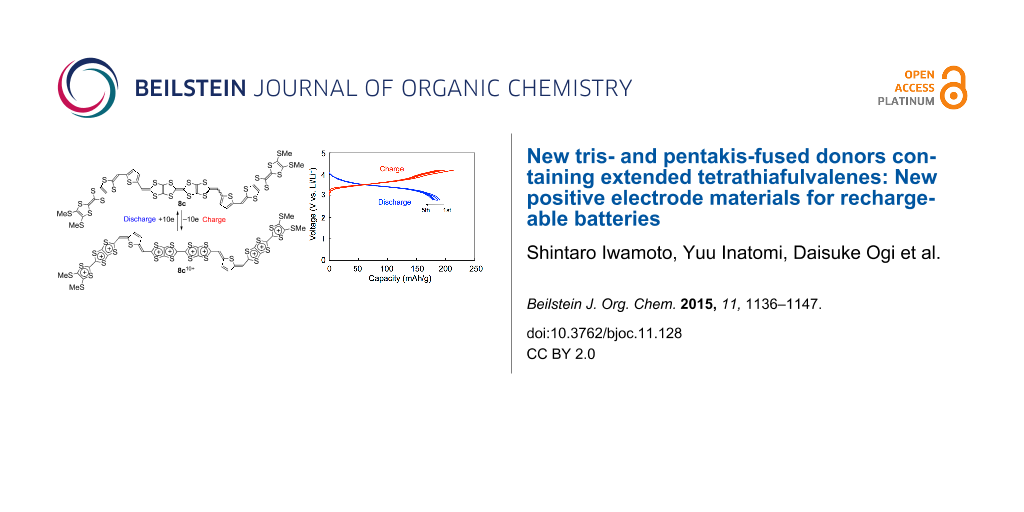
Abstract
Derivatives of tris-fused TTF extended with two ethanediylidenes (5), tris- and pentakis-fused TTFs extended with two thiophene-2,5-diylidenes (6–9) were successfully synthesized. Cyclic voltammograms of the tetrakis(n-hexylthio) derivative of 5 and 7 (5d, 7d) consisted of two pairs of two-electron redox waves and two pairs of one-electron redox waves. On the other hand, four pairs of two-electron redox waves and two pairs of one-electron redox waves were observed for the tetrakis(n-hexylthio) derivative of 9 (9d). Coin-type cells using the bis(ethylenedithio) derivatives of 5 (5b), 6 (6b) and the tetrakis(methylthio) derivatives of 5 (5c) and 8 (8c) as positive electrode materials showed initial discharge capacities of 157–190 mAh g−1 and initial energy densities of 535–680 mAh g−1. The discharge capacities after 40 cycles were 64–86% of the initial discharge capacities.

Graphical Abstract
Introduction
Tetrathiafulvalene (TTF, 1a) and its analogues have attracted much attention as potential components for organic functional materials as well as multi-electron redox systems [1-5]. Fused TTF oligomers [5] are of considerable interest as multi-electron redox systems, because the TTF units strongly interact with each other. For example, a bis-fused TTF, 2,5-bis(1,3-dithiol-3-ylidene)-1,3,4,6-tetrathiapentalene (BDT-TTP or simply TTP) exhibits four pairs of one-electron redox waves at +0.44, +0.62, +1.05 and +1.13 V (V vs SCE, in benzonitrile) [6]. The E2–E1 value is considerably larger than most dimeric TTF derivatives linked by σ-bond (typically 0.05–0.10 V) [7]. Fused TTF donors also play important roles in the development of highly functional materials. For example, TTP and its derivatives have yielded a large number of molecular conductors retaining metallic conductivity down to ≤4.2 K, because they have a tendency to construct two-dimensional molecular arrays through side-by-side sulfur···sulfur interaction [5,8]. On the other hand, a tris-fused TTF, 2,2’-bis[5-(1,3-dithiol-2-ylidene)-1,3,4,6-tetrathiapentanylidene] (TTPY, 2a) and its derivatives have afforded highly conducting SbF6− and iodine salts of σrt ≈ 10−1–101 S cm−1 on compressed pellets [9,10].
Recently, we reported that TTP and TTPY can be utilized as positive electrode materials for rechargeable batteries [11]. All organic molecules exhibiting multi-electron redox behaviour seem to be promising as active materials for rechargeable batteries. However, most organic molecules have a crucial disadvantage, that is, they dissolve in organic solvents used for electrolyte solutions. TTF cannot be used as an active electrode material for rechargeable batteries for the above reason, while bis(ethylenedithio)-TTF (BEDT-TTF, 1b) exhibits relatively good charge–discharge cycle performance because of its lower solubility in organic solvents [12]. However, the substitution of two ethylenedithio groups on TTF results in a significant decrease in the theoretical capacity (about half that of TTF). Utilization of polymerized materials is one of the solutions to decrease solubility. However, insertion of a linkage group, which is usually required to construct polymers, also results in considerable decrease in the theoretical capacity [13]. As for fused TTF oligomers, theoretical capacity rather increases as the number of TTF units increases because two carbons are shared in the two TTF units. TTP and TTPY are actually less soluble in organic solvents than TTF. In particular, TTPY is barely soluble in common solvents even in carbon disulfide. However, the maximum electrons cannot be utilized for TTP and TTPY batteries because TTP and TTPY dissolve in the electrolyte solutions in their maximum oxidation states (tetravalent for TTP and hexavalent for TTPY, respectively) [11].
