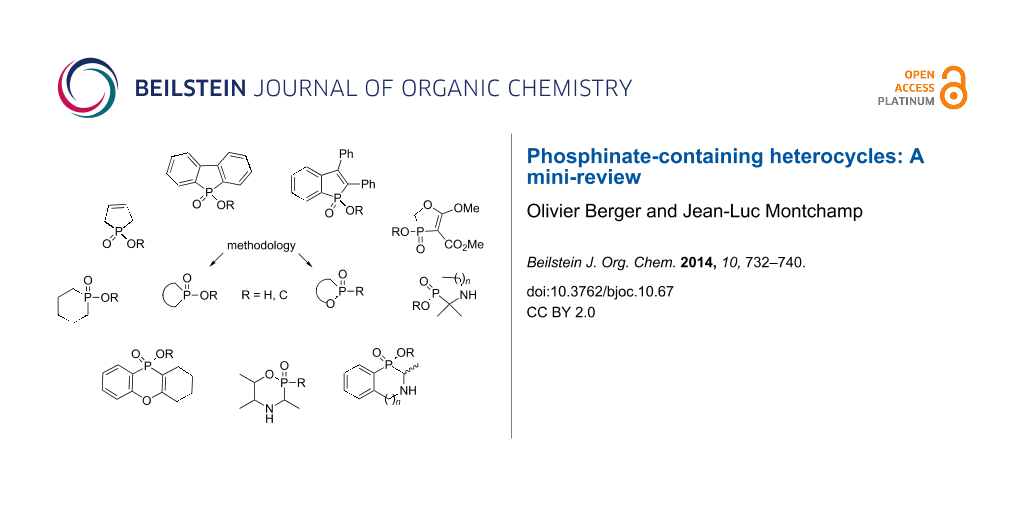
Abstract
This review provides an overview of recent efforts towards the synthesis of phosphinate heterocycles R1R2P(O)(OR). Our laboratory and others’ have been involved in this field and as a result new P–C, P–N, and P–O containing heterocyclic motifs are now available through a variety of methods. While developing rapidly, this area is still in its infancy so that biological testing of the compounds has not yet been conducted and applications are rare. The growing availability of synthetic methods will undoubtedly change this situation in the near future.

Graphical Abstract
Introduction
The preparation of P-heterocycles has been the subject of many studies over the years, and the field has been extensively reviewed [1-8]. Typically, accessing P-heterocycles involves multistep sequences with low overall yields [1-8]. In the past 20 years, significant effort has been devoted to synthetic and reactivity studies of a particular family of organophosphorus compounds: the phosphinates R1R2P(O)(OR) [9]. Because the phosphinic acid moiety P(O)OH can mimic carboxylic acids, its incorporation into heterocycles may offer new opportunities for the discovery of biologically active analogs. However, little or no biological data is available at this time. Selected recent synthetic work by us and others is presented below.
Review
Phospholes
Several compounds have been prepared in this series. Keglevich and coworkers realized the synthesis of phosphole derivatives 2a–f based on the McCormack reaction [10] followed by microwave-assisted esterification of the phosphinic acid using different alcohols in large excess (Scheme 1) [11,12]. Six phospholes 2a–f were prepared in yields up to 94%.
![[1860-5397-10-67-i1]](/bjoc/content/inline/1860-5397-10-67-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Montchamp and coworkers have synthesized phospholes 4a,b by ring closing metathesis using 2 or 5 mol % of 2nd generation Grubbs’ catalyst (Scheme 2) [13,14]. Two compounds 4a,b were prepared in 51% and 62% yields. The same approach was reported earlier by Mioskowski and coworkers [15,16] except the starting phosphinates 3a,b were prepared less efficiently by the sila-Arbuzov reaction of bis(trimethylsiloxy)phosphine (Me3SiO)2PH.
![[1860-5397-10-67-i2]](/bjoc/content/inline/1860-5397-10-67-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Phosphindoles
Montchamp and coworkers have synthesized a few phosphindoles. The first phosphindole 6 was simply obtained in 84% yield by reacting an α,ω-bisphosphonate derivative 5 with n-butyllithium in a phospha-Dieckmann condensation (Scheme 3) [17].
![[1860-5397-10-67-i3]](/bjoc/content/inline/1860-5397-10-67-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Phospha-Dieckmann condensation.
Scheme 3: Phospha-Dieckmann condensation.
Cyclohexyl 2-(biphenyl)-H-phosphinate 7 was cyclized using 2 mol % of Pd(OAc)2 in refluxing THF to produce another phosphindole 8 in 48% yield (Scheme 4) [18].
![[1860-5397-10-67-i4]](/bjoc/content/inline/1860-5397-10-67-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Palladium-catalyzed oxidative arylation.
