637&scale=1.0)
|
iPr | 18d (36)e | 19d (82)f | 20d (95) |
| 5c |
![[Graphic 13]](/bjoc/content/inline/1860-5397-10-66-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-10-66-i24.svg?max-width=637&scale=1.0)
|
– | – | 20e (79) |
aReagents and conditions: i) an equimolar mixture of ester 5 and Shiff base, DME, rt, 31P NMR control, under argon atmosphere; ii) 1 N HCl, rt, until clear solution; iii) H2, ethanol, catalysis 10% Pd/C, rt, normal pressure. bIsolated yields. cSymmetrical cyclic triazinanes (masked imines) were used to generate unstable imines. dYield was defined with 31P NMR. eDiastereomeric ratio: 18b (~7:2), 18c (~3:2), 18d (~3:2). fYields were defined as the sum of yields of compounds 19b–d, isolated from the reaction mixture and obtained after hydrolysis of intermediates 18b–d. gThe maximum yield was obtained with 2 mol equivalents of imine.
The syntheses of compounds 20a–e were performed with purification and characterization of the intermediates, wherever possible as summarized in Table 2.
Products 14b–e and 20b–e were obtained as racemic mixtures. As expected, the 31P NMR spectra of 14 and 20 display characteristic signals around 11–15 ppm with 2JFP couplings for CF3 bearing substrates. The CHF2 bearing also possessed 2JFP couplings of 75–95 Hz. The 19F NMR spectra were characterized by signals in the region −75 ppm for CF3 derivatives and −137 ppm for CHF2 ones. Some distinctive characteristics of the reactivity of ester 5 were observed. This ester readily reacted with azomethines, but in contrast to its nonfluorinated counterparts this ester generated mixtures of the adducts 18 and 19. The interaction of ester 5 with imine 16a gave adduct 18a (Table 2, entry 1) in high conversion yield but after purification over silica gel only the appropriate acid 19a was isolated. In the case of the reaction of ester 5 with imine 16e no adduct was formed. Ethyl phosphinates 18b–d were obtained as a mixture of two diastereoisomers, which were not separated but they are clearly observed by 1H NMR as separate signals for the CHF2 group. The adducts 18b–d were hydrolyzed to acids 19b–d in quantitative yields and did not form hydrochlorides similar to the CF3 aminophosphinic acids 13a–b and 17a–d. Hydrogenolysis of the 17a–d and 19a–d efficiently gave free acids 14 and 20, but required column ion-exchange chromatography to produce analytically pure products.
We then explored the addition of acid 1 to imine 21 [31], which is N-Boc protected, typically used for amino acid protection (Scheme 4). This produced acid 14b in one step, but in only 32% yield. The N-tert-butoxycarbonyl group was removed during the reaction due to the high acidity of the CF3 phosphinic acid group.
![[1860-5397-10-66-i4]](/bjoc/content/inline/1860-5397-10-66-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Interaction of the acid 1 with tert-butyl benzylidenecarbamate (21). Reagents and conditions: i) an equimolar mixture of reagents, DME, rt, 48 h, under argon atmosphere, 32%.
Scheme 4: Interaction of the acid 1 with tert-butyl benzylidenecarbamate (21). Reagents and conditions: i) an...
The variability of Schiff bases ensures access to a range of structurally diverse phosphinic acid analogues of amino acids in the relatively simple way. Thus, we investigated the hydrophosphinylation of some Shiff bases bearing a carboxylate functionality to obtain aminocarboxylic acids, containing pendant CF3 or CHF2 phosphinic acid linkages. Thus, phosphinic acids 1 and 6 reacted with the N-Boc-protected Schiff base of ethyl glyoxalate 22 [32] under mild conditions to produce the N-deprotected phosphinylglycines 23 and 24 in satisfactory yields (Scheme 5).
![[1860-5397-10-66-i5]](/bjoc/content/inline/1860-5397-10-66-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Interaction of the acids 1 and 6 with ethyl 2-[(tert-butoxycarbonyl)imino]acetate (22). Reagents and conditions: i) an equimolar mixture of reagents, DME, rt, 31P NMR control, under argon atmosphere, isolated yields: 23 (68%), 24 (61%).
Scheme 5: Interaction of the acids 1 and 6 with ethyl 2-[(tert-butoxycarbonyl)imino]acetate (22). Reagents an...
Attempts to convert ester 23 to the free acid failed. Removal of the ester group from 23 by acidolysis with HCl or HI was accompanied by cleavage of the P–C bond to give only (trifluoromethyl)phosphonic acid (10) after an ion-exchange chromatography. Attempts to remove the ester group in anhydrous base with 1 equivalent of sodium silanolate (Me3SiONa) at room temperature efficiently produced the highly stable sodium salt of acid 23. With an excess of Me3SiONa and heating to 50 °C, fluoroform liberation from 23 was observed to give fluorine-free products.
In contrast the CHF2-containing ester 24 was stable toward acidic hydrolysis under mild conditions, but in the presence of an excess of sodium silanolate, free phosphinylglycine 25 was obtained, but in a poor yield (Scheme 6).
