Abstract
Anodic fluorination of dithioacetals bearing electron-withdrawing ester, acetyl, amide, and nitrile groups at their α-positions was comparatively studied using various supporting poly(HF) salts like Et3N·nHF (n = 3–5) and Et4NF·nHF (n = 3–5). In the former two cases, the corresponding α-fluorination products or fluorodesulfurization products were obtained selectively depending on supporting poly(HF) salts used. In sharp contrast, in the latter two cases, fluorination product selectivity was strongly affected by the electron-withdrawing ability of α-substituents: A dithioacetal bearing a relatively weak electron-withdrawing amide group provided a fluorodesulfurization product selectively while a dithioacetal having a strongly electron-withdrawing nitrile group gave the α-fluorination product predominantly regardless of the poly(HF) salts used.

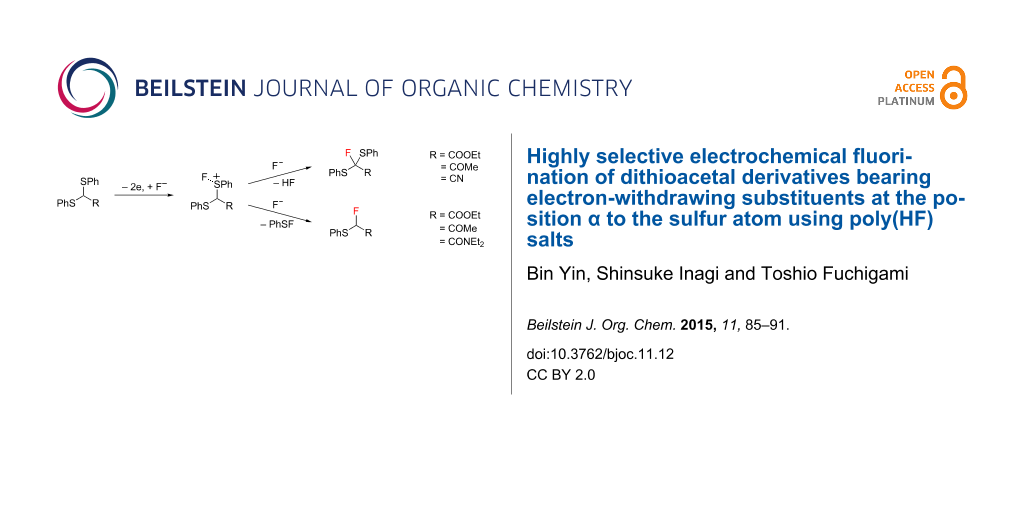
Graphical Abstract
Introduction
The introduction of fluorine atom(s) into organic molecules very often improves or enhances their desired characteristic physical and biological properties hence organofluorine compounds are highly useful for medicinal, agrochemical, and materials science [1-6]. In order to prepare new fluorine compounds, selective fluorination of organic compounds is becoming significantly important. Although the selective fluorination has been extensively studied, highly efficient and safe fluorination methods are still demanded [7,8]. With these facts in mind, we have developed a selective electrochemical fluorination using ionic liquid poly(HF) salts such as Et3N·nHF and Et4NF·nHF (n = 3–5) as supporting electrolyte and fluorine source [9-11], and we have systematically studied the anodic fluorination of various heteroatom-containing compounds including heterocycles and macromolecules so far [12-20].
More than 20 years ago, we reported the first successful example of the electrochemical selective fluorination of heteroatom-containing compounds such as α-(phenylthio)ester and its analogues as shown in Scheme 1 [21,22]. Furthermore, anodic fluorodesulfurization of dithioacetals was achieved by direct and indirect anodic oxidation in the presence of the poly(HF) salts [12-16,23-25] or alkali-metal fluorides like KF and CsF with PEG 200 [17]. The anodic fluorination of a dithioacetal derived from an aliphatic aldehyde provided the fluorodesulfurization product while a dithioacetal derived from an aromatic aldehyde provided the α-fluorination product (Scheme 2) [25]. These results suggest that the fluorinated product selectivity seems to be controlled by the easiness of the deprotonation of the cationic intermediate A. Namely, since the α-proton of the aromatic dithioacetal is more acidic compared to that of an aliphatic dithioacetal, the deprotonation of the former is faster than it is for the latter. Therefore, it can be stated that the product selectivity is controlled by the kinetic acidity of the cationic intermediate A [26,27].