Scheme 4: Palladium-catalyzed oxidative arylation.
A phosphindol-3-one 11 was prepared in 54% yield from butylphosphinate 9 by first methylation using DBU and iodomethane followed by a cross-coupling with ethyl 2-bromobenzoate (10) and then a Dieckmann-like condensation using LiHMDS (Scheme 5) [19].
![[1860-5397-10-67-i5]](/bjoc/content/inline/1860-5397-10-67-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Tandem cross-coupling/Dieckmann condensation.
Scheme 5: Tandem cross-coupling/Dieckmann condensation.
Tanaka and coworkers have synthesized chiral benzopyrano and naphthopyrano-fused helical phosphafluorenes 14a–d from dialkynyl phosphinate 12 and phenol-linked terminal tetrayne 13 at room temperature for only 1 h using a cationic rhodium(I)/(R)-tol-BINAP complex as a catalyst. Four helical phosphafluorenes 14a–d were prepared in yields up to 40% and enantiomeric excesses up to 73% (Scheme 6) [20].
![[1860-5397-10-67-i6]](/bjoc/content/inline/1860-5397-10-67-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Rhodium-catalyzed double [2 + 2 + 2] cycloaddition.
Scheme 6: Rhodium-catalyzed double [2 + 2 + 2] cycloaddition.
Chen and Duan have synthesized one phosphinoline 17 in 60% yield by the alkyne–arene annulation of ethyl phenyl-H-phosphinate (15) using 2 equivalents of Ag2O (Scheme 7) [21]. Miura et al. simultaneously reported the same reaction but with 4 equivalents of AgOAc instead, delivering the heterocycle 17 in 53% yield (Scheme 8) [22]. Both reactions used 4 equivalents of Ag(I) as well as an excess of H-phosphinate.
![[1860-5397-10-67-i7]](/bjoc/content/inline/1860-5397-10-67-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Silver oxide-mediated alkyne–arene annulation.
Scheme 7: Silver oxide-mediated alkyne–arene annulation.
![[1860-5397-10-67-i8]](/bjoc/content/inline/1860-5397-10-67-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Silver acetate-mediated alkyne–arene annulation.
Scheme 8: Silver acetate-mediated alkyne–arene annulation.
1,3-Oxaphospholes
Cristau and coworkers have achieved the direct synthesis of 1,3-oxaphospholes 20a–f (Scheme 9) by reacting chloroalkylphosphinic or phosphonic chlorides 18 with malonic diester 19 in the presence of two equivalents of sodium hydride [23,24]. 1,3-Oxaphospholes 20a–f were obtained in yields up to 70%.
![[1860-5397-10-67-i9]](/bjoc/content/inline/1860-5397-10-67-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Cyclization through phosphinylation/alkylation of malonate anion.
Scheme 9: Cyclization through phosphinylation/alkylation of malonate anion.
1-Aza-3-phospha-6-oxabicyclo[3.3.0]octanes
The synthesis of chiral bicyclic phosphinates 23a–k by domino hydrophosphinylation/Michael/Michael reaction was realized by Fourgeaud et al. (Scheme 10) [25].
![[1860-5397-10-67-i10]](/bjoc/content/inline/1860-5397-10-67-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Tandem hydrophosphinylation/Michael/Michael reaction of allenyl-H-phosphinates.
Scheme 10: Tandem hydrophosphinylation/Michael/Michael reaction of allenyl-H-phosphinates.
Several 1-oxa-3-aza-6-phosphabicyclo[3.3.0]octanes derivatives 23a–k were obtained in yields around 70% by reacting allenes 21 with imines 22 derived from (R)- or (S)-phenylglycinol, (S)-2-aminobutanol or ethanolamine. Diastereoisomeric ratios were generally close to 50:50. A model for this reaction’s diastereoselectivity was proposed.
Cyclo-PALA
Montchamp and coworkers have achieved the synthesis of 5- and 6-membered rings “cyclo-PALA” analogs which are 1,3-azaphospholidine and 1,4-azaphosphorine derivatives 26, 29 [26].
For the 5-membered ring 26, hydroxymethyl-H-phosphinic acid (24) underwent a sila-Arbuzov reaction with the bromide 25, the crude mixture was esterified with diphenyldiazomethane, cyclized using Mitsunobu conditions and then hydrogenolyzed to produce the five-membered amide 26 in 22% overall yield (Scheme 11).
![[1860-5397-10-67-i11]](/bjoc/content/inline/1860-5397-10-67-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: 5-Membered “cyclo-PALA” via intramolecular Mitsunobu reaction.
Scheme 11: 5-Membered “cyclo-PALA” via intramolecular Mitsunobu reaction.