Possible molecular modifications for TTPY to reduce solubility in electrolyte solvents are as follows; (i) introduction of rigid substituents such as the ethylenedithio group as mentioned above, (ii) use of a rigid extended-TTF unit, (iii) increase of the number of (extended) TTF units. As for the modification (ii), insertion of a π-spacer is sometimes useful. A vinylogous TTF (3, Figure 1) [14-16] shows lower solubility in ordinary organic solvents than TTF, although the thiophene-containing analog (4, Figure 1) [17-19] is more soluble than TTF. Increase of the TTF units might be the best way; however, the preparation of tetrakis- and/or pentakis-fused TTFs is not easy because of the low solubility of the precursor molecules. Insertion of thiophene spacers is a possible strategy for synthesizing fused TTF oligomers, because thiophene inserted precursors are more soluble than the TTF-type precursors as mentioned above. We succeeded in the synthesis of fused TTF pentamer and heptamer composed of the unit of 4 [20]. In this paper, we report the synthesis and electrochemical properties of vinyl extended TTPY analogue (5b–d) and tris- and pentakis-fused TTF analogues extended by the insertion of two thiophene rings (6b, 6d, 7d, 8c and 9d). We also report the charge–discharge properties of rechargeable batteries incorporating the methylthio and ethylenedithio derivatives of 5, 6 and 8 (5b, 5c, 6b and 8c) as a positive electrode material.
![[1860-5397-11-128-1]](/bjoc/content/figures/1860-5397-11-128-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of 1–9 and TTP.
Figure 1: Chemical structures of 1–9 and TTP.
Results and Discussion
Synthesis
The synthesis of new donors was carried out according to Scheme 1. A trimethylphosphite-mediate cross coupling between a 1,3-dithiole-2-thione (10) [21] and 1,3-dithiol-2-one (11) [22-24] gave a TTF derivative with two ethoxyphosphoryl groups (12) in 63% yield. We adopted the cross-coupling reaction between the 1,3-dithiole-2-thione and the 1,3-dithiol-2-one derivatives for the following reasons. The homo-coupling reaction of 10 afforded 12 in low yields, and purification by column chromatography was difficult because of undesired byproducts. The homo-coupling reaction using 11 might give 12 in a good yield, however, toxic and expensive mercury(II) acetate has to be used for the synthesis of 11. Thus, the cross-coupling reaction is useful for saving 11. The Horner–Wadsworth–Emmons reaction of 12 with 2 equiv of aldehydes 13b–d [16,25] in the presence of BuLi in THF at −78 °C gave the desired bis-adducts 5b–d in 54−85% yields. Similarly, 6b, 6c, 7d, 8c and 9d were obtained in 62–85% yields by the reaction of 12 with 14b,c, 15d [17-19] or 16c, 17d [26] in the presence of BuLi in THF. All the new donors were obtained as stable solids.
![[1860-5397-11-128-i1]](/bjoc/content/inline/1860-5397-11-128-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Theoretical calculations
We performed theoretical calculations of 5A, 6Aa and 7Aa by using the Gaussian 09 program based on the density functional theory (DFT) at the B3LYP/6-31G(d) level [27]. Their HOMO and HOMO–n (n = 1–2 and 1–4) of the trans isomers of 5a, 6a and 8a are shown in Figures 2–4, respectively. The shapes, energy levels and total energies of the trans and cis isomers were almost the same as each other. The HOMO of 5a was distributed over the whole molecules. Molecular orbital coefficients were largely observed in the vinylogous TTF moieties rather than in the central TTF moiety (Figure 2). In the HOMO–1, most molecular orbital coefficients were found on the bilateral vinylogous TTF moieties. The HOMO–2 was mainly located on the central TTF unit. In the bilateral vinylogous TTF moieties, small molecular orbital coefficients were observed. The shapes of the HOMO, HOMO-1 and HOMO-2 of the thiophene extended donor 6a resembled those of 5a (Figure 3). The HOMO of 8a spread mainly over the central TTF and the bilateral extended TTF moieties, and the TTF moieties at the both ends barely contributed to the HOMO (Figure 4). Similarly to 6a, larger molecular orbital coefficients of the sulfur atoms were found in the extended TTF moieties of 8a rather than in the central TTF moiety. The HOMO–1 was mainly distributed on the two extended TTF moieties. Small molecular orbital coefficients were observed in the TTF moieties at the both ends. In contrast, the HOMO–2 was hardly distributed on the extended TTF units, but was substantially located on the three TTF units. The TTF moieties at the both ends considerably contributed to the HOMO–3. The HOMO–4 is distributed mainly on the central TTF moiety, although small molecular orbital coefficients were observed in the other donor units.