![[1860-5397-11-12-i1]](/bjoc/content/inline/1860-5397-11-12-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Anodic fluorination of sulfides having an electron-withdrawing group.
Scheme 1: Anodic fluorination of sulfides having an electron-withdrawing group.
![[1860-5397-11-12-i2]](/bjoc/content/inline/1860-5397-11-12-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Anodic fluorination of dithioacetals.
Scheme 2: Anodic fluorination of dithioacetals.
With these facts in mind, we studied comparatively the anodic fluorination of dithioacetal derivatives having various electron-withdrawing groups at their α-positions using various poly(HF) salts [28].
Results and Discussion
Various α-substituted methyl phenyl sulfides, 1a, 1c, 1e, and 1g, and their α-phenylthio derivatives (dithioacetals) were prepared, and their oxidation potentials (![[Graphic 1]](/bjoc/content/inline/1860-5397-11-12-i5.svg?max-width=637&scale=1.18182) ) were measured by cyclic voltammetry in an anhydrous acetonitrile (MeCN) solution containing n-Bu4NBF4 (0.1 M) using a platinum disk working electrode and a saturated calomel electrode (SCE) as the reference electrode. All compounds exhibited irreversible multiple oxidation peaks and the first oxidation peak potentials are summarized in Table 1. It was expected that the introduction of an additional phenylthio group to the α-position of the sulfides would decrease their oxidation potentials. However, unexpectedly they are higher than those of the corresponding sulfides having a single phenylthio group. Although a detailed reason is not clear at present, the additional phenylthio group does not act as an electroauxiliary, but acts as an electron-withdrawing group. As shown in Table 1, the oxidation potentials of sulfide 1g and dithioacetal 1h having a stronger electron-withdrawing cyano group (Taft σ* = +1.30) [29] are much higher compared to those of 1a, 1b with an ester group (Taft σ* = +0.69) [29] and 1c, 1d with an acetyl group (Taft σ* = +0.60) [29], respectively. This indicates that the polar effect, namely electron-withdrawing effect of a substituent greatly affects the electron-transfer step from the substrate to the anode.
) were measured by cyclic voltammetry in an anhydrous acetonitrile (MeCN) solution containing n-Bu4NBF4 (0.1 M) using a platinum disk working electrode and a saturated calomel electrode (SCE) as the reference electrode. All compounds exhibited irreversible multiple oxidation peaks and the first oxidation peak potentials are summarized in Table 1. It was expected that the introduction of an additional phenylthio group to the α-position of the sulfides would decrease their oxidation potentials. However, unexpectedly they are higher than those of the corresponding sulfides having a single phenylthio group. Although a detailed reason is not clear at present, the additional phenylthio group does not act as an electroauxiliary, but acts as an electron-withdrawing group. As shown in Table 1, the oxidation potentials of sulfide 1g and dithioacetal 1h having a stronger electron-withdrawing cyano group (Taft σ* = +1.30) [29] are much higher compared to those of 1a, 1b with an ester group (Taft σ* = +0.69) [29] and 1c, 1d with an acetyl group (Taft σ* = +0.60) [29], respectively. This indicates that the polar effect, namely electron-withdrawing effect of a substituent greatly affects the electron-transfer step from the substrate to the anode.
Table 1:
First oxidation potentials, ![[Graphic 2]](/bjoc/content/inline/1860-5397-11-12-i6.svg?max-width=637&scale=1.18182) of compounds 1.
of compounds 1.
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-12-i7.svg?max-width=637&scale=1.0)
|
|||
| Substrate | X | R (σ* value)a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-12-i8.svg?max-width=637&scale=1.18182) (V vs SCE)b (V vs SCE)b
|
|---|---|---|---|
| 1a | H | COOEt (+ 0.69) | 1.60 |
| 1b | SPh | COOEt (+ 0.69) | 1.73 |
| 1c | H | COMe (+ 0.60) | 1.59 |
| 1d | SPh | COMe (+ 0.60) | 1.63 |
| 1e | H | CONEt2 | 1.60 |
| 1f | SPh | CONEt2 | 1.64 |
| 1g | H | CN (+ 1.30) | 1.85 |
| 1h | SPh | CN (+ 1.30) | 2.04 |
aFrom [29]. bSubstrate concentration: 5 mM; sweep rate: 100 mV/s; working electrode: Pt disk (Ø = 1 mm).