![[1860-5397-10-66-i6]](/bjoc/content/inline/1860-5397-10-66-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Transformation of the ester 24 into the appropriate free acid 25. Reagents and conditions: i) two fold excess of Me3SiONa, DME, 50 °C, 24 h, under argon atmosphere, ion-exchange chromatography, H2O, 35%.
Scheme 6: Transformation of the ester 24 into the appropriate free acid 25. Reagents and conditions: i) two f...
The reactions of substrates 1 and 6 with imine 26, which is available from valine [33], readily gave adducts 27 and 28, which were decarboxylated under acidolysis to afford the phosphinic acid analogues of valine 14d and 20d (Scheme 7).
![[1860-5397-10-66-i7]](/bjoc/content/inline/1860-5397-10-66-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction of the acids (1) and (6) with methyl 2-imino-3-methylbutanoate (26). Reagents and conditions: i) an equimolar mixture of reagents, DME, rt, 31P NMR control, under argon atmosphere, isolated yields: 27 (63%), 28 (40%).
Scheme 7: Reaction of the acids (1) and (6) with methyl 2-imino-3-methylbutanoate (26). Reagents and conditio...
The hydrophosphinylation of substrates with activated C=C double bonds
The high reactivity of (trifluoromethyl)phosphinic acid (1) with C=N double bonds prompted us to explore its reactivity towards activated C=C double bonds. By analogy with the synthesis of the phosphonic acid analogue of aspartic acid, developed by Chambers and Isbell [34], the P–H substrate 1 was reacted with N-Boc-protected aminoacrylate 29 [35], and this gave the precursor of the aspartic acid analogue 30 (Scheme 8).
![[1860-5397-10-66-i8]](/bjoc/content/inline/1860-5397-10-66-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Interaction of the acid 1 with ethyl 2-(tert-butoxycarbonylamino)acrylate (29). Reagents and conditions: i) an equimolar mixture of reagents, DME, rt, 31P NMR control, under argon atmosphere, 59%. ii) 5 N HCl, rt, 48 h, ion-exchange chromatography, H2O, 54%.
Scheme 8: Interaction of the acid 1 with ethyl 2-(tert-butoxycarbonylamino)acrylate (29). Reagents and condit...
Surprisingly, under the mild conditions of our experiment only the addition of acid 1 to the C=N double bond of the acrylic ester 29, occurred to produce the tertiary phosphinyl derivative of alanine 31 in a satisfactory yield. Ester 31 was then hydrolyzed to give the free acid 32 in a moderate yield.
For the synthesis of the aspartic acid analogue 34, a reaction between a mixture of the freshly prepared esters 3 and 4 and N-acetyl-protected aminoacrylic acid 33 was carried out [34] (Scheme 9).
![[1860-5397-10-66-i9]](/bjoc/content/inline/1860-5397-10-66-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Interaction of a mixture of the esters 3 and 4 with 2-acetamidoacrylic acid (33). Reagents and conditions: i) an equimolar mixture of 4 and 33 and 1.5 equiv of 3, rt, 31P NMR control, under argon atmosphere, isolated yields: 35 (7%), 36 (40%) (diastereomeric ratio ~8:7), 37 (18%). ii) 5 N HCl, rt, 24 h, 95%. iii) 5 N HCl, in an ampoule, 130 °C, 8 h, 34%.
Scheme 9: Interaction of a mixture of the esters 3 and 4 with 2-acetamidoacrylic acid (33). Reagents and cond...
It was thought that diester 3 might esterify amidoacrylic acid 33 [17,34] to produce ethyl 2-acetamidoacrylate and this compound in turn might add to monoester 4 to give the protected phosphinic acid analogue of aspartic acid 34. Unfortunately, the only addition of 4 to the C=N double bond occurred similar to the previous transformation illustrated in Scheme 8. Insoluble in the reaction mixture phosphinic acid 37 was filtered off and characterized. 31P NMR analysis of filtrate showed the presence of adduct 35 and the decarboxylation product 36 in an approximate 1:10 ratio along with starting esters 3 and 4 and (trifluoromethyl)phosphonic acid (10) (<5%). Products 35 and 36 were separated by chromatography and characterized. Ester 36 was obtained as a mixture of two diastereoisomers, which are clearly seen by 1H- and 19F NMR. This ester was then readily converted by acidolysis into the phosphinic acid analogue 37, of N-acetylalanine, which was isolated in the 56% from acid 33, and then transformed into free amino acid 14c.
We have been able to prepare the isomeric aspartic acid analogue 41 with phosphorous α- to the amino group by the analogy with the published method [17,36]. The synthesis of phosphinic acid 41 was accomplished by addition of acid 1 to the activated C=C double bond of malonate 38 followed by hydrolysis and decarboxylation to generate adduct 39 in two steps (Scheme 10).
![[1860-5397-10-66-i10]](/bjoc/content/inline/1860-5397-10-66-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Interaction of a mixture of the acid 1 with diethyl acetaminomethylenemalonate (38). Reagents and conditions: i) an equimolar mixture of reagents, acetonitrile, rt, 31P NMR control, under argon atmosphere, 44%. ii) 5 N HCl, reflux, 12 h, 82%. iii) 5 N HCl, in an ampoule, 130 °C, 5 h, 43%.