![[1860-5397-11-128-2]](/bjoc/content/figures/1860-5397-11-128-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular orbitals of 5a (trans isomer).
Figure 2: Molecular orbitals of 5a (trans isomer).
![[1860-5397-11-128-3]](/bjoc/content/figures/1860-5397-11-128-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular orbitals of 6a (trans isomer).
Figure 3: Molecular orbitals of 6a (trans isomer).
![[1860-5397-11-128-4]](/bjoc/content/figures/1860-5397-11-128-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular orbitals of 8a (trans isomer).
Figure 4: Molecular orbitals of 8a (trans isomer).
The orbital energies of the HOMO and HOMO–n (n = 1–2 and 1–4) for 5a, 6a and 8a are summarized in Table 1. The orbital energies of HOMO of 5a, 6a and 8a (−4.532 to −4.605 eV) are comparable to each other, and are higher by 0.18−0.25 eV than that of TTPY (−4.787 eV). If the oxidation relates to the orbital energy, the first oxidations of 5–9 might occur at lower potentials than TTPY. The energy differences between the HOMO and HOMO–1 of all the donors (0.041–0.113 eV) were smaller than that of TTPY (0.186 eV). In particular, the orbital energies of the HOMO and HOMO–1 of 8a (−4.602 and −4.643 eV, respectively) were close to each other, suggesting that the first four-electron oxidation of 8a might occur in a narrow potential range. The HOMO–2 of 5a and 6Aa (−5.257 and −5.129 eV, respectively) and HOMO–4 of 8a (−5.328 eV) were slightly higher than the orbital energy of the HOMO–2 of TTPY (−5.439 eV). These results suggest that the electrons at the HOMO–2 of 5a and 6a and HOMO–4 of 8a might be removed more easily than those at the HOMO–2 of TTPY.
Electrochemical properties
The redox behaviors of 5d, 7d and 9d were investigated by using cyclic voltammetry. Deconvoluted cyclic voltammograms of 5d, 7d and 9d measured in a carbon disulfide/benzonitrile (1:1, v/v) solution are shown in Figure 5. As for the tris-fused donors 5d and 7d, four pairs of redox waves were observed. The peak currents of the first and second redox waves were about double those of the others. The maximum number of electrons participating in the redox was six, considering that both donors have six redox-active 1,3-dithiol-2-ylidene (DT) sites. Thus, we think that the first and second redox waves correspond to two-electron redox processes and that the remaining waves correspond to one-electron transfer processes. The pentakis-fused donor 9d shows six pairs of redox waves. The peak currents of the last two pairs of redox waves were about half as large as those of the others. Considering that 9d has ten redox-active 1,3-dithiol-2-ylidene sites, it is suggested that the last two pairs of redox waves of 9d correspond to one-electron transfer process, while the others correspond to two-electron transfer processes.
![[1860-5397-11-128-5]](/bjoc/content/figures/1860-5397-11-128-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Deconvoluted cyclic voltammograms of (a) 5d, (b) 7d and (c) 9d.
Figure 5: Deconvoluted cyclic voltammograms of (a) 5d, (b) 7d and (c) 9d.
The redox potentials of 5d, 7d and 9d are summarized in Table 2 together with their related compounds. The first two-electron redox potentials of 5d (Em1 = −0.01 V) and 7d (Em1 = −0.04 V) were more negative by 0.13 and 0.16 V than the first redox potential of a TTPY derivative 2e (E1 = +0.12 V) measured under the identical conditions. The first redox waves of 5d and 7d involved two-electron transfer processes, and that E1 of the extended donors 3c and 18 (−0.06 V) was lower than that of the TTF derivative 1c (+0.03 V). These results suggest that two positive charges formed by the first two-electron oxidation process of 5d and 7d are presumably distributed mainly on each of the two extended TTF moieties so as to reduce on-site Coulomb repulsion (Scheme 2). Similarly, the two positive charges in 5d4+ and 7d4+ might be located mainly on each of the two extended TTF donors. Observation of two sequent one-electron redox waves in the higher potential region (+0.6 to +0.9 V) indicates that the central TTF moiety contributes to the remaining redox processes. The significant positive shifts of the E5 and E6 of 5d and 7d by 0.62–0.75 V and 0.41–0.63 V, respectively, compared to the E1 and E2 of 3c is probably due to the strong electron-withdrawing effect by two dicationic extended TTF units in the tetracationic states. In other words, the presence of five and six positive charges in the molecules induces significantly large on-site coulomb repulsion. The E5 and E6 of 7d are lower by 0.12–0.14 V than those of 5d, suggesting that 7d6+ is more stabilized than 5d6+. The thiophene spacers inserted between two cationic 1,3-dithiole rings might reduce the intramolecular coulomb repulsion in 7d6+.