For the six-membered “cyclo-PALA” 29, isoprenyl-H-phosphinic acid (27) reacted with the bromide 25 under sila-Arbuzov conditions, the crude phosphinic acid was esterified, using BnBr/Ag2O, ozonolyzed and then reduced with sodium borohydride to afford an alcohol intermediate 28. This product was cyclized using Mitsunobu conditions and finally hydrogenolyzed to deliver the 6-membered heterocycle 29 in 12% overall yield (Scheme 12) [26].
![[1860-5397-10-67-i12]](/bjoc/content/inline/1860-5397-10-67-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: 6-Membered “cyclo-PALA” via intramolecular Mitsunobu reaction.
Scheme 12: 6-Membered “cyclo-PALA” via intramolecular Mitsunobu reaction.
In this particular study phosphinates 26 and 29 were tested as inhibitors of aspartate transcarbamoylase (ATCase). 5-Membered 26 was completely inactive, whereas 6-membered 29 showed modest activity (Ki = 1 μM, 63 times less active than phosphonic acid N-phosphonacetyl-L-aspartate PALA, Ki = 16 nM).
1,3-Azaphosphorines and 1,3-azaphospholidines
Several 1,3-azaphosphorines and 1,3-azaphospholidines were synthesized by Montchamp and coworkers. The reaction of 2-aminoethyl-H-phosphinate 30a (n = 1) with carbonyl compounds 31 in refluxing butanol or concentrated hydrochloric acid took place smoothly to generate seven 1,3-azaphospholidines 32a–g in yields up to 55% (Scheme 13) [27,28].
![[1860-5397-10-67-i13]](/bjoc/content/inline/1860-5397-10-67-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Intramolecular Kabachnik–Fields reaction.
Scheme 13: Intramolecular Kabachnik–Fields reaction.
The reaction of 3-aminopropyl-H-phosphinate 30b with aldehydes 31 in refluxing butanol allowed the formation of eight 1,3-azaphosphorines 32h–o in yields up to 76% (Scheme 13).
Montchamp and coworkers also prepared two other examples of 1,3-azaphosphorines 35a,b (n = 1) in yields up to 61% by reacting ethyl-3-chloropropyl-H-phosphinate 33 with imines 34 in toluene at reflux (Scheme 14) [29].
![[1860-5397-10-67-i14]](/bjoc/content/inline/1860-5397-10-67-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Tandem Kabachnik–Fields/alkylation reaction.
Scheme 14: Tandem Kabachnik–Fields/alkylation reaction.
1,3-Azaphosphindoles and 1,3-benzazaphosphorines
Several compounds in this series were synthesized by Montchamp and coworkers using two different approaches. The first one is the reaction between an imine 34 and 2-bromophenyl-substituted H-phosphinate esters 36 in the presence of Cs2CO3, and catalytic Pd(PPh3)4 in refluxing toluene to generate the corresponding cyclized products 37a–h in yields up to 76% (Scheme 15) [29].
![[1860-5397-10-67-i15]](/bjoc/content/inline/1860-5397-10-67-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Tandem Kabacknik–Fields/C–N cross-coupling reaction.
Scheme 15: Tandem Kabacknik–Fields/C–N cross-coupling reaction.
The second way is the formation of the imine first by reacting an amine 39a,b with an aldehyde 38, then the phosphinate is introduced and the mixture stirred for 24 h at reflux to generate the corresponding H-phosphinate esters. Addition of DIPEA and catalytic Pd/dppf in a mixture DMF/DME to the intermediates generated the corresponding cyclized derivatives 40a,b in yields up to 53% (Scheme 16) [18].
![[1860-5397-10-67-i16]](/bjoc/content/inline/1860-5397-10-67-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Tandem Kabacknik–Fields/C-P cross-coupling reaction.
Scheme 16: Tandem Kabacknik–Fields/C-P cross-coupling reaction.
For these compounds, the authors were able to separate the different diastereoisomers generated during the reaction by simple column chromatography on silica gel.
1,4-Azaphosphorines
In this series, only a few examples have been reported in the literature. One derivative has been prepared by Manthey and coworkers in 50% yield as a precursor to a dihydroorotase inhibitor (Scheme 17) [30].
![[1860-5397-10-67-i17]](/bjoc/content/inline/1860-5397-10-67-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Heterocyclization via amide formation.
Scheme 17: Heterocyclization via amide formation.
In this example, the amino acid 41 was first cyclized using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (42) at pH 5.6 followed by protection of the carboxylic acid and phosphinic acid moieties by diphenylmethyl group using a slight excess of diphenyldiazomethane. The two diastereoisomers obtained were readily separable by column chromatography.
Another example has been synthesized in 45% yield by Montchamp and coworkers (Scheme 18) [14].
