Abstract
Cinnamic acid derivatives bearing a nitroxyl moiety (2,2,6,6-tetramethyl-1-oxyl-4-piperidyl 3-E-aryl acrylates) were synthesized in 30–100% yield using a Mizoroki–Heck cross-coupling reaction between 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl and iodobenzene derivatives in the presence of palladium(II) acetate coordinated with a tri(o-tolyl)phosphine ligand immobilized in a polyurea matrix.

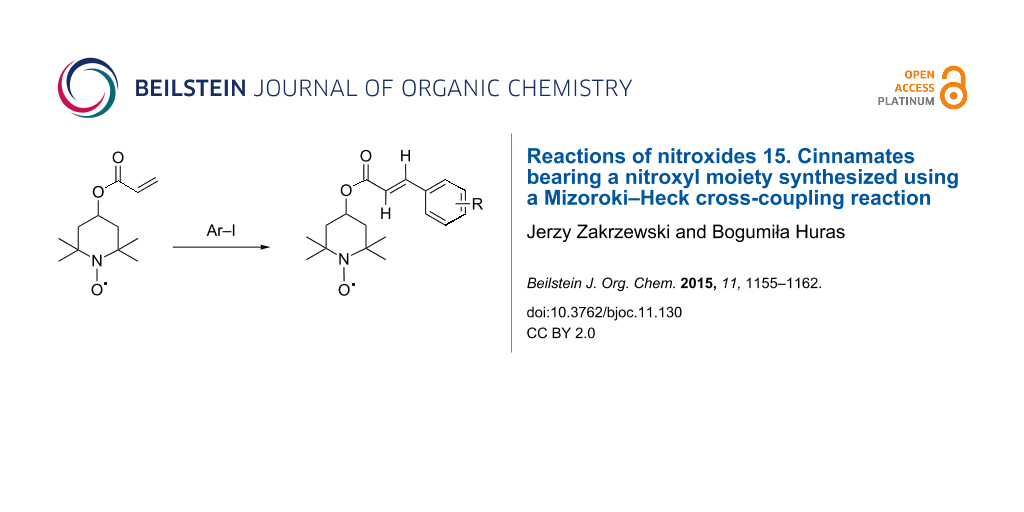
Graphical Abstract
Introduction
Cinnamic acid derivatives are known as biologically active compounds. Cinnamic acid and its hydroxy derivatives bearing a phenolic moiety such as coumaric, caffeic, ferulic, sinapinic, and chlorogenic acids [2-6], simple cinnamic acid derivatives (esters, amides, etc.), and prenylated cinnamates [4], have been proved to be effective as antioxidants [2,7], radical scavengers [2], antimicrobials [2,7,8], antibacterials [2], antivirals [2,7], and fungicides [2,7,8]. Cinnamic derivatives have also been recognized as active agents against tuberculosis and malaria [3,7], cardiovascular diseases [3], and cancer [4]. Cinnamates show depigmenting [4], antidiabetic, antihyperglycemic, anticholesterolemic, anti-inflammatory, hepatoprotective, CNS depressant, anxiolytic, and cytotoxic activity [7]. Cinnamate esters have been used as effective UV filters (especially in UVB region, 280–320 nm) in cosmetics [7,9], and as fragrance materials [7]. Fragrance material reviews on cinnamic acid derivatives were extensively described in Food and Chemical Toxicology (especially in 2007) and in Food and Cosmetics Toxicology (in the 70s). The most recent reviews in this series were published in 2011 [10,11].
Cinnamic acid derivatives can be synthesized using Perkin, Knoevenagel, Claisen [7], and Wittig [12] condensation reactions. Since the discovery of catalytic coupling reactions, cinnamic derivatives have also been obtained using the Mizoroki–Heck cross-coupling reaction between aryl halides and an olefin [7,13-17]. Because cinnamates themselves are also olefins, they can serve as cinnamic substrates to synthesize more complex cinnamates [18,19]. Cinnamic acid derivatives are also formed when saturated aliphatic acids (instead of unsaturated ones as acrylates) are reacted with simple aromatic compounds (as benzene) in the presence of palladium(II) chloride [20]. Due to the important biological activity of cinnamates, the incorporation of a spin label moiety, particularly a nitroxyl fragment, is interesting, because it allows for the study of cinnamates using spin labeling methods.