At first, anodic fluorination of ethyl α,α-bis(phenylthio)acetate (1b) [30,31] was carried out at platinum plate electrodes in an undivided cell using various solvents in the presence of Et3N·3HF as supporting salt and fluorine source. A constant current was passed until the starting material 1b was completely consumed (monitored by TLC). As shown in Table 2, the anodic fluorination of 1b proceeded to give the corresponding α-fluoro product 2b in a good yield regardless of the solvent used. Thus, it was found that the solvents did not affect the yield of 2b in contrast to the current efficiency. When the reaction was performed in DME as the solvent, anodic decomposition of DME took place simultaneously with the anodic fluorination of 1b, which resulted in low current efficiency. In all cases, fluorodesulfurization product 3b [22] was detected in considerable yield. When CH2Cl2 and MeNO2 were used, a small amount of ethyl α,α-difluoro-α-(phenylthio)acetate (4b) [22] was also detected (Table 2, entries 3 and 4). As a blank test, the electrolytic solution of 1b was mechanically stirred without electrolysis overnight and 1b was mostly recovered (Table 2, entry 5). Therefor electrolysis is necessary for the fluorination to take place.
Table 2: Anodic fluorination of 1b in various solvents containing Et3N·3HFa.
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-12-i9.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Solvent | Charge passed (F/mol) | Yield (%)b,c | Total yield (%) | ||
|---|---|---|---|---|---|---|
| 2b | 3b | 4b | ||||
| 1 | MeCN | 3.0 | 74 (70) | 9 | 0 | 83 |
| 2 | DME | 5.0 | 74 | 9 | 0 | 83 |
| 3 | CH2Cl2 | 2.5 | 73 | 5 | 4 | 82 |
| 4 | MeNO2 | 2.2 | 73 | 7 | 1 | 81 |
| 5d | MeCN | – | – | – | – | – |
aConstant current (8 mA/cm2) electrolysis was carried out in 0.3 M Et3N·3HF/solvent. bDetermined by 19F NMR. cIsolated yield given in parentheses. dMechanical stirring was performed overnight at ambient temperature without electrolysis.
Among the solvents tested for electrolysis, we decided to use MeCN for further studies on the anodic fluorination based on a good current efficiency and the formation of only one byproduct.
Next, anodic fluorination of 1b was carried out in MeCN using various poly(HF) salts until the substrate was completely consumed and the results are shown in Figure 1. As mentioned earlier, the anodic fluorination of 1b using Et3N·3HF provided α-fluorinated product 2b selectively in good yield along with a small amount of fluorodesulfurization product 3b. In contrast, the fluorodesulfurization reaction was significantly promoted with increasing HF content of the poly(HF) salts; especially the use of Et3N·5HF gave predominantly the fluorodesulfurization product 3b in 85% yield (Figure 1a). Previously, we obtained 3b in 75% yield by constant potential anodic oxidation of ethyl α-(phenylthio)acetate in a similar electrolytic solution [22]. A comparable dependency of product selectivity on supporting poly(HF) salts was also observed in a series of Et4NF·nHF (n = 3–5) although the product yields are moderate (Figure 1b).
![[1860-5397-11-12-1]](/bjoc/content/figures/1860-5397-11-12-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Dependency of fluorinated product selectivity on a series of fluoride salts (a) Et3N·nHF (n = 3–5) and (b) Et4NF·nHF (n = 3–5).
Figure 1: Dependency of fluorinated product selectivity on a series of fluoride salts (a) Et3N·nHF (n = 3–5) ...