![[1860-5397-10-67-i18]](/bjoc/content/inline/1860-5397-10-67-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Cyclization via reductive amination.
Scheme 18: Cyclization via reductive amination.
To prepare the required phosphinate 45 a double allylation of H3PO2 was performed using 2 equivalents of cinnamyl alcohol 44 in the presence of 2 mol % of Pd/Xanthpos followed by an esterification using benzyl bromide. Ozonolysis, and reductive amination using excess benzylamine in the presence of sodium cyanoborohydride completed the synthesis.
Phosphorines
Two phosphorines 47a,b were obtained by Montchamp and coworkers via the cyclization of 5-bromopentyl-H-phosphinate esters 46a,b in the presence of LiHMDS in 71% and 74% yields for the butyl and ethyl esters respectively (Scheme 19) [28,31].
![[1860-5397-10-67-i19]](/bjoc/content/inline/1860-5397-10-67-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Another phosphorine 49 was obtained by Montchamp and coworkers in 57% yield via the cyclization through conjugate addition of ethyl 7-(ethoxy-H-phosphinoyl)-3-methyl-2-heptenoate (48) in the presence of potassium tert-butoxide (Scheme 20) [28].
![[1860-5397-10-67-i20]](/bjoc/content/inline/1860-5397-10-67-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Cyclization through intramolecular Michael addition.
Scheme 20: Cyclization through intramolecular Michael addition.
A phosphorino[3’,4’:4,5]furo[2,3-d]-1,3-dioxole 51 was synthesized in 36% yield by Tattersall and coworkers by realizing a double Arbuzov-type reaction between bis(trimethylsiloxy)phosphine and the dibromide 50 followed by the esterification of the phosphinic acid using diazomethane (Scheme 21) [32]. The heterocyclization step followed methodology initially introduced by Frost et al [33].
![[1860-5397-10-67-i21]](/bjoc/content/inline/1860-5397-10-67-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Double Arbuzov reaction of bis(trimethylsiloxy)phosphine.
Scheme 21: Double Arbuzov reaction of bis(trimethylsiloxy)phosphine.
Compound 51 was subsequently converted into the corresponding analog of cyclic AMP, but no biological activity was reported.
1,2-Oxaphosphorines
Gouverneur and coworkers have realized the synthesis of several 1,2-oxaphosphorine derivatives 53a–k using diastereoselective ring closing metathesis with 2 to 4 mol % of various catalysts (Scheme 22) [34].
![[1860-5397-10-67-i22]](/bjoc/content/inline/1860-5397-10-67-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Diastereoselective ring-closing metathesis.
Scheme 22: Diastereoselective ring-closing metathesis.
During this work, they obtained 11 different compounds in yields up to 100% and diastereomeric excesses up to 86%. The starting phosphinates 52a–k were prepared using classical chemistry involving Grignard addition to EtOP(O)Cl2.
Phenoxaphosphine
Scheme 23 shows the synthesis of one phenoxaphosphine 56 in 55% yield by Li and coworkers via the reaction between diethyl 2-oxocyclohexylphosphonate (54) and benzyne generated from 2-(trimethylsilyl)phenyl triflate (55) and cesium fluoride [35].
![[1860-5397-10-67-i23]](/bjoc/content/inline/1860-5397-10-67-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: 2-Ketophosphonate/benzene annulation.
Scheme 23: 2-Ketophosphonate/benzene annulation.
1,4,2-Oxazaphosphinane
This series of compounds is only represented by few examples all generated through methodology developed by Pirat and coworkers. Scheme 24 shows the synthesis of a H-phosphinate intermediate 59 in 65% yield via the reaction between the imine 57 of the racemic 1,2-diphenylethanolamine with benzaldehyde and methyl phosphinate (58) followed by the cyclization through a base catalyzed transesterification [23,36].
![[1860-5397-10-67-i24]](/bjoc/content/inline/1860-5397-10-67-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Tandem Kabachnik–Fields/transesterification reaction.
Scheme 24: Tandem Kabachnik–Fields/transesterification reaction.
This versatile intermediate 59 was reacted with aldehydes, imines, olefins and aryl bromides or aryl iodides to generate a wide range of phosphinates.
The same authors have also prepared another H-phosphinate intermediate 61 in 71% yield (Scheme 25) [37].
![[1860-5397-10-67-i25]](/bjoc/content/inline/1860-5397-10-67-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Tandem Kabachnik–Fields/transesterification reaction with oxazolidine.
Scheme 25: Tandem Kabachnik–Fields/transesterification reaction with oxazolidine.