To accomplish the synthesis of cinnamates bearing a nitroxyl moiety, we applied a Mizoroki–Heck cross-coupling reaction, using 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3, an olefin component bearing a nitroxyl moiety), recently applied in the Morita–Baylis–Hillmann reaction [21]. The use of nitroxides in cross-coupling reactions is described only in a limited number of papers [22-32]. To the best of our knowledge, there are no systematic studies on the use of nitroxides in the Mizoroki–Heck cross-coupling reaction.
Despite the observation that an unpaired electron in nitroxides does not participate in organic reactions has been well known since the beginning of the nitroxide progress in 60's [33] the reactions of nitroxides involving an unpaired electron are also recognized (e.g., reductions, acidic medium, carbene addition, etc.). Because we would like to check the possibility of performing the Mizoroki–Heck cross-coupling reaction with nitroxides, herein the synthesis of cinnamates bearing a nitroxyl moiety is presented using aryl iodides as exemplary test compounds.
Results and Disscusion
4-Acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3) was obtained in 90–95% yield by the reaction of acryloyl chloride (2) with 2,2,6,6-tetramethyl-4-piperidinol-1-oxyl (1) in the presence of triethylamine [21,34-36].
The couplings of 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3) with iodobenzene (4a) and 4-methyliodobenzene (4b) were used as the test reactions to check the effectiveness of various palladium catalyst systems.
The use of Pd(OAc)2/Ph3P/Bu3N [37] resulted in a low yield of the target products. No products were obtained when other catalyst systems: Pd[PPh3]4 [38], Pd(CF3COO)2/tri(2-furylphosphine) and Pd(acetylacetonate)2/tri(2-furylphosphine) [39] were tested. As a rule, unreacted 3 was identified, and always isolated. A number of unidentified products were detected by means of TLC.
Finally, the target cinnamates bearing a nitroxyl moiety (5a–i) were obtained using the coupling of 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3) with a series of iodobenzene derivatives (4a–i) in the presence of palladium(II) acetate coordinated with tri(o-tolyl)phosphine ligand immobilized in a polyurea matrix (commercially available as PdEnCatTOTP30™) [40-45] (Scheme 1, Table 1).
![[1860-5397-11-130-i1]](/bjoc/content/inline/1860-5397-11-130-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cinnamates bearing a nitroxyl moiety 5a–i from 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3) and iodobenzene derivatives; (i) CH2=CH–COCl (2), NEt3, benzene, 90–95%; (ii) R–Ar–I 4a–i, PdEnCat TOTP30, Bu4N+AcO−, toluene, 80–100 °C, 30–100%.
Scheme 1: Cinnamates bearing a nitroxyl moiety 5a–i from 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3...
Table 1: Cinnamates bearing a nitroxyl moiety (5a–i).
| Compound | R | Reaction temperature [°C] | Reaction time [h] | Yield [%] | mp [°C] |
|---|---|---|---|---|---|
| 5a | H | 80 | 22 | 42.4 | 97–98 |
| 5b | 4-CH3 | 100 | 2–20 | 30.4 | oil |
| 5c | 3,4-diCl | 80 | 25 | 62.7 | 118–120 |
| 5d | 4-CH3O | 85 | 24 | 75.8 | 70–74 |
| 5e | 2-NO2 | 90 | 20 | 76.4 | 107–110 |
| 5f | 3-NO2 | 80–90 | 20 | 99.9 | 121–123 |
| 5g | 4-NO2 | 80–85 | 23 | 55.3 | 148–149 |
| 5h | 2-CH3-4-NO2 | 80–85 | 27 | 88.3 | 109–112 |
| 5i | 2-Cl-4-NO2 | 80–85 | 27 | 56.2 | 107–110 |
The best results were obtained when an electron-withdrawing substituent NO2 was present in the benzene ring. The reactions of iodobenzene bearing 4-F, 4-CF3, 2,4-diNO2 substituents were unsuccessful. In the case of the reaction of 4b leading to the cinnamate 5b, the results were unrepeatable (the times and yields of the reactions). Unidentified impurities together with the product 5b were observed (13C NMR).