According to these results, we carried out the anodic fluorination of other dithioacetals bearing different electron-withdrawing substituents such as acetyl, amide, and cyano groups under similar conditions. The results are summarized in Table 3. In the case of α,α-bis(phenylthio)acetone (1d) [32], the use of Et3N·3HF and Et4NF·3HF resulted in predominant α-fluorination to provide the corresponding monofluorinated product 2d in good to moderate yields (Table 3, entries 1 and 3). On the contrary, when higher HF content salts such as Et3N·5HF and Et4NF·5HF were used, fluorodesulfurization product 3d [22] was obtained almost exclusively in moderate or good yield (Table 3, entries 2 and 4). Regardless of poly(HF) salts, difluorinated product 4d [33] was always formed due to the further oxidation of products 2d and 3d. In contrast, anodic fluorination of N,N-diethyl-α,α-bis(phenylthio)acetamide (1f) with Et3N·3HF required a large excess amount of electricity to consume the starting substrate 1f, and fluorodesulfurization took place exclusively to provide the corresponding mono- and difluorinated products 3f and 4f [22,34] with the same ratio in rather low yields (Table 3, entry 5). The longer electrolysis caused the formation of complicated products. This result is quite different from the case of 1b and 1d (Table 2, entry 1 and Table 3, entry 1). Such different anodic behavior may be attributable to different pKa values of the α-proton of the substrates. It is known that the acidity of the α-proton of N,N-diethylacetoamide is 4 to 5 times lower than that of acetone and ethyl acetate [35]. Therefore, the acidity of the α-proton of 1f having an amide group would be much lower compared to that of 1b and 1d having an ester and acetyl group, respectively. Thus, it is reasonable that no deprotonation of the cationic intermediate of 1f took place. Moreover, when a higher HF content poly(HF) salt like Et3N·5HF was used, fluorodesulfurization product 3f was exclusively formed in good yield. This tendency is quite similar to the result of anodic fluorination of 1b and 1d (Figure 1a and Table 3, entries 2 and 4). Thus, it was found that due to the low acidity of the α-proton of 1f, fluorodesulfurization took place prior to α-fluorination even in the presence of Et3N·3HF containing the free base Et3N. In sharp contrast to these cases, α,α-bis(phenylthio)acetonitrile (1h) [36] bearing a strongly electron-withdrawing cyano group underwent α-fluorination exclusively to produce α-fluorinated product 2h in excellent yields regardless of the poly(HF) salts (Table 3, entries 7–12). In case of 1h, no difluoro product was formed at all, which is probably due to a much higher oxidation potential of 2h compared to that of the starting substrate 1h. In support of this, we have already shown that introduction of one fluorine atom to the α-position of α-(phenylthio)acetonitrile increased the oxidation potential by 0.36 V [22].
Table 3: Anodic fluorination of dithioacetal derivative 1 in acetonitrilea.
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-12-i10.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R | Supporting electrolyte | Charge passed (F/mol) | Yield (%)b,c | ||
|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||
| 1 | COMe (1d) | Et3N·3HF | 3.0 | 80 (66) | – | 5 |
| 2 | COMe (1d) | Et3N·5HF | 2.5 | – | 63 | 3 |
| 3 | COMe (1d) | Et4NF·3HF | 2.7 | 60 | – | 10 |
| 4 | COMe (1d) | Et4NF·5HF | 2.5 | 6 | 78 (70) | 1 |
| 5 | CONEt2 (1f) | Et3N·3HF | 5.0 | 0 | 18 | 17 |
| 6 | CONEt2 (1f) | Et3N·5HF | 3.0 | 0 | 72 (63) | 1 |
| 7 | CN (1h) | Et3N·3HF | 3.0 | 98 (87) | 0 | 0 |
| 8 | CN (1h) | Et3N·4HF | 2.8 | 98 | 0 | 0 |
| 9 | CN (1h) | Et3N·5HF | 2.5 | 90 | 0 | 0 |
| 10 | CN (1h) | Et4NF·3HF | 2.7 | 94 | 0 | 0 |
| 11 | CN (1h) | Et4NF·4HF | 2.5 | 95 | 0 | 0 |
| 12 | CN (1h) | Et4NF·5HF | 2.5 | 93 | 0 | 0 |
aConstant current (8 mA/cm2) electrolysis was carried out using 0.3 M supporting fluoride salt. bDetermined by 19F NMR. cIsolated yields are given in parentheses.
A plausible mechanism for the anodic fluorination of dithioacetals 1b, 1d, and 1f is shown in Scheme 3. The fluorination reaction is initiated by electron transfer from a sulfur atom of the substrate to generate the corresponding radical cation B, which traps a fluoride ion to afford radical C. This is followed by a further oxidation to give cationic intermediate D. In the cases of 1b and 1d having electron-withdrawing ester and acetyl groups, the α-protons are acidic enough and can be cleaved by either base, free Et3N (from Et3N·3HF) [37] or fluoride ions (from Et4NF·3HF). The resulting cation E reacts with a fluoride ion to form 2b and 2d. Further anodic fluorodesulfurization occurs to provide the corresponding difluorinated products 4b and 4d, respectively. On the other hand, the desulfurization of intermediate D followed by reaction with fluoride provides the corresponding fluorodesulfurization products 3b, 3d, and 3f.