Table 2: Redox potentials of 5d, 7d, 9d and their related compounds (V vs Fc/Fc+, in benzonitrile/carbon disulfide 1:1, v/v).
| Donor | E1 | E2 | E3 | E4 | E5 | E6 | E7 | E8 | E9 | E10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Em1a | Em2a | Em3a | Em4a | |||||||
| 5d | −0.01 | +0.19 | +0.79 | +0.89 | ||||||
| 7d | −0.04 | +0.09 | +0.65 | +0.77 | ||||||
| 9d | −0.07 | +0.16 | +0.38 | +0.57 | +0.81 | +0.92 | ||||
| 2e | +0.12 | +0.19 | +0.39 | +0.87 | ||||||
| 1c | +0.03 | +0.36 | ||||||||
| 3c | −0.06 | +0.05 | ||||||||
| 18 | −0.06 | +0.02 | ||||||||
| 19 | +0.03 | +0.36 | +0.56 | |||||||
a Em1 = (E1+E2)/2. Em2 = (E3+E4)/2. Em3 = (E5+E6)/2. Em4 = (E7+E8)/2.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-128-i4.svg?max-width=637&scale=1.18182)
![[1860-5397-11-128-i2]](/bjoc/content/inline/1860-5397-11-128-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible redox processes of 5d and 7d.
Scheme 2: Plausible redox processes of 5d and 7d.
As for 9d, two positive charges in 9d2+ are probably distributed mainly on each of the two thiophene-inserted TTF moieties, since the first redox wave of 9d corresponds to a simultaneous two-electron transfer process, and the Em1 (−0.07 V) is comparable to the E1 of 18 (−0.06 V) as shown in Scheme 3. The Em2 of 9d (+0.16 V) is lower by 0.20 V than the E3 of 19 (+0.36 V) [28], suggesting that the second redox process is contributed by two extended TTF moieties similarly to the first redox process. On the other hand, both the third and fourth redox waves involve two-electron transfer, and their potentials (Em3 = +0.38 V, Em4 = +0.57 V, respectively) are comparable to the E3 and E4 of 19 (E3 = +0.36 V, E4 = +0.56 V, respectively). Therefore, it is indicated that positive charges formed by the third and fourth redox processes are distributed over two TTF moieties at the both ends. The central TTF moiety contributes to the remaining two stages of the one-electron redox processes at +0.81 and +0.92 V, similarly to 5d and 7d.
![[1860-5397-11-128-i3]](/bjoc/content/inline/1860-5397-11-128-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Charge and discharge properties of rechargeable batteries
In order to examine the cell performance, IEC R2016 coin-type cells were fabricated using a positive electrode incorporating 5, 6 and 8 (5b, 5c, 6b and 8c) as positive electrode materials. The applied current densities were 40 mA g−1 (0.2 C rate) and 100 mA g−1 (0.5 C rate), respectively. The nominal charge capacity of a battery or an electrode is expressed as a C-rate. A 0.2 C rate means that the full discharge capacity reached in 5 h. Cyclic voltammetry in the solid state was carried out prior to the charge–discharge test so as to determine the turning back voltage (Supporting Information File 1, Figure S4). The electrodes incorporating 5b and 5c exhibited three indistinct oxidation peaks at 3.4, 3.6 V and 4.1 V. Multi-scan cycle voltammetry revealed a significant decay of redox waves at 4.1 V presumably due to dissolution of the oxidative species of 5b and 5c in the electrolyte solution. In contrast, there was no distinct decay of the redox waves at 3.6 V, suggesting that the oxidative species of 5b and 5c formed at 3.6 V barely dissolved in the electrolyte solution. In contrast, no distinct dissolution was observed for the 6b and 8c cells even at 4.2 V. Thus, we determined the turn-back voltages as 3.8 V for the 5b and 5c cells, and 4.2 V for the 6b and 8c cell.
The results are summarized in Table 3, and the first five charge–discharge curves of 5b/Li and 6b/Li cells cycled at room temperature are shown in Figure 6. A 5c cell also exhibited charge–discharge curves similar to 5b/Li and 6b/Li cells. No distinct plateau was observed in both the charge and discharge processes for all the cells in spite of observation of well-separated redox waves in a solution. This is possibly due to the apparent overlap of the redox processes in the solid state (see also Figure S4 in Supporting Information File 1) [11]. The initial discharge capacities of 5b/Li, 5c/Li and 6b/Li cells were 157, 168 and 158 mAh g−1, respectively. They correspond to 93 and 99% of the theoretical capacity for the five-electron redox of 5b and 5c, and 94% of the theoretical capacity for the six-electron redox of 6b, respectively. The first discharge capacities observed are comparable to those of the positive active materials for commercially available lithium ion batteries on the market (150–170 mAh g−1). Cycle-life performances for 5b/Li, 5c/Li and 6b/Li cells are shown in Figure 7. In all cases, the discharge capacities decreased gradually as the number of cycles increased. The discharge capacities after 40 cycles were 86, 73 and 74% of the first discharge capacities for 5b/Li, 5c/Li and 6b/Li cells, respectively. The decrease in capacities for the cells using organic electrode materials might be attributed to elution of the positive electrode materials into the electrolyte solution. The result that the 5b cell shows higher cycle performance than the other cells is consistent with the lower solubility of 5b with rigid ethylenedithio substituents than 5c with flexible methylthio substituents.