No products in the case of 4-F, 4-CF3, 2,4-diNO2 substituents and unrepeatability in the case of cinnamate 5b can be caused by means of a type of a heterogeneous catalyst which is immobilized on a solid support. In addition to the widely highlighted unquestionable advantages of immobilized, heterogeneous catalysts (easy separation from the reaction mixture and the possibility of re-use), such catalysts are rigidly anchored on a carrier and may cause hindered interaction with substrates. Some of the catalytic sites may be buried within the polymer matrix and cannot participate in the reactions [46]. This disadvantage may cause various unwanted effects. As examples, no clear relationship between the structures of the aryl iodides and the yields were observed, as described in [47], or a catalyst that accepts only electron-rich aryl iodides [48].
The structure of the synthesized cinnamates bearing a nitroxyl moiety were confirmed using EIMS, ESIMS, HREIMS, HRESIMS, 1H NMR, 13C NMR, and IR spectra (Experimental part and Supporting Information File 1). Recording of the NMR spectra of the nitroxides required removing of the unpaired electron. This was achieved by adding a drop of a reducing agent – in fact the spectra of the corresponding hydroxylamines were recorded. In this research phenylhydrazine has been used [49,50], however, sometimes using of hydrazobenzene [51,52] or pentafluorophenyl hydrazine [35] as reducing agents have been reported, as well.
The E geometry of 5a–i was confirmed by 1H NMR spectroscopy. The values of the coupling constants of the doublets visible in the 1H NMR spectra of 5a–i belonging to the double bond of 5a–i remain in the range 15.5–16.0 Hz. These values confirm the E geometry of 5a–i. In Table 2 the shifts and the coupling constants of the double bond are presented.
Table 2: Chemical shifts and coupling constants of the hydrogen atoms belonging to the double bond of 5a–i: OOC–CH(B)=CH(D)–C6H4(3)–R.
| 5 | R | δ H(B) [ppm] | δ H(D) [ppm] | JDB [Hz] |
|---|---|---|---|---|
| a | H | 6.41 | 7.67 | 16.0 |
| b | 4-CH3 | 6.35 | 7.64 | 15.8 |
| c | 3,4-diCl | 6.38 | 7.40 | 15.5 |
| d | OCH3 | 6.28 | 7.62 | 16.0 |
| e | 2-NO2 | 6.33 | 8.10 | 16.0 |
| f | 3-NO2 | 6.53 | 7.69 | 16.0 |
| g | 4-NO2 | 6.52 | 7.32 | 16.0 |
| h | 2-CH3-4-NO2 | 6.42 | 7.90 | 15.9 |
| i | 2-Cl-4-NO2 | 6.51 | 8.03 | 16.0 |
The substituent in 2-position of the cinnamates 5e, 5h, 5i causes the noticeable shift of HD hydrogen to the lower field. This observation suggests that conformation X, where the substituent in 2-position and the HD atom are placed close to each other, is more populated than conformation Y (Figure 1).
![[1860-5397-11-130-1]](/bjoc/content/figures/1860-5397-11-130-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Conformations of the substituent in 2-position of the cinnamates 5e, 5h, 5i.
Figure 1: Conformations of the substituent in 2-position of the cinnamates 5e, 5h, 5i.
For cinnamate 5f (3-NO2), the direct measurement of the chemical shift of the hydrogen atom HD (OOC-CH(B)=CH(D)-C6H4-NO2(m)) was not possible, due to the presence of more than one equidistant (16 Hz) peak close to 7.5 ppm. Irradiation of HB (δ 6.53 ppm) allowed to distinguish the signal of HD at 7.69 ppm.
A molecular peak was present in all mass spectra of the synthesized products 5a–5i. Except in the case of 5c,i it was clearly visible (5–25%). In all of the mass spectra, the abundant peak of the cinnamyl acyl cation ArCH=CHCO+ was present. This was a base peak in the case of 5a,d.
Characteristic signals for 4-XO-substituted 2,2,6,6-tetramethylpiperidine-1-oxyl moiety (TEMPOL, TEMPOL esters, etc.) were observed at m/z = 154, 124, 109 [53,54]. The signals at m/z 124, and 109 are abundant. The signal at m/z 154 was assigned to the structure, resulting from elimination of a XOH from the position 4 and 3 of the piperidine ring. The subsequent loss of a NO group (M = 30) and a CH3 group (M = 15), respectively, generates ions at m/z 124 and 109 (Scheme 2). The peaks at m/z 154, 124 and 109 were thoroughly analyzed for the acrylate 3, as an example.