![[1860-5397-11-12-i3]](/bjoc/content/inline/1860-5397-11-12-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible reaction mechanism for anodic fluorination of 1b, 1d, and 1f.
Scheme 3: Plausible reaction mechanism for anodic fluorination of 1b, 1d, and 1f.
When high HF content salts like Et3N·5HF and Et4NF·nHF (n = 4, 5) are used, the higher concentration of HF in the electrolytic solution would increase the amount of D rather than E in an equilibrium between them as shown in Scheme 4. Namely, the deprotonation of D would be retarded due to high concentration of protons in the solution, and consequently the C–S bond cleavage seems to take place more favorably than a deprotonation. A similar effect on the suppression of defluorination of CF3-enolate anion in the presence of a large amount of fluoride ions has been reported [38].
![[1860-5397-11-12-i4]](/bjoc/content/inline/1860-5397-11-12-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism for suppression of the elimination of HF (deprotonation) and preferable desulfurization of D at high concentrations of HF in an electrolytic solution.
Scheme 4: Mechanism for suppression of the elimination of HF (deprotonation) and preferable desulfurization o...
On the other hand, it is known that the acidity of α-protons of acetoamides is much lower compared to that of acetate and acetone as mentioned. Therefore, it is understandable that the anodic fluorination of 1f having a weakly electron-withdrawing amide group resulted in fluorodesulfurization to provide 3f even when Et3N·3HF containing free base, Et3N was used. As mentioned, the yield of monofluorodesulfurization product 3f increased markedly by using high HF content salt, Et3N·5HF.
In sharp contrast, in the case of substrate 1h having a cyano group, α-fluorination without desulfurization always took place even when Et3N·5HF and Et4NF·5HF were used. This can be explained in terms of fast deprotonation of cationic intermediate D promoted by a strongly electron-withdrawing cyano group as shown in Scheme 3.
Conclusion
The regioselective anodic fluorination of ethyl α,α-bis(phenylthio)acetate and its acetone, acetoamide, and acetonitrile analogues was successfully carried out using various poly(HF) salts such as Et3N·nHF and Et4NF·nHF (n = 3–5) to provide α-fluoro and/or fluorodesulfurization products. The fluorinated product selectivity was found to depend on substituents and supporting poly(HF) salts. The unique product selectivity was tentatively explained in terms of electron-withdrawing ability (Taft σ*) of substituents and HF content of the used supporting poly(HF) salts.
Supporting Information
| Supporting Information File 1: General methods, synthetic procedures, characterzation data of all new compounds including copies of 1H NMR, 13C NMR and 19F NMR spectra. | ||
| Format: PDF | Size: 387.7 KB | Download |
Acknowledgements
We gratefully acknowledge Morita Chemical Industries Co., Ltd for supplying triethylamine poly(hydrogen fluoride) [Et3N·nHF (n = 3–5)] and tetraethylammonium fluoride poly(hydrogen fluoride) [Et4NF·nHF (n = 3–5)] that were used as supporting salt fluorine source. We also thank technical staffs working at Center for Advanced Materials Analysis (Suzukakedai Campus) of Tokyo Institute of Technology for analyzing new compounds. One of the authors (Y. B.) acknowledges Ministry of Education, Culture, Sports, Science & Technology of Japan for awarding national scholarship to conduct this research.
References
-
Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley: Chichester, 2009. doi:10.1002/9781444312096
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, 2008. doi:10.1002/9780470281895
Return to citation in text: [1] -
Uneyama, K. Organofluorine Chemistry; Blackwell Publishing: Oxford, 2006. doi:10.1002/9780470988589
Return to citation in text: [1] -
O’Hagen, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003
Return to citation in text: [1] -
Hiyama, T., Ed. Organofluorine Compounds. Chemistry and Applications; Springer: Berlin, 2000. doi:10.1007/978-3-662-04164-2
Return to citation in text: [1] -
Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h
Return to citation in text: [1] -
Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 103–109. doi:10.1002/ejoc.200800872
Return to citation in text: [1] -
Fuchigami, T.; Atobe, M.; Inagi, S. Fundamental and Applications of Organic Electrochemistry. Synthesis, Materials, Devices; Wiley: Chichester, 2014.