This oxazaphosphinane 61 was synthesized in two steps at room temperature, first, by a nucleophilic attack of methyl hypophosphite on oxazolidine 60 followed by an intramolecular cyclization, this time without base catalyzed transesterification. The authors explained this difference of reactivity by the Thorpe–Ingold effect [38]. Indeed, the presence of four methyl groups allows the hydroxy function to be spatially closer to the reactive phosphinate, facilitating the intramolecular cyclization of this product.
Conclusion
Phosphinate heterocycles are becoming routine products in the literature. Classical approaches such as the McCormack reaction of conjugated dienes, the sila-Arbuzov reaction of bis(trimethylsiloxy)phosphine with dihalides, etc. continue to be useful. However, novel approaches in both the preparation of acyclic precursors and the reactions to achieve their heterocyclization, have led to more efficient synthesis and broader structural diversity. While, like with any other P-heterocycles the phosphinates can be employed for the synthesis of novel phosphine ligands, their potential for the discovery of novel biologically active motifs is tantalizing.
Acknowledgements
This material is based in part upon work supported by the National Science Foundation under Grant No. 1262254. We also acknowledge the following coworkers for prior contributions to this field: Laëtitia Coudray, Patrice Ribière, Clemence Queffélec, Isabelle Abrunhosa-Thomas, Christelle Petit, Laurent Gavara, Stephanie Ortial, Henry C. Fisher, Fabien Gelat, and Karla Bravo-Altamirano.
References
-
Mathey, F., Ed. Phosphorus-Carbon Heterocyclic Chemistry: The Rise of a New Domain; Elsevier: Oxford, 2001.
Return to citation in text: [1] [2] -
Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley and Sons: New York, 2000.
Return to citation in text: [1] [2] -
Dillon, K. B.; Mathey, F.; Nixon, J. F. Phosphorus: the Carbon Copy; John Wiley and Sons: Chichester, 1998.
Return to citation in text: [1] [2] -
Katritzky, A. R.; Rees, C. W.; Scriven, E. F. C., Eds. Comprehensive Heterocyclic Chemistry II; Pergamon: New York, 1996.
Return to citation in text: [1] [2] -
Mathey, F. Chem. Rev. 1990, 90, 997–1025. doi:10.1021/cr00104a004
Return to citation in text: [1] [2] -
Quin, L. D. The Heterocyclic Chemistry of Phosphorus; John Wiley and Sons: Chichester, 1981.
Return to citation in text: [1] [2] -
Venkataramu, S. D.; MacDonell, G. D.; Purdum, W. R.; El-Deek, M.; Berlin, K. D. Chem. Rev. 1977, 77, 121–181. doi:10.1021/cr60306a001
Return to citation in text: [1] [2] -
Berlin, K. D.; Hellwege, D. M. Top. Phosphorus Chem. 1969, 6, 1–186.
Return to citation in text: [1] [2] -
Montchamp, J.-L. Acc. Chem. Res. 2014, 47, 77–87. doi:10.1021/ar400071v
Return to citation in text: [1] -
McCormack, W. B. Org. Synth. 1973, 5, 787.
Return to citation in text: [1] -
Kiss, N. Z.; Böttger, E.; Drahos, L.; Keglevich, G. Heteroat. Chem. 2013, 24, 283–288. doi:10.1002/hc.21092
Return to citation in text: [1] -
Keglevich, G.; Kiss, N. Z.; Mucsi, Z.; Körtvélyesi, T. Org. Biomol. Chem. 2012, 10, 2011–2018. doi:10.1039/c2ob06972e
Return to citation in text: [1] -
Bravo-Altamirano, K.; Abrunhosa-Thomas, I.; Montchamp, J.-L. J. Org. Chem. 2008, 73, 2292–2301. doi:10.1021/jo702542a