![[1860-5397-11-130-i2]](/bjoc/content/inline/1860-5397-11-130-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The formation of the fragment ions at m/z 154, 124, 109.
Scheme 2: The formation of the fragment ions at m/z 154, 124, 109.
Proposed elemental composition of the fragmentation ions at m/z 154, 124, 109 for the acrylate 3 were confirmed by the measurement of their exact mass:
- m/z 154 (calculated C9H16NO: 154.12319, found: 154.12390),
- m/z 124 (calculated for C9H16: 124.12520, found: 124.12515),
- m/z 109 (calculated for C8H13: 109.10173, found: 109.10079).
The signal at m/z 124 is the base one in the case of 5b,e,g,h, and that at m/z 109 in the case of 5c,f,i. The m/z values and relative intensities of the discussed signals are collected in Table 3.
Table 3: The relative intensities of the molecular, cinnamic acyl, m/z 154, m/z 124, and m/z 109 ions.
| 5 | M [m/z (int.%)] | ArCH=CHCO [m/z (int.%)] |
m/z 154
int. % |
m/z 124
int. % |
m/z 109
int. % |
|---|---|---|---|---|---|
| a | 302 (10) | 131 (100, C6H5CH=CHCO) | 27 | 89 | 69 |
| b | 316 (24) | 145 (91, 4-CH3C6H4CH=CHCO) | 46 | 100 | 75 |
| c | 370 (2) | 199 (64, 3,4-Cl2CH3C6H3CH=CHCO) | 16 | 88 | 100 |
| d | 332 (19) | 161 (100, 4-CH3OC6H4CH=CHCO) | 20 | 61 | 44 |
| e | 347 (23) | 176 (63, 2-NO2C6H4CH=CHCO) | 25 | 100 | 87 |
| f | 347 (5) | 176 (85, 3-NO2C6H4CH=CHCO) | 13 | 83 | 100 |
| g | 347 (19) | 176 (89, 4-NO2C6H4CH=CHCO) | 19 | 100 | 90 |
| h | 361 (15) | 190 (74, 2-CH3-4-NO2C6H4CH=CHCO) | 29 | 100 | 82 |
| i | 381 (2) | 210 (41, 2-Cl-4-NO2C6H4CH=CHCO) | 15 | 89 | 100 |
In conclusion, we showed that 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3) can be used as starting material in the synthesis of new cinnamates containing a nitroxyl group in a Mizoroki–Heck cross-coupling reaction.
Experimental
General
The protocols for the synthesis of 4-acryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (3) and its precursor 2,2,6,6-tetramethyl-4-hydroxypiperidin-1-oxyl (TEMPOL, 1) were done according to [21]. The identity of 3 was additionally confirmed by 1H and 13C NMR performed in the presence of PhNHNH2. 1H NMR (200 MHz, δ, CDCl3, TMS, in the presence of PhNHNH2) 1.23 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3), 1.24 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.65 (t, J = 11.8 Hz, 2H, HHC-CH(O-)-CHH), 1.96 (ddt, J = 12.8 Hz, J = 4.2 Hz, J = 1.5 Hz, 2H, HHC-CH(O-)-CHH), 5.13 (tt, J = 11.2 Hz, J = 4.3 Hz, 1H, CH-OC(=O)), 5.81 (dd, J = 10.3 Hz, J = 1.6 Hz, 1H, CHH=CH-COO (cis)), 6.09 (dd, J = 17.2 Hz, J = 10.3 Hz, 1H, CH2=CH-COO), 6.39 (dd, J = 17.2 Hz, J = 1.6 Hz, 1H, CHH=CH-COO (trans)); 13C NMR (50 MHz, δ, CDCl3, TMS, in the presence of PhNHNH2) 20.79 (2×CH3), 31.86 (2×CH3), 43.92 (2×CH2), 59.89 (2×C(CH3)2), 66.99 (CH-O-), 128.94 (COCH=CH2), 130.84 (COCH=CH2), 165.95 (C=O).