Scheme 10: Interaction of a mixture of the acid 1 with diethyl acetaminomethylenemalonate (38). Reagents and c...
Conclusion
In conclusion, we have presented a variety of approaches to novel fluorinated (1-aminoalkyl)phosphinic acids starting from the appropriate fluorinated P–H compounds with CF3 or CHF2 groups attached to phosphorus. Three-component one pot Mannich-type reactions of CF3(H)P(O)(OH) with dibenzylamine and aldehydes were investigated. Also nucleophilic addition of CF3(H)P(O)(OH) or CHF2(H)P(O)(OEt) to Shiff bases, aminoacrylates and acetaminomethylenemalonate have been used to prepare the title compounds.
Experimental
All reactions with P–H compounds were performed under an argon atmosphere. Flash chromatography was carried out using Merck silica gel 60 (230–400 mesh ASTM) and Aldrich ion-exchange resin Dowex WX-50. The NMR spectra were recorded on Varian VXR-300 or Bruker Avance DRX-500 spectrometers for 1H (TMS); on a Bruker Avance DRX-500 spectrometer for 13C {H} (TMS); on Varian Gemini-200 or Varian VXR-300 spectrometers for 19F (CFCl3) and for 31P (H3PO4).
Synthesis of starting materials
(Trifluoromethyl)phosphinic acid (1). To an emulsion of water (3.2 g, 180 mmol) in anhydrous hexane (20 mL), cooled to −78 °C CF3PCl2 [27] (16.5 g, 96.5 mmol) was added under stirring and the temperature was slowly raised to −10 °C, when hydrolysis started. The reaction mixture was allowed to come to 0 °C at such a rate to avoid a vigorous reaction (~3 h) and then to room temperature and stirring was continued overnight. Hexane was evaporated under reduced pressure and the residue was distilled to give 1 as a colorless liquid (10.84 g, 84%), bp 35 °C (0.05 mm Hg); 1H NMR (300 MHz, DMSO-d6) δH 7.05 (dq, 1JHP = 638.9 Hz, 1H, 3JHF = 4.2 Hz, PH), 13.6 (1H, s, OH); 31P NMR (121 MHz) δP 6.1 (dq, 1JPH = 639 Hz, 2JPF = 82 Hz); 19F NMR (188 MHz) δF −76.8 (dd, 2JFP = 82 MHz, 3JFH = 4 Hz). Caution: Safety precautions are necessary, because CF3PCl2 reacts violently with air. Care must be taken not to warm the reaction system rapidly, because rapid volatilization of gaseous HCl will be accompanied by carrying off CF3PCl2, which can inflame.
(Difluoromethyl)phosphinic acid (6). A mixture of 5 [18] (8 g, 56 mmol) and NaHCO3 (7 g, 83 mmol) in ether (50 mL) was stirred overnight at room temperature to produce a bulky precipitate of CHF2P(O)H(O)−Na+ [31P NMR (121 MHz, H2O): δP 11.9 (dtd, 1JPH = 570 Hz, 2JPF = 87 Hz, 2JPH = 25 Hz]. This precipitate was filtered, thoroughly washed with ether, solved in water (25 mL) and passed down an ion-exchange column. Water from the resulting solution was evaporated under reduced pressure and the residue was kept in vacuo (0.05 mmHg) for 24 h at room temperature to give 6 as a viscous colorless undistillable liquid (7.19 g, 78%); Anal. calcd for CH3F2O2P: C, 10.35; H, 2.61; P, 26.71; found: C, 10.48; H, 2.70; P, 26.59; 1H NMR (300 MHz, DMSO-d6) δH 6.15 (tdd, 2JHF = 48.6 Hz, 2JHP = 24.5 Hz, 3JHH = 1.5 Hz, 1H, CHF2), 6.9 (dm, 1JHP = 566.5 Hz, 1H, PH), 12.7 (s, 1H, OH); 31P NMR (121 MHz) δP 12.1 (dtd, 1JPH = 566 Hz, 2JPF = 86 Hz, 2JPH = 25 Hz); 19F NMR (188 MHz) δF 9.6 (dd, 2JFP = 86 Hz, 2JFH = 49 Hz ).
Three-component reactions
The general procedure for the condensation of the acid 1 with dibenzylamine and aldehydes (I). An equimolar mixture of 1 (2.68 g, 20 mmol) and dibenzylamine (3.94 g, 20 mmol) in 1 N HCl (20 mL) was heated at 80 °C under stirring. In the course of ~1 h aldehydes 12a,b were added with a syringe and the reaction mixture was kept at this temperature for additional 1 h. The resulting mixture was left overnight at room temperature to produce the precipitate, which was filtered, washed with acetone–water (10:1) and dried to afford 13a or 13b. The filtrate was evaporated to the dryness, the residue was triturated with acetone–water (10:1) to give an additional quantity of 13a,b.