Return to citation in text: [1] -
Coudray, L.; Bravo-Altamirano, K.; Montchamp, J.-L. Org. Lett. 2008, 10, 1123–1126. doi:10.1021/ol8000415
Return to citation in text: [1] [2] -
Bujard, M.; Gouverneur, V.; Mioskowski, C. J. Org. Chem. 1999, 64, 2119–2123. doi:10.1021/jo981795j
Return to citation in text: [1] -
Briot, A.; Bujard, M.; Gouverneur, V.; Nolan, S. P.; Mioskowski, C. Org. Lett. 2000, 2, 1517–1519. doi:10.1021/ol005651e
Return to citation in text: [1] -
Gavara, L.; Gelat, F.; Montchamp, J.-L. Tetrahedron Lett. 2013, 54, 817–820. doi:10.1016/j.tetlet.2012.11.119
Return to citation in text: [1] -
Berger, O.; Petit, C.; Deal, E. L.; Montchamp, J.-L. Adv. Synth. Catal. 2013, 355, 1361–1373. doi:10.1002/adsc.201300069
Return to citation in text: [1] [2] -
Gavara, L.; Petit, C.; Montchamp, J.-L. Tetrahedron Lett. 2012, 53, 5000–5003. doi:10.1016/j.tetlet.2012.07.019
Return to citation in text: [1] -
Fukawa, N.; Osaka, T.; Noguchi, K.; Tanaka, K. Org. Lett. 2010, 12, 1324–1327. doi:10.1021/ol100227k
Return to citation in text: [1] -
Chen, Y.-R.; Duan, W.-L. J. Am. Chem. Soc. 2013, 135, 16754–16757. doi:10.1021/ja407373g
Return to citation in text: [1] -
Unoh, Y.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2013, 52, 12975–12979. doi:10.1002/anie.201307211
Return to citation in text: [1] -
Cristau, H.-J.; Pirat, J.-L.; Virieux, D.; Monbrun, J.; Ciptadi, C.; Bekro, Y.-A. J. Organomet. Chem. 2005, 690, 2472–2481. doi:10.1016/j.jorganchem.2004.11.035
Return to citation in text: [1] [2] -
Virieux, D.; Ciptadi, C.; Bekro, Y.-A.; Cristau, H.-J. Eur. J. Org. Chem. 2004, 3205–3211. doi:10.1002/ejoc.200400179
Return to citation in text: [1] -
Fourgeaud, P.; Daydé, B.; Volle, J.-N.; Vors, J.-P.; Van der Lee, A.; Pirat, J.-L.; Virieux, D. Org. Lett. 2011, 13, 5076–5079. doi:10.1021/ol2019345
Return to citation in text: [1] -
Coudray, L.; Pennebaker, A. F.; Montchamp, J.-L. Bioorg. Med. Chem. 2009, 17, 7680–7689. doi:10.1016/j.bmc.2009.09.045
Return to citation in text: [1] [2] -
Queffélec, C.; Ribière, P.; Montchamp, J.-L. J. Org. Chem. 2008, 73, 8987–8991. doi:10.1021/jo801768y
Return to citation in text: [1] -
Ortial, S.; Fisher, H. C.; Montchamp, J.-L. J. Org. Chem. 2013, 78, 6599–6608. doi:10.1021/jo4008749
Return to citation in text: [1] [2] [3] -
Queffélec, C.; Montchamp, J.-L. Org. Biomol. Chem. 2010, 8, 267–273. doi:10.1039/b917428a
Return to citation in text: [1] [2] -
Manthey, M. K.; Huang, D. T. C.; Bubb, W. A.; Christopherson, R. I. J. Med. Chem. 1998, 41, 4550–4555. doi:10.1021/jm970814z
Return to citation in text: [1] -
Abrunhosa-Thomas, I.; Sellers, C. E.; Montchamp, J.-L. J. Org. Chem. 2007, 72, 2851–2856. doi:10.1021/jo062436o
Return to citation in text: [1] -
Regan, A. C.; Sciammetta, N.; Tattersall, P. I. Tetrahedron Lett. 2000, 41, 8211–8215. doi:10.1016/S0040-4039(00)01437-4
Return to citation in text: [1] -
Montchamp, J.-L.; Tian, F.; Frost, J. W. J. Org. Chem. 1995, 60, 6076–6081. doi:10.1021/jo00124a018
Return to citation in text: [1] -
Dunne, K. S.; Bisaro, F.; Odell, B.; Paris, J.-M.; Gouverneur, V. J. Org. Chem. 2005, 70, 10803–10809. doi:10.1021/jo0518708
Return to citation in text: [1] -
Liu, Y.-L.; Liang, Y.; Pi, S.-F.; Li, J.-H. J. Org. Chem. 2009, 74, 5691–5694. doi:10.1021/jo900847u
Return to citation in text: [1] -