PdEnCatTOTP30 was purchased from Aldrich. Iodobenzene derivatives and catalysts were purchased from Aldrich, Alfa-Aesar and Fluorochem. The Pd(PPh3)4 was synthesized according to the known procedure [38]. The experiments were performed in a 25–50 mL round-bottom two necked flask, equipped with a magnetic stirrer and a reflux condenser under anhydrous argon atmosphere (inlet through the top of the condenser equipped with a short drying column packed with drying silica, outlet by a needle placed in a septum in a side neck of the flask). Most of the products were obtained as red solids. TLC was performed on silica gel Merck aluminium roll 5562 or aluminium sheets 5554. Column chromatography was performed using Merck 1.09385.1000 or Zeochem 60 hyd 40–63 μm silica gel (0.040–0.063 mm, 230–400 mesh), respectively. TLC visualisation: UV 254 nm light and/or iodine vapours. MS (EI, 70 eV, m/z, int. (%)) data were recorded using AMD 604 and Agilent Technologies 5975 B mass spectrometers. HRMS (EI) data were recorded using an AMD 604 mass spectrometer. MS and HRMS (ESI, positive ion, CH3OH as a solvent) were recorded using a Micromass LCT apparatus. IR (cm−1) data were recorded using a FTIR Jasco 420 spectrometer. 1H and 13C NMR data were collected using a Varian UNITYplus 200 spectrometer. NMR spectra were performed with a drop of phenylhydrazine (in fact the spectra of corresponding hydroxylamines were recorded) [49].
Cinnamates 5a–i; general procedure using PdEnCatTOTP30 as a catalyst
4-Acryloyloxy-2,2,6,6-tetramethylpiperidin-1-oxyl (3, 0.113 g, 0.5 mmol), Aryl iodide (4a–i, 0.5 mmol), Bu4N+AcO− (hygroscopic) (0.3 g), PdEnCatTOTP30 (0.0625 g, 0.025 mol, 5 mol %), and toluene (2 mL) were placed in a flask, stirred and heated at 80–100 °C for 20–27 h under argon. The particular temperatures and times of the reactions are summarized in Table 1. After approximately 6 h, a second portion of a catalyst (0.0625 g, 0.025 mol, 5 mol %) was added. The progress of the reaction was monitored by TLC (silica, hexane/ethyl acetate 9:1). Upon completion of the reaction, the solids were filtered off, the dark filtrate was concentrated under reduced pressure and subjected to column chromatography (hexane/ethyl acetate 9:1, benzene/ethyl acetate 95:5, benzene/methanol 95:5 as possible mobile phases). The red zone was collected, the eluate was evaporated under reduced pressure to yield red oils (solidified in a refrigerator), or directly red crystals of 5a–i.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-phenylacrylate (5a)
42.4%; mp 97–98 °C; MS (EI, 70 eV, m/z, int [%]) 302 (10, M+), 272 (7), 207 (19), 179 (6), 178 (10), 154 (27), 140 (20), 139 (14), 131 (100, ArCH=CHCO), 124 (89), 109 (69), 103 (41), 82 (15), 81 (12), 77 (21), 69 (13), 68 (9), 67 (9), 55 (10), 41 (18); HRMS (EI, 70 eV, m/z, int [%]): calcd for C18H24NO3: 302.1756, found: 302.1750; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.26 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3), 1.28 