[(Dibenzylamino)methyl](trifluoromethyl)phosphinic acid (13a). Following the general procedure (I) using 3.2 mL of 37% aqueous formaldehyde solution (20 mmol) 13a was obtained as a white solid (3.57 g, 52%), mp 229 °C; Anal. calcd for C16H17F3NO2P: C, 55.98; H, 4.99; N, 4.08; found: C, 55.69; H, 5.28; N. 4.15; 1H NMR (300 MHz, DMSO-d6) δH 2.94 (d, 2JHP = 9.3 Hz, 2H, CH2P), 4.45 (s, 4H, CH2Ph), 7.47–7.59 (m, 10H, Harom.); 31P NMR (121 MHz) δP 3.8 (qt, 2JPF = 81 Hz, 2JPH = 9 Hz); 19F NMR (188 MHz) δF −73.8 (d, 2JFP = 81 Hz).
The general procedure for N-deprotection of compounds with N-Bn function under the catalytic hydrogenation conditions (II). To a solution of compounds, containing N-Bn fragment (5 mmol) in ethanol (10 mL) 10% Pd/C (0.05 g) was added, and the mixture was hydrogenated at room temperature and normal pressure. After ~3 h the precipitation commenced, and water (5 mL) was added to dissolve this precipitate. The hydrogenation was then continued with a fresh portion of the catalyst (0.05 g) for a further 3 h. Last procedure was repeated whenever necessary and the reaction was left overnight. To the resulting mixture water was added until a white solid was fully dissolved, and the catalyst was then filtered off. The filtrate was evaporated to dryness; the residue was dissolved in acetone and allowed to stand at 5 °C until complete precipitation. The formed solid was filtered, washed with acetone and dried to give compounds with the free NH2 function.
(Aminomethyl)(trifluoromethyl)phosphinic acid (14a). Following the general procedure (II) 14a was obtained as a white powder (0.78 g, 95%); mp 192 °C; Anal. calcd for C2H5F3NO2P: C, 14.73; H, 3.09; N, 8.59; found: C, 14.69; H, 2.89; N. 8.42; 1H NMR (300 MHz, D2O) δH 3.14 (d, 2JHP = 11.4 Hz); 31P NMR (121 MHz) δP 12.2 (qt, 2JPF = 96 Hz, 2JPH = 11 Hz); 19F NMR (188 MHz) δF −76.1 (d, 2JFP = 96 Hz); 13C NMR (125 MHz) δC 34.8 (d, 1JCP = 105.6 Hz, CH2), 122,1 (qd, 1JCF = 316.0 Hz, 1JCP = 179.9 Hz, CF3).
Hydrophosphinylation of azomethines
The general procedure for the addition of acid 1 and ester 5 to substrates with the C=N double bond (III). An equimolar mixture of an imine and 1 or 5 in DME (10 mL for 5 mmol) was stirred at room temperature under 31P NMR control until the 31P signals of starting P–H compounds disappeared. Sometimes an appropriate adduct precipitated and this was filtered off. The reaction mixture or the filtrate was then evaporated to dryness and the residue was worked up as described below for the individual substances.
Ethyl [(benzylamino)(phenyl)methyl](difluoromethyl)phosphinate (18b, Table 2, entry 2). Following the general procedure (III) a crude solid, obtained from 5 (0.49 g, 3.4 mmol) and 16b (0.68g, 3.4 mmol) was extracted with boiling hexane (3 × 30 mL), this extract was evaporated to the dryness to afford 18b as a yellowish solid (0.67 g, 58%); mp 85–93 °C, as a mixture of two diastereoisomers in an approximately 1:3.5 ratio due to 1H NMR (300 MHz, CDCl3) δH 0.97 (t, 3JHH = 7.6 Hz, 0.7H, CH3, minor isomer), 1.25 (t, 3JHH = 7.6 Hz, 2.3H, CH3, major isomer), 2.17 (br s, 1H, NH), 3.46 (d, JAB = 12.6 Hz, 0.22H, CH2Ph, minor isomer), 3.51 (d, JAB = 12.6 Hz, 0.77H, CH2Ph, major isomer), 3.78 (d, JAB = 12.6 Hz, 1H, CH2Ph), 3.9 (dm, 2JHP = 16.9 Hz, 0.22H, PCH, minor isomer), 4.08 (d, 2JHP = 17.1 Hz, 0.8H, PCH, major isomer), 4.1–4.25 (m, 2H, OCH2), 5.88 (td, 2JHF = 49.2 Hz, 2JHP = 27.8 Hz, 0.8H, CHF2, major isomer), 6.25 (td, 2JHF = 49.3 Hz, 2JHP 27.6 Hz, 0.2H, CHF2, minor isomer), 7.15–7.40 (m, 10H, Harom); 31P NMR (81 MHz) δP 30.8 (m); 19F NMR (188 MHz) δF −132 to −140.5 (complex multiplet). To the viscous residue after extraction of 18b water (20 mL) was added, resulting solution was decolorized with activated charcoal, filtrated and allowed to stand at 5 °C until crystallization completed, producing [(benzylamino)(phenyl)methyl](difluoromethyl)phosphinic acid (19b) as a white solid (0.32 g, 30%); mp 247 °C; Anal. calcd for C15H16F2NO2P: C, 57.88; H, 5.18; N, 4.50; found: C, 57.91; H, 5.04; N, 4.48; 1H NMR (300 MHz, DMSO-d6) δH 3.95 (d, JAB = 12.9 Hz, 1H, CH2Ph), 4.04 (d, 2JHP = 10.2 Hz, 1H, PCH), 4.10 (d, JAB = 12.9 Hz, 1H, CH2Ph), 5.63 (td, 2JHF = 49.2 Hz, 2JHP = 21.9 Hz, 1H, CHF2), 7.30–7.42 (m, 10H, Harom); 31P NMR (81 MHz) δP 13.4 (tm, 2JPF 68 Hz). An additional quantity of 19b was obtained by hydrolysis of 18b (0.67 g, 2 mmol) with 1N HCl (15 mL) at room temperature until the starting ester has dissolved. The resulting solution was evaporated to dryness at reduced pressure and the residue was recrystallized from water to give 19b (0.6 g, 97%). The overall yield of 19b is 0.92 g (87%).