Cristau, H.-J.; Monbrun, J.; Tillard, M.; Pirat, J.-L. Tetrahedron Lett. 2003, 44, 3183–3186. doi:10.1016/S0040-4039(03)00446-5
Return to citation in text: [1] -
Volle, J.-N.; Kaloyanov, N.; Saada, M. C.; Virieux, D.; Pirat, J.-L. Tetrahedron Lett. 2007, 48, 4695–4697. doi:10.1016/j.tetlet.2007.05.014
Return to citation in text: [1] -
Jung, M. E.; Piizzi, G. Chem. Rev. 2005, 105, 1735–1766. doi:10.1021/cr940337h
Return to citation in text: [1]
| 34. | Dunne, K. S.; Bisaro, F.; Odell, B.; Paris, J.-M.; Gouverneur, V. J. Org. Chem. 2005, 70, 10803–10809. doi:10.1021/jo0518708 |
| 35. | Liu, Y.-L.; Liang, Y.; Pi, S.-F.; Li, J.-H. J. Org. Chem. 2009, 74, 5691–5694. doi:10.1021/jo900847u |
| 23. | Cristau, H.-J.; Pirat, J.-L.; Virieux, D.; Monbrun, J.; Ciptadi, C.; Bekro, Y.-A. J. Organomet. Chem. 2005, 690, 2472–2481. doi:10.1016/j.jorganchem.2004.11.035 |
| 36. | Cristau, H.-J.; Monbrun, J.; Tillard, M.; Pirat, J.-L. Tetrahedron Lett. 2003, 44, 3183–3186. doi:10.1016/S0040-4039(03)00446-5 |
| 1. | Mathey, F., Ed. Phosphorus-Carbon Heterocyclic Chemistry: The Rise of a New Domain; Elsevier: Oxford, 2001. |
| 2. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley and Sons: New York, 2000. |
| 3. | Dillon, K. B.; Mathey, F.; Nixon, J. F. Phosphorus: the Carbon Copy; John Wiley and Sons: Chichester, 1998. |
| 4. | Katritzky, A. R.; Rees, C. W.; Scriven, E. F. C., Eds. Comprehensive Heterocyclic Chemistry II; Pergamon: New York, 1996. |
| 5. | Mathey, F. Chem. Rev. 1990, 90, 997–1025. doi:10.1021/cr00104a004 |
| 6. | Quin, L. D. The Heterocyclic Chemistry of Phosphorus; John Wiley and Sons: Chichester, 1981. |
| 7. | Venkataramu, S. D.; MacDonell, G. D.; Purdum, W. R.; El-Deek, M.; Berlin, K. D. Chem. Rev. 1977, 77, 121–181. doi:10.1021/cr60306a001 |
| 8. | Berlin, K. D.; Hellwege, D. M. Top. Phosphorus Chem. 1969, 6, 1–186. |
| 11. | Kiss, N. Z.; Böttger, E.; Drahos, L.; Keglevich, G. Heteroat. Chem. 2013, 24, 283–288. doi:10.1002/hc.21092 |
| 12. | Keglevich, G.; Kiss, N. Z.; Mucsi, Z.; Körtvélyesi, T. Org. Biomol. Chem. 2012, 10, 2011–2018. doi:10.1039/c2ob06972e |
| 25. | Fourgeaud, P.; Daydé, B.; Volle, J.-N.; Vors, J.-P.; Van der Lee, A.; Pirat, J.-L.; Virieux, D. Org. Lett. 2011, 13, 5076–5079. doi:10.1021/ol2019345 |
| 26. | Coudray, L.; Pennebaker, A. F.; Montchamp, J.-L. Bioorg. Med. Chem. 2009, 17, 7680–7689. doi:10.1016/j.bmc.2009.09.045 |
| 22. | Unoh, Y.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2013, 52, 12975–12979. doi:10.1002/anie.201307211 |
| 1. | Mathey, F., Ed. Phosphorus-Carbon Heterocyclic Chemistry: The Rise of a New Domain; Elsevier: Oxford, 2001. |
| 2. | Quin, L. D. A Guide to Organophosphorus Chemistry; John Wiley and Sons: New York, 2000. |
| 3. | Dillon, K. B.; Mathey, F.; Nixon, J. F. Phosphorus: the Carbon Copy; John Wiley and Sons: Chichester, 1998. |
| 4. | Katritzky, A. R.; Rees, C. W.; Scriven, E. F. C., Eds. Comprehensive Heterocyclic Chemistry II; Pergamon: New York, 1996. |
| 5. | Mathey, F. Chem. Rev. 1990, 90, 997–1025. doi:10.1021/cr00104a004 |
| 6. | Quin, L. D. The Heterocyclic Chemistry of Phosphorus; John Wiley and Sons: Chichester, 1981. |
| 7. | Venkataramu, S. D.; MacDonell, G. D.; Purdum, W. R.; El-Deek, M.; Berlin, K. D. Chem. Rev. 1977, 77, 121–181. doi:10.1021/cr60306a001 |