Table 3: Charge–discharge parameters for the rechargeable batteries using 5, 6 and 8.
| 5b/Li | 5c/Li | 6b/Li | 8c/Li | |
|---|---|---|---|---|
| Theoretical capacities for maximum electrons utilization indicated in parentheses (mAh g−1). | 203(6) | 203(6) | 169(6) | 205(10) |
| 1st Discharge capacity (mAh g−1) | 157 | 168 | 158 | 190 |
| Number of electron per molecule participating discharge | 5 | 5 | 6 | 10 |
| Average voltage for 1st discharge (V) | 3.41 | 3.40 | 3.44 | 3.58 |
| 1st Energy density (mWh g-1) | 535 | 571 | 544 | 680 |
| 40th Discharge capacity/1st discharge capacity (%) | 86 | 73 | 74 | 64 |
![[1860-5397-11-128-6]](/bjoc/content/figures/1860-5397-11-128-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a) Galvanostatic charge-discharge curves of (a) 5c/Li and (b) 6b/Li cells.
Figure 6: (a) Galvanostatic charge-discharge curves of (a) 5c/Li and (b) 6b/Li cells.
![[1860-5397-11-128-7]](/bjoc/content/figures/1860-5397-11-128-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Cycle-life performances for 5b/Li, 5c/Li and 6b/Li cells.
Figure 7: Cycle-life performances for 5b/Li, 5c/Li and 6b/Li cells.
Figure 8 shows the first five charge–discharge curves of a 8c/Li cell cycled at room temperature and their cycle performances up to 40 cycles. The initial charge and discharge capacities were 202 and 190 mAh g−1, respectively. They correspond to 99% and 93% of the theoretical capacity for ten-electron utilization (205 mAh g−1). This result strongly indicates that ten-electron redox per molecule participates in the charge and discharge processes. Similarly to the 5b, 5c and 6b cells, the 8c/Li cell shows no distinct plateau in both the charge and discharge processes in spite of observation of six pairs of redox waves in a solution. The initial discharge capacity of the 8c/Li cell (190 mAh g−1) is higher than those of the cathode active materials for commercially available lithium ion batteries. The discharge capacity after 40 cycles (121 mAh g−1) was 64% of the first discharge capacity. The high cycle-life performance in spite of utilization of the highest oxidation state of +10 might be because of strong van der Waals force between the large π-electron frameworks of the thiophene-inserted pentakis-fused TTF.
![[1860-5397-11-128-8]](/bjoc/content/figures/1860-5397-11-128-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: (a) Galvanostatic charge–discharge curves, and (b) cycle-life performances for a 8c/Li cell.
Figure 8: (a) Galvanostatic charge–discharge curves, and (b) cycle-life performances for a 8c/Li cell.
The energy densities for the first discharge process (1st energy densities) calculated by multiples of the initial discharge capacity and the average voltage are also summarized in Table 3. The first energy densities of 5b, 5c and 6b cells were 535–571 mWh g−1, which are comparable to that of TTPY (543 mWh g−1 for four-electron utilization). On the other hand, the first energy density of 8c cell (680 mWh g−1) is larger which is larger by 110–150 mWh g−1 than those of the others. This value is also superior to the energy densities of most inorganic cathode materials for LIBs [29,30], 20/Li (605 mWh g−1) [31], and is slightly smaller than that of 21/Li (700 mWh g−1) [32] (Figure 9).
![[1860-5397-11-128-9]](/bjoc/content/figures/1860-5397-11-128-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Molecular structures of 20 and 21.
Figure 9: Molecular structures of 20 and 21.
Conclusion
A TTF derivative with two phosphonate groups (12) is a useful building block for the synthesis of odd-numbered fused TTF donors containing extended TTF units. We have demonstrated that some derivatives of 5, 6 and 8 can be utilized as positive electrode materials for rechargeable batteries. The 5b/Li cell showed considerably higher cycle performance when the number of electrons is suppressed to five per molecule (5/6 of the maximum electrons). It is noted that the 6b and 8c cells showed good cycle performance in spite of utilizing the maximum amounts of electrons (six and ten electrons, respectively). The 8c cell exhibited significantly high energy density (680 mWh g−1) at the first discharge thanks to ten electrons utilization. The information obtained from the present work could be helpful in the molecular design and synthesis of new positive electrode materials for rechargeable batteries. We are engaged in the synthesis of the other derivatives of 5, 6 and 8 containing unsubstituted derivatives and higher homologues of 5, which are expected to exhibit higher charge–discharge performance than the materials described in this paper.
Supporting Information
| Supporting Information File 1: Experimental details and spectroscopic data, optimized structures of 5a, 6a and 8a (trans isomers) and cycle-life performances for rechargeable batteries using 5c, 5b and 6b. | ||
| Format: PDF | Size: 466.8 KB | Download |
Acknowledgements
This work is partially supported by a Grant-in-Aid for Scientific Research (No. 23550155), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, MEXT program "Elements Strategy Initiative to Form Core Research Center" (since 2012), and Grant-in-Aid for Research Promotion, Ehime University.