Return to citation in text: [1] -
Fuchigami, T. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; Marcel Dekker: New York, 2001; Chapter 25.
Return to citation in text: [1] -
Childs, W. V.; Christensen, L.; Klink, F. W. In Organic Electrochemistry, 3rd ed.; Lund, H.; Baizer, M. M., Eds.; Marcel Dekker: New York, 1991; Chapter 24.
Return to citation in text: [1] -
Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Org. Lett. 2010, 12, 644–646. doi:10.1021/ol9028836
Return to citation in text: [1] [2] -
Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Adv. Synth. Catal. 2010, 352, 2757–2760. doi:10.1002/adsc.201000501
Return to citation in text: [1] [2] -
Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/C1CC12414E
Return to citation in text: [1] [2] -
Takahashi, K.; Furusawa, T.; Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Electrochim. Acta 2012, 77, 47–53. doi:10.1016/j.electacta.2012.05.049
Return to citation in text: [1] [2] -
Takahashi, K.; Inagi, S.; Fuchigami, T. J. Electrochem. Soc. 2013, 160, G3046–G3052. doi:10.1149/2.005307jes
Return to citation in text: [1] [2] -
Sawamura, T.; Takahashi, K.; Inagi, S.; Fuchigami, T. Angew. Chem., Int. Ed. 2012, 51, 4413–4416. doi:10.1002/anie.201200438
Return to citation in text: [1] [2] -
Inagi, S.; Hayashi, S.; Fuchigami, T. Chem. Commun. 2009, 1718–1720. doi:10.1039/B822510A
Return to citation in text: [1] -
Hayashi, S.; Inagi, S.; Fuchigami, T. Macromolecules 2009, 42, 3755–3760. doi:10.1021/ma900358x
Return to citation in text: [1] -
Shaaban, M. R.; Ishii, H.; Fuchigami, T. J. Org. Chem. 2000, 65, 8685–8689. doi:10.1021/jo001129u
Return to citation in text: [1] -
Fuchigami, T.; Shimojo, M.; Konno, A.; Nakagawa, K. J. Org. Chem. 1990, 55, 6074–6075. doi:10.1021/jo00312a006
Return to citation in text: [1] -
Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Fuchigami, T.; Sano, M. J. Electroanal. Chem. 1996, 414, 81–84. doi:10.1016/0022-0728(96)04642-6
Return to citation in text: [1] -
Fuchigami, T.; Mitomo, K.; Ishii, H.; Konno, A. J. Electroanal. Chem. 2001, 507, 30–33. doi:10.1016/S0022-0728(01)00440-5
Return to citation in text: [1] -
Yoshiyama, T.; Fuchigami, T. Chem. Lett. 1992, 21, 1995–1998. doi:10.1246/cl.1992.1995
Return to citation in text: [1] [2] -
Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 1992, 25, 233–240. doi:10.1021/ar00017a005
Return to citation in text: [1] -
Fuchigami, T.; Ichikawa, S. J. Org. Chem. 1994, 59, 607–615. doi:10.1021/jo00082a018
Return to citation in text: [1] -
Preliminary results of this work were presented at 225th ECS Meeting, 2014, Abstr. No. 820.
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] [2] [3] [4] -
Tabti, B.; Gourmala, C.; Bounouara Bouderba, H.; Mulengi, J. K. J. Soc. Alger. Chim. 1996, 6, 199–206.
Return to citation in text: [1] -
Ritter, R. H.; Cohen, T. J. Am. Chem. Soc. 1986, 108, 3718–3725. doi:10.1021/ja00273a028
Return to citation in text: [1] -
Keiko, N. A.; Funtikova, E. A.; Stepanova, L. G.; Chuvashev, Yu. A.; Larina, L. I. Russ. J. Org. Chem. 2002, 38, 970–976. doi:10.1023/A:1020893310626
Return to citation in text: [1] -
Gouault, S.; Guérin, C.; Lemoucheux, L.; Lequeux, T.; Pommelet, J.-C. Tetrahedron Lett. 2003, 44, 5061–5064. doi:10.1016/S0040-4039(03)01134-1
Return to citation in text: [1] -
Yagupolski, L.; Korinko, V. A. Zh. Obshch. Khim. 1969, 39, 1747.