| 8. | Berlin, K. D.; Hellwege, D. M. Top. Phosphorus Chem. 1969, 6, 1–186. |
| 23. | Cristau, H.-J.; Pirat, J.-L.; Virieux, D.; Monbrun, J.; Ciptadi, C.; Bekro, Y.-A. J. Organomet. Chem. 2005, 690, 2472–2481. doi:10.1016/j.jorganchem.2004.11.035 |
| 24. | Virieux, D.; Ciptadi, C.; Bekro, Y.-A.; Cristau, H.-J. Eur. J. Org. Chem. 2004, 3205–3211. doi:10.1002/ejoc.200400179 |
| 18. | Berger, O.; Petit, C.; Deal, E. L.; Montchamp, J.-L. Adv. Synth. Catal. 2013, 355, 1361–1373. doi:10.1002/adsc.201300069 |
| 20. | Fukawa, N.; Osaka, T.; Noguchi, K.; Tanaka, K. Org. Lett. 2010, 12, 1324–1327. doi:10.1021/ol100227k |
| 17. | Gavara, L.; Gelat, F.; Montchamp, J.-L. Tetrahedron Lett. 2013, 54, 817–820. doi:10.1016/j.tetlet.2012.11.119 |
| 21. | Chen, Y.-R.; Duan, W.-L. J. Am. Chem. Soc. 2013, 135, 16754–16757. doi:10.1021/ja407373g |
| 15. | Bujard, M.; Gouverneur, V.; Mioskowski, C. J. Org. Chem. 1999, 64, 2119–2123. doi:10.1021/jo981795j |
| 16. | Briot, A.; Bujard, M.; Gouverneur, V.; Nolan, S. P.; Mioskowski, C. Org. Lett. 2000, 2, 1517–1519. doi:10.1021/ol005651e |
| 37. | Volle, J.-N.; Kaloyanov, N.; Saada, M. C.; Virieux, D.; Pirat, J.-L. Tetrahedron Lett. 2007, 48, 4695–4697. doi:10.1016/j.tetlet.2007.05.014 |
| 13. | Bravo-Altamirano, K.; Abrunhosa-Thomas, I.; Montchamp, J.-L. J. Org. Chem. 2008, 73, 2292–2301. doi:10.1021/jo702542a |
| 14. | Coudray, L.; Bravo-Altamirano, K.; Montchamp, J.-L. Org. Lett. 2008, 10, 1123–1126. doi:10.1021/ol8000415 |
| 19. | Gavara, L.; Petit, C.; Montchamp, J.-L. Tetrahedron Lett. 2012, 53, 5000–5003. doi:10.1016/j.tetlet.2012.07.019 |
| 38. | Jung, M. E.; Piizzi, G. Chem. Rev. 2005, 105, 1735–1766. doi:10.1021/cr940337h |
| 29. | Queffélec, C.; Montchamp, J.-L. Org. Biomol. Chem. 2010, 8, 267–273. doi:10.1039/b917428a |
| 26. | Coudray, L.; Pennebaker, A. F.; Montchamp, J.-L. Bioorg. Med. Chem. 2009, 17, 7680–7689. doi:10.1016/j.bmc.2009.09.045 |
| 27. | Queffélec, C.; Ribière, P.; Montchamp, J.-L. J. Org. Chem. 2008, 73, 8987–8991. doi:10.1021/jo801768y |
| 28. | Ortial, S.; Fisher, H. C.; Montchamp, J.-L. J. Org. Chem. 2013, 78, 6599–6608. doi:10.1021/jo4008749 |
| 32. | Regan, A. C.; Sciammetta, N.; Tattersall, P. I. Tetrahedron Lett. 2000, 41, 8211–8215. doi:10.1016/S0040-4039(00)01437-4 |
| 33. | Montchamp, J.-L.; Tian, F.; Frost, J. W. J. Org. Chem. 1995, 60, 6076–6081. doi:10.1021/jo00124a018 |
| 28. | Ortial, S.; Fisher, H. C.; Montchamp, J.-L. J. Org. Chem. 2013, 78, 6599–6608. doi:10.1021/jo4008749 |
| 31. | Abrunhosa-Thomas, I.; Sellers, C. E.; Montchamp, J.-L. J. Org. Chem. 2007, 72, 2851–2856. doi:10.1021/jo062436o |
| 28. | Ortial, S.; Fisher, H. C.; Montchamp, J.-L. J. Org. Chem. 2013, 78, 6599–6608. doi:10.1021/jo4008749 |
| 30. | Manthey, M. K.; Huang, D. T. C.; Bubb, W. A.; Christopherson, R. I. J. Med. Chem. 1998, 41, 4550–4555. doi:10.1021/jm970814z |
| 14. | Coudray, L.; Bravo-Altamirano, K.; Montchamp, J.-L. Org. Lett. 2008, 10, 1123–1126. doi:10.1021/ol8000415 |
| 29. | Queffélec, C.; Montchamp, J.-L. Org. Biomol. Chem. 2010, 8, 267–273. doi:10.1039/b917428a |
| 18. | Berger, O.; Petit, C.; Deal, E. L.; Montchamp, J.-L. Adv. Synth. Catal. 2013, 355, 1361–1373. doi:10.1002/adsc.201300069 |
© 2014 Berger and Montchamp; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)