See Supporting Information for details of the syntheses, characteristics and NMR spectra of all new compounds.
Supporting Information
Experimental procedures and full characterization data for all new compounds including elemental analysis and 1H, 31P, 19F and 13C NMR are provided in the Supporting Information.
| Supporting Information File 1: Experimental procedures, elemental analysis and NMR data. | ||
| Format: PDF | Size: 219.4 KB | Download |
| Supporting Information File 2: NMR spectra of the most typical compounds. | ||
| Format: PDF | Size: 2.2 MB | Download |
| Supporting Information File 3: NMR spectra of the most typical compounds (continuation). | ||
| Format: PDF | Size: 915.5 KB | Download |
References
-
Kukhar, V. P.; Hudson, H. R., Eds. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; John Willey & Sons, Ltd: Chichester, U.K., 2000.
Return to citation in text: [1] -
Ordóñez, M.; Viveros-Ceballos, J. L.; Cativiela, C.; Arizpe, A. Curr. Org. Synth. 2012, 9, 310–341. doi:10.2174/157017912801270595
Return to citation in text: [1] -
Ordóñez, M.; Sayago, F. J.; Cativela, C. Tetrahedron 2012, 68, 6369–6412. doi:10.1016/j.tet.2012.05.008
Return to citation in text: [1] -
Kudzin, Z. H.; Kudzin, M. H.; Drabowicz, J.; Stevens, C. V. Curr. Org. Chem. 2011, 15, 2015–2071. doi:10.2174/138527211795703612
Return to citation in text: [1] -
Palacios, F.; Alonso, C.; de los Santos, J. M. Chem. Rev. 2005, 105, 899–932. doi:10.1021/cr040672y
Return to citation in text: [1] [2] -
Fields, S. C. Tetrahedron 1999, 55, 12237–12273. doi:10.1016/S0040-4020(99)00701-2
Return to citation in text: [1] -
Welch, J. T.; Eswarakrishman, S., Eds. Fluorine in Bioorganic Chemistry; John Willey & Sons, Inc.: New York, 1991.
Return to citation in text: [1] -
Hiyama, T., Ed. Organofluorine Compounds. Chemistry and Applications; Springer: Berlin, 2000.
Return to citation in text: [1] -
Banks, R. E.; Smart, B. E.; Tatlow, J. C., Eds. Organofluorine Chemistry. Principles and Commercial Applications; Plenum: New York, 1994.
Return to citation in text: [1] -
Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q
Return to citation in text: [1] -
Bayer, E.; Gugel, K. H.; Hägele, M.; Hagenmaier, M.; Jessipow, S.; König, A. W.; Zähner, H. Helv. Chim. Acta 1972, 55, 224–239. doi:10.1002/hlca.19720550126
Return to citation in text: [1] [2] -
Mater, L. Phosphorus Sulfur Relat. Elem. 1983, 14, 295–322. doi:10.1080/03086648308073262
Return to citation in text: [1] -
Popoff, L. C.; Huber, L. K.; Block, B. P.; Morton, P. D.; Riordan, R. P. J. Org. Chem. 1963, 28, 2898–2900. doi:10.1021/jo01045a516
Return to citation in text: [1] -
Oleksyszyn, J.; Tyka, R.; Mastalerz, P. Synthesis 1978, 479–480. doi:10.1055/s-1978-24792
Return to citation in text: [1] [2] -
Wasielewski, C.; Antczak, K.; Rachon, J. Pol. J. Chem. 1978, 52, 1315.
Chem. Abstr. 1978, 89, 163655u.
Return to citation in text: [1] -
Subotkowski, W.; Tyka, R.; Mastalerz, P. Pol. J. Chem. 1980, 54, 503.
Chem. Abstr. 1980, 94, 30845n.
Return to citation in text: [1] -
Soroka, M.; Mastalerz, P. Rocz. Chem. 1976, 50, 661.
Chem. Abstr. 1976, 80, 121058k.
Return to citation in text: [1] [2] [3] -
Froestl, W.; Mickel, S. J.; Hall, R. G.; von Sprecher, G.; Strub, D.; Bauman, P. A.; Brugger, F.; Gentsch, C.; Jaekel, J.; Olpe, H.-R.; Rihs, G.; Vassout, A.; Waldmeier, P. C.; Bittiger, H. J. Med. Chem. 1995, 38, 3297–3312. doi:10.1021/jm00017a015
Return to citation in text: [1] [2] [3] [4] -
Kabachnic, M. I.; Medvedev, T. Y. Dokl. Akad. Nauk SSSR 1952, 83, 689.