Return to citation in text: [1] -
McMurry, J. E.; Simanek, E. E. Fundamentals of Organic Chemistry, 6th ed.; Brooks–Cole Publishing Co, 2006; Chapter 11.
Return to citation in text: [1] -
Ishibashi, H.; Okada, M.; Sato, K.; Ikeda, M.; Ishiyama, K.; Tamura, Y. Chem. Pharm. Bull. 1985, 33, 90–95. doi:10.1248/cpb.33.90
Return to citation in text: [1] -
Hou, Y.; Higashiya, S.; Fuchigami, T. J. Org. Chem. 1999, 64, 3346–3349. doi:10.1021/jo981979y
Return to citation in text: [1] -
Ishikawa, N.; Yokozawa, T. Bull. Chem. Soc. Jpn. 1983, 56, 724–726. doi:10.1246/bcsj.56.724
Return to citation in text: [1]
| 1. | Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley: Chichester, 2009. doi:10.1002/9781444312096 |
| 2. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 3. | Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, 2008. doi:10.1002/9780470281895 |
| 4. | Uneyama, K. Organofluorine Chemistry; Blackwell Publishing: Oxford, 2006. doi:10.1002/9780470988589 |
| 5. | O’Hagen, D. J. Fluorine Chem. 2010, 131, 1071–1081. doi:10.1016/j.jfluchem.2010.03.003 |
| 6. | Hiyama, T., Ed. Organofluorine Compounds. Chemistry and Applications; Springer: Berlin, 2000. doi:10.1007/978-3-662-04164-2 |
| 21. | Fuchigami, T.; Shimojo, M.; Konno, A.; Nakagawa, K. J. Org. Chem. 1990, 55, 6074–6075. doi:10.1021/jo00312a006 |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 30. | Tabti, B.; Gourmala, C.; Bounouara Bouderba, H.; Mulengi, J. K. J. Soc. Alger. Chim. 1996, 6, 199–206. |
| 31. | Ritter, R. H.; Cohen, T. J. Am. Chem. Soc. 1986, 108, 3718–3725. doi:10.1021/ja00273a028 |
| 12. | Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Org. Lett. 2010, 12, 644–646. doi:10.1021/ol9028836 |
| 13. | Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Adv. Synth. Catal. 2010, 352, 2757–2760. doi:10.1002/adsc.201000501 |
| 14. | Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/C1CC12414E |
| 15. | Takahashi, K.; Furusawa, T.; Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Electrochim. Acta 2012, 77, 47–53. doi:10.1016/j.electacta.2012.05.049 |
| 16. | Takahashi, K.; Inagi, S.; Fuchigami, T. J. Electrochem. Soc. 2013, 160, G3046–G3052. doi:10.1149/2.005307jes |
| 17. | Sawamura, T.; Takahashi, K.; Inagi, S.; Fuchigami, T. Angew. Chem., Int. Ed. 2012, 51, 4413–4416. doi:10.1002/anie.201200438 |
| 18. | Inagi, S.; Hayashi, S.; Fuchigami, T. Chem. Commun. 2009, 1718–1720. doi:10.1039/B822510A |
| 19. | Hayashi, S.; Inagi, S.; Fuchigami, T. Macromolecules 2009, 42, 3755–3760. doi:10.1021/ma900358x |
| 20. | Shaaban, M. R.; Ishii, H.; Fuchigami, T. J. Org. Chem. 2000, 65, 8685–8689. doi:10.1021/jo001129u |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 9. | Fuchigami, T.; Atobe, M.; Inagi, S. Fundamental and Applications of Organic Electrochemistry. Synthesis, Materials, Devices; Wiley: Chichester, 2014. |
| 10. | Fuchigami, T. In Organic Electrochemistry, 4th ed.; Lund, H.; Hammerich, O., Eds.; Marcel Dekker: New York, 2001; Chapter 25. |
| 11. | Childs, W. V.; Christensen, L.; Klink, F. W. In Organic Electrochemistry, 3rd ed.; Lund, H.; Baizer, M. M., Eds.; Marcel Dekker: New York, 1991; Chapter 24. |