References
-
Yamada, J.; Sugimoto, T., Eds. TTF Chemistry–Fundamental and Applications of Tetrathiafulvalene; Kodansha-Springer: Tokyo, 2004.
Return to citation in text: [1] -
Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n
Return to citation in text: [1] -
Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::AID-ANIE1372>3.0.CO;2-I
Return to citation in text: [1] -
Gorgues, A.; Hudhomme, P.; Sallé, M. Chem. Rev. 2004, 104, 5151–5184. doi:10.1021/cr0306485
Return to citation in text: [1] -
Misaki, Y. Sci. Technol. Adv. Mater. 2009, 10, 024301. doi:10.1088/1468-6996/10/2/024301
Return to citation in text: [1] [2] [3] -
Misaki, Y.; Matsui, T.; Kawakami, K.; Nishikawa, H.; Yamabe, T.; Shiro, M. Chem. Lett. 1993, 22, 1337–1340. doi:10.1246/cl.1993.1337
Return to citation in text: [1] -
Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5114. doi:10.1021/cr030651o
Return to citation in text: [1] -
Misaki, Y.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka, S. Chem. Lett. 1994, 23, 1653–1656. doi:10.1246/cl.1994.1653
Return to citation in text: [1] -
Nishikawa, H.; Kawauchi, S.; Misaki, Y.; Yamabe, T. Chem. Lett. 1996, 25, 43–44. doi:10.1246/cl.1996.43
Return to citation in text: [1] -
Misaki, Y.; Kawakami, K.; Higuchi, N.; Nishikawa, H.; Yamabe, T. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 1996, 284, 337–344. doi:10.1080/10587259608037936
Return to citation in text: [1] -
Inatomi, Y.; Hojo, N.; Yamamoto, T.; Watanabe, S.; Misaki, Y. ChemPlusChem 2012, 77, 973–976. doi:10.1002/cplu.201200197
Return to citation in text: [1] [2] [3] -
Inatomi, Y.; Hojo, N.; Yamamoto, T.; Shimada, M.; Watanabe, S. presentation No. 167. In ECS Meeting Abstracts 2008, MA2008-1, 213th ECS meeting, Phoenix, May 22, 2008; The Electrochemical Society.
Return to citation in text: [1] -
Oyaizu, K.; Suga, T.; Yoshimura, K.; Nishide, H. Macromolecules 2008, 41, 6646–6652. doi:10.1021/ma702576z
Return to citation in text: [1] -
Yoshida, Z.; Kawase, T.; Awaji, H.; Sugimoto, I.; Sugimoto, T.; Yoneda, S. Tetrahedron Lett. 1983, 24, 3469–3472. doi:10.1016/S0040-4039(00)86015-3
Return to citation in text: [1] -
Sugimoto, T.; Awaji, H.; Sugimoto, I.; Misaki, Y.; Kawase, T.; Yoneda, S.; Yoshida, Z.; Kobayashi, T.; Anzai, H. Chem. Mater. 1989, 1, 535–547. doi:10.1021/cm00005a015
Return to citation in text: [1] -
Moore, A. J.; Bryce, M. R.; Ando, D. J.; Hursthouse, M. B. J. Chem. Soc., Chem. Commun. 1991, 320–322. doi:10.1039/c39910000320
Return to citation in text: [1] [2] -
Hansen, T. K.; Lakshmikantham, M. V.; Cava, M. P.; Niziurski-Mann, R. E.; Jensen, F.; Becher, J. J. Am. Chem. Soc. 1992, 114, 5035–5039. doi:10.1021/ja00039a013
Return to citation in text: [1] [2] -
Takahashi, K.; Nihira, T.; Yoshifuji, M.; Tomitani, K. Bull. Chem. Soc. Jpn. 1993, 66, 2330–2334. doi:10.1246/bcsj.66.2330
Return to citation in text: [1] [2] -
Benahmed-Gasmi, A. S.; Frère, P.; Garrigues, B.; Gorgues, A.; Jubault, M.; Carlier, R.; Texier, F. Tetrahedron Lett. 1992, 33, 6457–6460. doi:10.1016/S0040-4039(00)79014-9
Return to citation in text: [1] [2] -
Misaki, Y.; Kubo, A.; Matsuda, W.; Fueno, H.; Tanaka, K. Curr. Appl. Phys. 2006, 6, 934–938. doi:10.1016/j.cap.2005.01.043
Return to citation in text: [1] -
Misaki, Y.; Nishikawa, H.; Kawakami, K.; Uehara, T.; Yamabe, T. Tetrahedron Lett. 1992, 33, 4321–4324. doi:10.1016/S0040-4039(00)74250-X
Return to citation in text: [1] -