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.74 (t, J = 12.0 Hz, 2H, HHC-CH(O-)-CHH), 2.02 (ddd, J = 12.8 Hz, J = 4.4 Hz, J = 1.2 Hz, 2H, HHC-CH(O-)-CHH), 5.20 (tt, J = 11.2 Hz, J = 4.3 Hz, 1H, CH-OC(=O)), 6.41 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO), 7.67 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.87 (2×CH3), 31.67 (2×CH3), 43.87 (2×CH2), 60.46 (2×C(CH3)2), 66.76 (CH-O-), 118.51 (CH), 128.30 (CH), 129.10 (CH), 130.52 (CH), 134.57 (C), 145.00 (CH), 166.68 (C=O); IR (cm−1, KBr) 2976, 2937, 1712, 1639, 1450, 1308, 1168, 1008, 978, 859, 765, 709, 685.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(4-methylphenyl)acrylate (5b)
30.4%, oil; MS (EI, 70 eV, m/z, int [%]) 316 (24, M+), 286 (15), 230 (6), 163 (13), 162 (21), 154 (46), 145 (91, ArCH=CHCO), 140 (62), 139 (27), 124 (100), 117 (43), 115 (49), 109 (75), 98 (14), 91 (32), 82 (37), 81 (22), 69 (31), 68 (17), 67 (18), 65 (10), 60 (8), 59 (9), 58 (11), 57 (13), 56 (19), 55 (25), 43 (35), 41 (39); MS (ESI, m/z, int [%]): 340 (15, M + 23 + H), 318 (M + 2H); HRMS (EI, 70 eV, m/z, int [%]): calcd for C19H26NO3: 316.1913, found: 316.1926; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.85 (t, J = 12.1 Hz, 2H, HHC-CH(O-)-CHH), 2.02–2.17 (m, 2H, HHC-CH(O-)-CHH), 2.37 (s, 3H, CH3), 5.23 (tt, J = 11.1 Hz, J = 4.4 Hz, 1H, CH-OC(=O)), 6.35 (d, J = 15.8 Hz, 1H, Ar-CH=CH-COO), 7.64 (d, J = 15.8 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2): 21.35 (impurity), 21.69 (2×CH3), 23.04 (ArCH3), 29.84 (2×CH3), 43.01 (2×CH2), 62.60 (2×C(CH3)2), 65.67 (CH-O-), 115.32 (impurity), 117.03 (CH), 128.34 (CH), 129.86 (CH), 141.12 (C), 145.37 (CH), 166.73 (C=O), 179.47 (impurity); IR (cm−1, film): 2976, 1711, 1635, 1166, 815.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(3,4-dichlorophenyl)acrylate (5c)
62.7%; pink crystals, mp 118–120 °C; MS (EI, 70 eV, m/z, int [%]) 370 (2, M+), 201 (40), 199 (64, ArCH=CHCO), 173 (12), 171 (19), 154 (16), 136 (42), 124 (88), 109 (100), 101 (8), 99 (11), 82 (16), 81 (19), 69 (18), 67 (21), 56 (17), 55 (25), 41 (65); MS (ESI, m/z, int [%]): 395 (20), 393 (90, M + Na), 304 (100); HRMS (ESI): calcd for C18H22NO3Cl2Na: 393.0874, found, 393.0890; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.27 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.29 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.75 (t, J = 12.0 Hz, 2H, HHC-CH(O-)-CHH), 2.02 (dd, J = 12.8 Hz, J = 4.4 Hz, 2H, HHC-CH(O-)-CHH), 5.20 (tt, J = 11.3 Hz, J = 4.3 Hz, 1H, CH-OC(=O)), 6.38 (d, J = 15.5 Hz, 1H, Ar-CH=CH-COO), 7.40 (d, J = 15.5 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.87 (2×CH3), 31.54 (2×CH3), 43.72 (2×CH2), 60.70 (2×C(CH3)2), 67.04 (CH-O-), 117.10 (C), 120.36 (CH), 127.23 (CH), 129.85 (CH), 131.10 (CH), 133.45 (C), 134.59 (C), 142.24 (CH), 166.02 (C=O); IR (cm−1, KBr) 2975, 1706, 1638, 1474, 1319, 1267, 1173, 1150, 1206, 979, 822.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(4-methoxyphenyl)acrylate (5d)