| 29. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 7. | Umemoto, T.; Singh, R. P.; Xu, Y.; Saito, N. J. Am. Chem. Soc. 2010, 132, 18199–18205. doi:10.1021/ja106343h |
| 8. | Kishi, Y.; Inagi, S.; Fuchigami, T. Eur. J. Org. Chem. 2009, 103–109. doi:10.1002/ejoc.200800872 |
| 29. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 26. | Yoon, U. C.; Mariano, P. S. Acc. Chem. Res. 1992, 25, 233–240. doi:10.1021/ar00017a005 |
| 27. | Fuchigami, T.; Ichikawa, S. J. Org. Chem. 1994, 59, 607–615. doi:10.1021/jo00082a018 |
| 29. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 25. | Yoshiyama, T.; Fuchigami, T. Chem. Lett. 1992, 21, 1995–1998. doi:10.1246/cl.1992.1995 |
| 29. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 17. | Sawamura, T.; Takahashi, K.; Inagi, S.; Fuchigami, T. Angew. Chem., Int. Ed. 2012, 51, 4413–4416. doi:10.1002/anie.201200438 |
| 12. | Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Org. Lett. 2010, 12, 644–646. doi:10.1021/ol9028836 |
| 13. | Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Adv. Synth. Catal. 2010, 352, 2757–2760. doi:10.1002/adsc.201000501 |
| 14. | Fuchigami, T.; Inagi, S. Chem. Commun. 2011, 47, 10211–10223. doi:10.1039/C1CC12414E |
| 15. | Takahashi, K.; Furusawa, T.; Sawamura, T.; Kuribayashi, S.; Inagi, S.; Fuchigami, T. Electrochim. Acta 2012, 77, 47–53. doi:10.1016/j.electacta.2012.05.049 |
| 16. | Takahashi, K.; Inagi, S.; Fuchigami, T. J. Electrochem. Soc. 2013, 160, G3046–G3052. doi:10.1149/2.005307jes |
| 23. | Fuchigami, T.; Sano, M. J. Electroanal. Chem. 1996, 414, 81–84. doi:10.1016/0022-0728(96)04642-6 |
| 24. | Fuchigami, T.; Mitomo, K.; Ishii, H.; Konno, A. J. Electroanal. Chem. 2001, 507, 30–33. doi:10.1016/S0022-0728(01)00440-5 |
| 25. | Yoshiyama, T.; Fuchigami, T. Chem. Lett. 1992, 21, 1995–1998. doi:10.1246/cl.1992.1995 |
| 28. | Preliminary results of this work were presented at 225th ECS Meeting, 2014, Abstr. No. 820. |
| 32. | Keiko, N. A.; Funtikova, E. A.; Stepanova, L. G.; Chuvashev, Yu. A.; Larina, L. I. Russ. J. Org. Chem. 2002, 38, 970–976. doi:10.1023/A:1020893310626 |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 37. | Hou, Y.; Higashiya, S.; Fuchigami, T. J. Org. Chem. 1999, 64, 3346–3349. doi:10.1021/jo981979y |
| 38. | Ishikawa, N.; Yokozawa, T. Bull. Chem. Soc. Jpn. 1983, 56, 724–726. doi:10.1246/bcsj.56.724 |
| 36. | Ishibashi, H.; Okada, M.; Sato, K.; Ikeda, M.; Ishiyama, K.; Tamura, Y. Chem. Pharm. Bull. 1985, 33, 90–95. doi:10.1248/cpb.33.90 |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 34. | Yagupolski, L.; Korinko, V. A. Zh. Obshch. Khim. 1969, 39, 1747. |
| 35. | McMurry, J. E.; Simanek, E. E. Fundamentals of Organic Chemistry, 6th ed.; Brooks–Cole Publishing Co, 2006; Chapter 11. |
| 22. | Fuchigami, T.; Shimojo, M.; Konno, A. J. Org. Chem. 1995, 60, 3459–3464. doi:10.1021/jo00116a037 |
| 33. | Gouault, S.; Guérin, C.; Lemoucheux, L.; Lequeux, T.; Pommelet, J.-C. Tetrahedron Lett. 2003, 44, 5061–5064. doi:10.1016/S0040-4039(03)01134-1 |
© 2015 Yin et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)