Misaki, Y.; Higuchi, N.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka, S. Angew. Chem., Int. Ed. Engl. 1995, 34, 1222–1225. doi:10.1002/anie.199512221
Return to citation in text: [1] -
Misaki, Y.; Ohta, T.; Higuchi, N.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka, S. J. Mater. Chem. 1995, 5, 1571–1579. doi:10.1039/jm9950501571
Return to citation in text: [1] -
Takahashi, K.; Tanioka, H.; Fueno, H.; Misaki, Y.; Tanaka, K. Chem. Lett. 2002, 31, 1002–1003. doi:10.1246/cl.2002.1002
Return to citation in text: [1] -
Moore, A. J.; Bryce, M. R. Tetrahedron Lett. 1992, 33, 1373–1376. doi:10.1016/S0040-4039(00)91626-5
Return to citation in text: [1] -
Nakamura, K.; Shirahata, T.; Miyamoto, H.; Misaki, Y. Heterocycles 2011, 83, 2115–2126. doi:10.3987/COM-11-12277
Return to citation in text: [1] -
Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Misaki, Y.; Sasaki, T.; Ohta, T.; Fujiwara, H.; Yamabe, T. Adv. Mater. 1996, 8, 804–807. doi:10.1002/adma.19960081006
Return to citation in text: [1] -
Aravindan, V.; Gnanaraj, J.; Lee, Y.-S.; Madhavi, S. J. Mater. Chem. A 2013, 1, 3518–3539. doi:10.1039/c2ta01393b
Return to citation in text: [1] -
Thackeray, M. M.; Wolverton, C.; Isaacs, E. D. Energy Environ. Sci. 2012, 5, 7854–7863. doi:10.1039/c2ee21892e
Return to citation in text: [1] -
Kato, M.; Ogi, D.; Yao, M.; Misaki, Y. Chem. Lett. 2013, 42, 1556–1558. doi:10.1246/cl.130841
Return to citation in text: [1] -
Kato, M.; Senoo, K.; Yao, M.; Misaki, Y. J. Mater. Chem. A 2014, 2, 6747–6754. doi:10.1039/c3ta14920j
Return to citation in text: [1]
| 17. | Hansen, T. K.; Lakshmikantham, M. V.; Cava, M. P.; Niziurski-Mann, R. E.; Jensen, F.; Becher, J. J. Am. Chem. Soc. 1992, 114, 5035–5039. doi:10.1021/ja00039a013 |
| 18. | Takahashi, K.; Nihira, T.; Yoshifuji, M.; Tomitani, K. Bull. Chem. Soc. Jpn. 1993, 66, 2330–2334. doi:10.1246/bcsj.66.2330 |
| 19. | Benahmed-Gasmi, A. S.; Frère, P.; Garrigues, B.; Gorgues, A.; Jubault, M.; Carlier, R.; Texier, F. Tetrahedron Lett. 1992, 33, 6457–6460. doi:10.1016/S0040-4039(00)79014-9 |
| 26. | Nakamura, K.; Shirahata, T.; Miyamoto, H.; Misaki, Y. Heterocycles 2011, 83, 2115–2126. doi:10.3987/COM-11-12277 |
| 1. | Yamada, J.; Sugimoto, T., Eds. TTF Chemistry–Fundamental and Applications of Tetrathiafulvalene; Kodansha-Springer: Tokyo, 2004. |
| 2. | Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n |
| 3. | Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::AID-ANIE1372>3.0.CO;2-I |
| 4. | Gorgues, A.; Hudhomme, P.; Sallé, M. Chem. Rev. 2004, 104, 5151–5184. doi:10.1021/cr0306485 |
| 5. | Misaki, Y. Sci. Technol. Adv. Mater. 2009, 10, 024301. doi:10.1088/1468-6996/10/2/024301 |
| 5. | Misaki, Y. Sci. Technol. Adv. Mater. 2009, 10, 024301. doi:10.1088/1468-6996/10/2/024301 |
| 8. | Misaki, Y.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka, S. Chem. Lett. 1994, 23, 1653–1656. doi:10.1246/cl.1994.1653 |
| 22. | Misaki, Y.; Higuchi, N.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka, S. Angew. Chem., Int. Ed. Engl. 1995, 34, 1222–1225. doi:10.1002/anie.199512221 |
| 23. | Misaki, Y.; Ohta, T.; Higuchi, N.; Fujiwara, H.; Yamabe, T.; Mori, T.; Mori, H.; Tanaka, S. J. Mater. Chem. 1995, 5, 1571–1579. doi:10.1039/jm9950501571 |
| 24. | Takahashi, K.; Tanioka, H.; Fueno, H.; Misaki, Y.; Tanaka, K. Chem. Lett. 2002, 31, 1002–1003. doi:10.1246/cl.2002.1002 |
| 7. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5114. doi:10.1021/cr030651o |