Chem. Abstr. 1952, 46, 421c.
Return to citation in text: [1] [2] -
Fields, E. K. J. Am. Chem. Soc. 1952, 74, 1528–1531. doi:10.1021/ja01126a054
Return to citation in text: [1] [2] -
Moedritzer, K.; Irani, R. R. J. Org. Chem. 1966, 31, 1603–1607. doi:10.1021/jo01343a067
Return to citation in text: [1] [2] -
Redmore, D. In The chemistry of P-C-N systems; Griffith, E. J.; Graison, M., Eds.; Topics in Phosphorus Chemistry, Vol. 8; John Willey & Sons, Inc.: New York, 1976; pp 515–585.
Return to citation in text: [1] -
Emeléus, H. J.; Haszeldine, R. N.; Paul, R. C. J. Chem. Soc. 1955, 563–574. doi:10.1039/JR9550000563
Return to citation in text: [1] [2] [3] -
Maslennikov, I. G.; Lavrent`ev, A. N.; Hovanskaya, N. V.; Lebedev, V. B.; Sochilin, E. G. Zh. Obshch. Khim. 1979, 49, 1498–1500.
J. Gen. Chem. USSR 1979, 49, 1307–1309; Chem. Abstr. 1979, 91, 140924h.
Return to citation in text: [1] [2] -
Golovanov, A. V.; Maslennikov, I. G.; Lavrent`ev, A. N. Zh. Obshch. Khim. 1988, 58, 1525–1529.
J. Gen. Chem. USSR 1988, 58, 1357–1360; Chem. Abstr. 1988, 108, 21973h.
Return to citation in text: [1] [2] [3] -
Bennet, F. W.; Emeléus, H. J.; Haszeldine, R. N. J. Chem. Soc. 1953, 1565–1571. doi:10.1039/JR9530001565
Return to citation in text: [1] -
Volbach, W.; Ruppert, I. Tetrahedron Lett. 1983, 24, 5509–5512. doi:10.1016/S0040-4039(00)94125-X
Return to citation in text: [1] [2] [3] -
Pudovik, A. N.; Konovalova, I. V. Synthesis 1979, 81–96. doi:10.1055/s-1979-28566
Return to citation in text: [1] -
Mahmood, T.; Shreeve, J. M. Inorg. Chem. 1986, 25, 3128–3131. doi:10.1021/ic00238a006
Return to citation in text: [1] -
Blackburn, G. M.; Brown, D.; Martin, S. J.; Parratt, M. J. J. Chem. Soc., Perkin Trans. 1 1987, 181–186. doi:10.1039/p19870000181
Return to citation in text: [1] -
Vidal, J.; Guy, L.; Sterin, S.; Collet, A. J. Org. Chem. 1993, 58, 4791–4793. doi:10.1021/jo00070a007
Return to citation in text: [1] -
Jung, M. E.; Shishido, K.; Light, L.; Davis, L. Tetrahedron Lett. 1981, 22, 4607–4610. doi:10.1016/S0040-4039(01)82993-2
Return to citation in text: [1] -
Poisel, H.; Schmidt, U. Chem. Ber. 1975, 108, 2547–2553. doi:10.1002/cber.19751080809
Return to citation in text: [1] -
Chambers, J. R.; Isbell, A. F. J. Org. Chem. 1964, 29, 832–836. doi:10.1021/jo01027a015
Return to citation in text: [1] [2] [3] -
Ramesh, R.; De, K.; Chandrasekaran, S. Tetrahedron 2007, 63, 10534–10542. doi:10.1016/j.tet.2007.07.094
Return to citation in text: [1] -
Merret, J. H.; Spurden, W. C.; Thomas, W. A.; Tong, B. P.; Whitcombe, I. W. A. J. Chem. Soc., Perkin Trans. 1 1988, 61–67. doi:10.1039/P19880000061
Return to citation in text: [1]
| 28. | Pudovik, A. N.; Konovalova, I. V. Synthesis 1979, 81–96. doi:10.1055/s-1979-28566 |
| 29. | Mahmood, T.; Shreeve, J. M. Inorg. Chem. 1986, 25, 3128–3131. doi:10.1021/ic00238a006 |
| 30. | Blackburn, G. M.; Brown, D.; Martin, S. J.; Parratt, M. J. J. Chem. Soc., Perkin Trans. 1 1987, 181–186. doi:10.1039/p19870000181 |
| 1. | Kukhar, V. P.; Hudson, H. R., Eds. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; John Willey & Sons, Ltd: Chichester, U.K., 2000. |
| 2. | Ordóñez, M.; Viveros-Ceballos, J. L.; Cativiela, C.; Arizpe, A. Curr. Org. Synth. 2012, 9, 310–341. doi:10.2174/157017912801270595 |
| 3. | Ordóñez, M.; Sayago, F. J.; Cativela, C. Tetrahedron 2012, 68, 6369–6412. doi:10.1016/j.tet.2012.05.008 |
| 4. | Kudzin, Z. H.; Kudzin, M. H.; Drabowicz, J.; Stevens, C. V. Curr. Org. Chem. 2011, 15, 2015–2071. doi:10.2174/138527211795703612 |
| 5. | Palacios, F.; Alonso, C.; de los Santos, J. M. Chem. Rev. 2005, 105, 899–932. doi:10.1021/cr040672y |
| 6. | Fields, S. C. Tetrahedron 1999, 55, 12237–12273. doi:10.1016/S0040-4020(99)00701-2 |
| 12. | Mater, L. Phosphorus Sulfur Relat. Elem. 1983, 14, 295–322. doi:10.1080/03086648308073262 |
| 19. |
Kabachnic, M. I.; Medvedev, T. Y. Dokl. Akad. Nauk SSSR 1952, 83, 689.