75.8%; mp 70–74 °C; MS (EI, 70 eV, m/z, int [%]) 332 (19, M+), 302 (6), 284 (2), 267 (5), 246 (4), 178 (32), 161 (100, ArCH=CHCO), 154 (20), 140 (8), 139 (9), 133 (17), 124 (61), 109 (44); MS (ESI, m/z, int [%]): 355 (100, M + Na); HRMS (ESI): calcd for C19H26NO4Na: 355.1760, found, 355.1753; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.24 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.25 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.67 (t, J = 11.9 Hz, 2H, HHC-CH(O-)-CHH), 2.00 (ddd, J = 11.2 Hz, J = 4.4 Hz, J = 1.4 Hz, 2H, HHC-CH(O-)-CHH), 1.67 (t, J = 11.9 Hz, 2H, HHC-CHOCO-CHH), 2.00 (ddd, J = 1.4 Hz, J = 4.4 Hz, J = 11.2 Hz, 2H, HHC-CHOCO-CHH), 3.83 (s, 3H, OCH3), 5.18 (tt, J = 11.2 Hz, J = 4.4 Hz, 1H, CH-OC(=O)), 6.28 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO), 7.62 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.79 (2×CH3), 31.96 (2×CH3), 44.12 (2×CH2), 55.57 (OCH3), 59.90 (2×C(CH3)2), 66.68 (CH-O-), 114.51 (CH), 116.03 (CH), 127.33, 129.94 (CH), 144.60 (CH), 161.57, 167.02 (C=O); IR (cm−1, KBr) 1707, 1632, 1604, 1515, 1290, 1255, 1163, 984, 827.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(2-nitrophenyl)acrylate (5e)
76.4%; mp 107–110 °C; MS (EI, 70 eV, m/z, int [%]) 347 (23, M+), 317 (5), 176 (63, ArCH=CHCO), 154 (25), 140 (11), 139 (17), 130 (61), 124 (100), 109 (87), 102 (21); MS (ESI, m/z, int [%]): 370 (100, M + Na); HRMS (ESI): calcd for C18H23N2O5Na: 370.1505, found, 370.1492; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.24 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.25 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.69 (t, J = 11.8 Hz, 2H, HHC-CH(O-)-CHH), 2.01 (ddd, J = 12.0 Hz, J = 3.4 Hz, J = 1.3 Hz, 2H, HHC-CH(O-)-CHH), 1.69 (t, J = 11.8 Hz, 2H, HHC-CHOCO-CHH), 2.01 (dtd, J = 1.4 Hz, J = 3.4 Hz, J = 12.0 Hz, 2H, HHC-CHOCO-CHH), 5.20 (tt, J = 11.3 Hz, J = 4.3 Hz, 1H, CH-OC(=O)), 6.33 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO), 8.10 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.82 (2×CH3) 31.91 (2×CH3) 43.96 (2×CH2) 59.84 (2×C(CH3)2) 67.55 (CH-O) 123.60 (CH) 125.14 (CH) 129.34 (C) 130.49 (CH) 130.80 (C) 133.73 (CH) 140.27 (CH) 148.00 (CH) 165.51 (C=O); IR (cm−1, KBr) 2977, 1717, 1632, 1523, 1341, 1292, 1272, 1171, 1009, 980, 866, 789, 758.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(3-nitrophenyl)acrylate (5f)
99.9%; mp 121–123 °C; MS (EI, 70 eV, m/z, int [%]) 347 (5, M+), 176 (85, ArCH=CHCO), 160 (6), 154 (13), 130 (17), 129 (17), 124 (83), 118 (12), 109 (100), 102 (73), 91 (13), 82 (17), 81 (22), 76 (21), 69 (18), 68 (12), 67 (25), 57 (14), 56 (18), 55 (27), 43 (19). 41 (62); MS (ESI, m/z, int [%]): 370 (100, M + Na); HRMS (ESI): calcd for C18H23N2O5Na: 370.1505, found, 370.1489; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2), 1.249 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.254 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.69 (t, J = 12.0 Hz, 2H, HHC-CHOCO-CHH), 2.01 (ddd, J = 1.4 Hz, J = 4.4 Hz, J = 11.2 Hz, 2H, HHC-CHOCO-CHH), 5.21 (tt, J = 11.4 Hz, J = 4.4 Hz, 1H, CH-OC(=O)), 6.53 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO), 7.69 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.75 (2×CH3), 32.05 (2×CH3), 44.05 (2×CH2), 59.76 (2×C(CH3)2), 67.55 (CH-O-), 121.77 (CH), 122.63 (CH), 124.74 (CH), 130.16 (CH), 133.85 (CH), 136.32 (C), 142.00 (CH), 148.87 (C), 165.83 (C=O); IR (cm−1, KBr) 3080, 2971, 1709, 1642, 1526, 1358, 1330, 1191, 983, 813, 671.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(4-nitrophenyl)acrylate (5g)