| 16. | Moore, A. J.; Bryce, M. R.; Ando, D. J.; Hursthouse, M. B. J. Chem. Soc., Chem. Commun. 1991, 320–322. doi:10.1039/c39910000320 |
| 25. | Moore, A. J.; Bryce, M. R. Tetrahedron Lett. 1992, 33, 1373–1376. doi:10.1016/S0040-4039(00)91626-5 |
| 6. | Misaki, Y.; Matsui, T.; Kawakami, K.; Nishikawa, H.; Yamabe, T.; Shiro, M. Chem. Lett. 1993, 22, 1337–1340. doi:10.1246/cl.1993.1337 |
| 20. | Misaki, Y.; Kubo, A.; Matsuda, W.; Fueno, H.; Tanaka, K. Curr. Appl. Phys. 2006, 6, 934–938. doi:10.1016/j.cap.2005.01.043 |
| 32. | Kato, M.; Senoo, K.; Yao, M.; Misaki, Y. J. Mater. Chem. A 2014, 2, 6747–6754. doi:10.1039/c3ta14920j |
| 5. | Misaki, Y. Sci. Technol. Adv. Mater. 2009, 10, 024301. doi:10.1088/1468-6996/10/2/024301 |
| 21. | Misaki, Y.; Nishikawa, H.; Kawakami, K.; Uehara, T.; Yamabe, T. Tetrahedron Lett. 1992, 33, 4321–4324. doi:10.1016/S0040-4039(00)74250-X |
| 13. | Oyaizu, K.; Suga, T.; Yoshimura, K.; Nishide, H. Macromolecules 2008, 41, 6646–6652. doi:10.1021/ma702576z |
| 14. | Yoshida, Z.; Kawase, T.; Awaji, H.; Sugimoto, I.; Sugimoto, T.; Yoneda, S. Tetrahedron Lett. 1983, 24, 3469–3472. doi:10.1016/S0040-4039(00)86015-3 |
| 15. | Sugimoto, T.; Awaji, H.; Sugimoto, I.; Misaki, Y.; Kawase, T.; Yoneda, S.; Yoshida, Z.; Kobayashi, T.; Anzai, H. Chem. Mater. 1989, 1, 535–547. doi:10.1021/cm00005a015 |
| 16. | Moore, A. J.; Bryce, M. R.; Ando, D. J.; Hursthouse, M. B. J. Chem. Soc., Chem. Commun. 1991, 320–322. doi:10.1039/c39910000320 |
| 29. | Aravindan, V.; Gnanaraj, J.; Lee, Y.-S.; Madhavi, S. J. Mater. Chem. A 2013, 1, 3518–3539. doi:10.1039/c2ta01393b |
| 30. | Thackeray, M. M.; Wolverton, C.; Isaacs, E. D. Energy Environ. Sci. 2012, 5, 7854–7863. doi:10.1039/c2ee21892e |
| 12. | Inatomi, Y.; Hojo, N.; Yamamoto, T.; Shimada, M.; Watanabe, S. presentation No. 167. In ECS Meeting Abstracts 2008, MA2008-1, 213th ECS meeting, Phoenix, May 22, 2008; The Electrochemical Society. |
| 17. | Hansen, T. K.; Lakshmikantham, M. V.; Cava, M. P.; Niziurski-Mann, R. E.; Jensen, F.; Becher, J. J. Am. Chem. Soc. 1992, 114, 5035–5039. doi:10.1021/ja00039a013 |
| 18. | Takahashi, K.; Nihira, T.; Yoshifuji, M.; Tomitani, K. Bull. Chem. Soc. Jpn. 1993, 66, 2330–2334. doi:10.1246/bcsj.66.2330 |
| 19. | Benahmed-Gasmi, A. S.; Frère, P.; Garrigues, B.; Gorgues, A.; Jubault, M.; Carlier, R.; Texier, F. Tetrahedron Lett. 1992, 33, 6457–6460. doi:10.1016/S0040-4039(00)79014-9 |
| 31. | Kato, M.; Ogi, D.; Yao, M.; Misaki, Y. Chem. Lett. 2013, 42, 1556–1558. doi:10.1246/cl.130841 |
| 11. | Inatomi, Y.; Hojo, N.; Yamamoto, T.; Watanabe, S.; Misaki, Y. ChemPlusChem 2012, 77, 973–976. doi:10.1002/cplu.201200197 |
| 28. | Misaki, Y.; Sasaki, T.; Ohta, T.; Fujiwara, H.; Yamabe, T. Adv. Mater. 1996, 8, 804–807. doi:10.1002/adma.19960081006 |
| 9. | Nishikawa, H.; Kawauchi, S.; Misaki, Y.; Yamabe, T. Chem. Lett. 1996, 25, 43–44. doi:10.1246/cl.1996.43 |
| 10. | Misaki, Y.; Kawakami, K.; Higuchi, N.; Nishikawa, H.; Yamabe, T. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 1996, 284, 337–344. doi:10.1080/10587259608037936 |
| 11. | Inatomi, Y.; Hojo, N.; Yamamoto, T.; Watanabe, S.; Misaki, Y. ChemPlusChem 2012, 77, 973–976. doi:10.1002/cplu.201200197 |
| 11. | Inatomi, Y.; Hojo, N.; Yamamoto, T.; Watanabe, S.; Misaki, Y. ChemPlusChem 2012, 77, 973–976. doi:10.1002/cplu.201200197 |
© 2015 Iwamoto et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)