Chem. Abstr. 1952, 46, 421c. |
| 20. | Fields, E. K. J. Am. Chem. Soc. 1952, 74, 1528–1531. doi:10.1021/ja01126a054 |
| 21. | Moedritzer, K.; Irani, R. R. J. Org. Chem. 1966, 31, 1603–1607. doi:10.1021/jo01343a067 |
| 34. | Chambers, J. R.; Isbell, A. F. J. Org. Chem. 1964, 29, 832–836. doi:10.1021/jo01027a015 |
| 11. | Bayer, E.; Gugel, K. H.; Hägele, M.; Hagenmaier, M.; Jessipow, S.; König, A. W.; Zähner, H. Helv. Chim. Acta 1972, 55, 224–239. doi:10.1002/hlca.19720550126 |
| 5. | Palacios, F.; Alonso, C.; de los Santos, J. M. Chem. Rev. 2005, 105, 899–932. doi:10.1021/cr040672y |
| 22. | Redmore, D. In The chemistry of P-C-N systems; Griffith, E. J.; Graison, M., Eds.; Topics in Phosphorus Chemistry, Vol. 8; John Willey & Sons, Inc.: New York, 1976; pp 515–585. |
| 17. |
Soroka, M.; Mastalerz, P. Rocz. Chem. 1976, 50, 661.
Chem. Abstr. 1976, 80, 121058k. |
| 34. | Chambers, J. R.; Isbell, A. F. J. Org. Chem. 1964, 29, 832–836. doi:10.1021/jo01027a015 |
| 10. | Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q |
| 18. | Froestl, W.; Mickel, S. J.; Hall, R. G.; von Sprecher, G.; Strub, D.; Bauman, P. A.; Brugger, F.; Gentsch, C.; Jaekel, J.; Olpe, H.-R.; Rihs, G.; Vassout, A.; Waldmeier, P. C.; Bittiger, H. J. Med. Chem. 1995, 38, 3297–3312. doi:10.1021/jm00017a015 |
| 34. | Chambers, J. R.; Isbell, A. F. J. Org. Chem. 1964, 29, 832–836. doi:10.1021/jo01027a015 |
| 7. | Welch, J. T.; Eswarakrishman, S., Eds. Fluorine in Bioorganic Chemistry; John Willey & Sons, Inc.: New York, 1991. |
| 8. | Hiyama, T., Ed. Organofluorine Compounds. Chemistry and Applications; Springer: Berlin, 2000. |
| 9. | Banks, R. E.; Smart, B. E.; Tatlow, J. C., Eds. Organofluorine Chemistry. Principles and Commercial Applications; Plenum: New York, 1994. |
| 18. | Froestl, W.; Mickel, S. J.; Hall, R. G.; von Sprecher, G.; Strub, D.; Bauman, P. A.; Brugger, F.; Gentsch, C.; Jaekel, J.; Olpe, H.-R.; Rihs, G.; Vassout, A.; Waldmeier, P. C.; Bittiger, H. J. Med. Chem. 1995, 38, 3297–3312. doi:10.1021/jm00017a015 |
| 35. | Ramesh, R.; De, K.; Chandrasekaran, S. Tetrahedron 2007, 63, 10534–10542. doi:10.1016/j.tet.2007.07.094 |
| 15. |
Wasielewski, C.; Antczak, K.; Rachon, J. Pol. J. Chem. 1978, 52, 1315.
Chem. Abstr. 1978, 89, 163655u. |
| 17. |
Soroka, M.; Mastalerz, P. Rocz. Chem. 1976, 50, 661.
Chem. Abstr. 1976, 80, 121058k. |
| 32. | Jung, M. E.; Shishido, K.; Light, L.; Davis, L. Tetrahedron Lett. 1981, 22, 4607–4610. doi:10.1016/S0040-4039(01)82993-2 |
| 14. | Oleksyszyn, J.; Tyka, R.; Mastalerz, P. Synthesis 1978, 479–480. doi:10.1055/s-1978-24792 |
| 11. | Bayer, E.; Gugel, K. H.; Hägele, M.; Hagenmaier, M.; Jessipow, S.; König, A. W.; Zähner, H. Helv. Chim. Acta 1972, 55, 224–239. doi:10.1002/hlca.19720550126 |
| 33. | Poisel, H.; Schmidt, U. Chem. Ber. 1975, 108, 2547–2553. doi:10.1002/cber.19751080809 |
| 14. | Oleksyszyn, J.; Tyka, R.; Mastalerz, P. Synthesis 1978, 479–480. doi:10.1055/s-1978-24792 |
| 21. | Moedritzer, K.; Irani, R. R. J. Org. Chem. 1966, 31, 1603–1607. doi:10.1021/jo01343a067 |
| 13. | Popoff, L. C.; Huber, L. K.; Block, B. P.; Morton, P. D.; Riordan, R. P. J. Org. Chem. 1963, 28, 2898–2900. doi:10.1021/jo01045a516 |
| 16. |
Subotkowski, W.; Tyka, R.; Mastalerz, P. Pol. J. Chem. 1980, 54, 503.
Chem. Abstr. 1980, 94, 30845n. |
| 31. | Vidal, J.; Guy, L.; Sterin, S.; Collet, A. J. Org. Chem. 1993, 58, 4791–4793. doi:10.1021/jo00070a007 |
| 26. | Bennet, F. W.; Emeléus, H. J.; Haszeldine, R. N. J. Chem. Soc. 1953, 1565–1571. doi:10.1039/JR9530001565 |
| 23. | Emeléus, H. J.; Haszeldine, R. N.; Paul, R. C. J. Chem. Soc. 1955, 563–574. doi:10.1039/JR9550000563 |
| 17. |
Soroka, M.; Mastalerz, P. Rocz. Chem. 1976, 50, 661.
Chem. Abstr. 1976, 80, 121058k. |
| 36. | Merret, J. H.; Spurden, W. C.; Thomas, W. A.; Tong, B. P.; Whitcombe, I. W. A. J. Chem. Soc., Perkin Trans. 1 1988, 61–67. doi:10.1039/P19880000061 |
| 24. |
Maslennikov, I. G.; Lavrent`ev, A. N.; Hovanskaya, N. V.; Lebedev, V. B.; Sochilin, E. G. Zh. Obshch. Khim. 1979, 49, 1498–1500.
J. Gen. Chem. USSR 1979, 49, 1307–1309; Chem. Abstr. 1979, 91, 140924h. |
| 25. |
Golovanov, A. V.; Maslennikov, I. G.; Lavrent`ev, A. N. Zh. Obshch. Khim. 1988, 58, 1525–1529.
J. Gen. Chem. USSR 1988, 58, 1357–1360; Chem. Abstr. 1988, 108, 21973h. |
| 27. | Volbach, W.; Ruppert, I. Tetrahedron Lett. 1983, 24, 5509–5512. doi:10.1016/S0040-4039(00)94125-X |
| 18. | Froestl, W.; Mickel, S. J.; Hall, R. G.; von Sprecher, G.; Strub, D.; Bauman, P. A.; Brugger, F.; Gentsch, C.; Jaekel, J.; Olpe, H.-R.; Rihs, G.; Vassout, A.; Waldmeier, P. C.; Bittiger, H. J. Med. Chem. 1995, 38, 3297–3312. doi:10.1021/jm00017a015 |
| 18. | Froestl, W.; Mickel, S. J.; Hall, R. G.; von Sprecher, G.; Strub, D.; Bauman, P. A.; Brugger, F.; Gentsch, C.; Jaekel, J.; Olpe, H.-R.; Rihs, G.; Vassout, A.; Waldmeier, P. C.; Bittiger, H. J. Med. Chem. 1995, 38, 3297–3312. doi:10.1021/jm00017a015 |
| 19. |
Kabachnic, M. I.; Medvedev, T. Y. Dokl. Akad. Nauk SSSR 1952, 83, 689.
Chem. Abstr. 1952, 46, 421c. |
| 20. | Fields, E. K. J. Am. Chem. Soc. 1952, 74, 1528–1531. doi:10.1021/ja01126a054 |
| 23. | Emeléus, H. J.; Haszeldine, R. N.; Paul, R. C. J. Chem. Soc. 1955, 563–574. doi:10.1039/JR9550000563 |
| 25. |
Golovanov, A. V.; Maslennikov, I. G.; Lavrent`ev, A. N. Zh. Obshch. Khim. 1988, 58, 1525–1529.
J. Gen. Chem. USSR 1988, 58, 1357–1360; Chem. Abstr. 1988, 108, 21973h. |
| 23. | Emeléus, H. J.; Haszeldine, R. N.; Paul, R. C. J. Chem. Soc. 1955, 563–574. doi:10.1039/JR9550000563 |
| 24. |
Maslennikov, I. G.; Lavrent`ev, A. N.; Hovanskaya, N. V.; Lebedev, V. B.; Sochilin, E. G. Zh. Obshch. Khim. 1979, 49, 1498–1500.
J. Gen. Chem. USSR 1979, 49, 1307–1309; Chem. Abstr. 1979, 91, 140924h. |
| 25. |
Golovanov, A. V.; Maslennikov, I. G.; Lavrent`ev, A. N. Zh. Obshch. Khim. 1988, 58, 1525–1529.
J. Gen. Chem. USSR 1988, 58, 1357–1360; Chem. Abstr. 1988, 108, 21973h. |
| 27. | Volbach, W.; Ruppert, I. Tetrahedron Lett. 1983, 24, 5509–5512. doi:10.1016/S0040-4039(00)94125-X |
| 27. | Volbach, W.; Ruppert, I. Tetrahedron Lett. 1983, 24, 5509–5512. doi:10.1016/S0040-4039(00)94125-X |
© 2014 Pavlenko et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)