55.3%; mp 148–149 °C; MS (EI, 70 eV, m/z, int [%]) 347 (19, M+), 317 (6), 261 (3), 176 (89, ArCH=CHCO), 160 (7), 154 (19), 140 (14), 139 (15), 130 (24), 124 (100), 109 (90), 102 (29); HRMS (EI, 70 eV, m/z, int [%]): calcd for C18H23N2O5: 347.16070, found, 347.16009; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.25 (s, 12H, 4×CH3), 1.68 (t, J = 12.0 Hz, 2H, HHC-CH(O-)-CHH), 2.00 (dd, J = 12.6 Hz, J = 4.0 Hz, 2H, HHC-CH(O-)-CHH), 1.68 (t, J = 12.0 Hz, 2H, HHC-CHOCO-CHH), 2.00 (dd, J = 4.0 Hz, J = 12.6 Hz, 2H, HHC-CHOCO-CHH), 5.21 (tt, J = 11.3 Hz, J = 4.4 Hz, 1H, CH-OC(=O)), 6.52 (dd, J = 16.0 Hz, J = 0.6 Hz, 1H, Ar-CH=CH-COO), 7.32 (dd, J = 16.0 Hz, J = 0.6 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.73 (2×CH3), 32.01 (2×CH3), 44.02 (2×CH2), 59.79 (2×C(CH3)2), 67.62 (CH-O-), 122.89 (CH), 124.37 (CH), 128.54 (C), 128.84 (CH), 140.69 (C), 141.92 (CH), 165.71 (C=O); IR (cm−1, KBr) 1705, 1638, 1519, 1344, 1177, 850.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(2-methyl-4-nitrophenyl)acrylate (5h)
88.3%; mp 109–112 °C; MS (EI, 70 eV, m/z, int [%]) 361 (15, M+), 347 (4), 331 (7), 191 (32), 190 (74, ArCH=CHCO), 174 (11), 161 (6), 160 (10), 154 (29), 144 (37), 140 (57), 139 (26), 124 (100), 116 (47), 115 (60), 109 (82), 98 (12), 95 (11), 89 (10), 82 (36), 81 (30), 69 (33), 68 (20), 67 (27), 58 (9), 57 (12), 56 (21), 55 (30), 43 (15), 42 (12), 41 (45); HRMS (EI, 70 eV, m/z, int [%]): calcd for C19H25N2O5: 361.1763, found, 361.1761; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.26 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.28 (s, 6H, (CH3)(CH3)CN(OH)C(CH3)(CH3)), 1.74 (t, J = 12.0 Hz, 2H, HHC-CH(O-)-CHH), 2.03 (ddd, J = 11.2 Hz, J = 4.4 Hz, J = 1.4 Hz, 2H, HHC-CH(O-)-CHH), 2.51 (ArCH3), 5.22 (tt, J = 11.3 Hz, J = 4.3 Hz, 1H, CH-OC(=O)), 6.42 (d, J = 15.9 Hz, 1H, Ar-CH=CH-COO), 7.90 (d, J = 15.9 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.10 (ArCH3), 20.90 (2×CH3), 31.66 (2×CH3), 43.81 (2×CH2), 60.32 (2×C(CH3)2), 67.47 (CH-O-), 121.62 (CH), 123.52 (CH), 125.72 (CH), 127.52 (CH), 128.95 (C), 139.22 (C), 139.93 (C), 140.21 (CH), 165.86 (C=O); IR (cm−1, KBr) 1715, 1519, 1347, 1170.
2,2,6,6-Tetramethyl-1-oxyl-4-piperidyl 3-E-(2-chloro-4-nitrophenyl)acrylate (5i)
56.2%; mp 107–110 °C; MS (EI, 70 eV, m/z, int [%]) 383 (1), 382 (1), 381 (2, M+), 212 (14), 211 (9), 210 (41, ArCH=CHCO), 194 (4), 192 (8), 166 (4), 164 (11), 154 (15), 140 (17), 139 (14), 138 (9), 136 (23), 124 (89), 109 (100), 101 (13), 100 (19), 99 (7), 98 (9), 95 (8), 89 (13), 82 (19), 81 (22), 75 (12), 74 (12), 69 (19), 68 (13), 67 (23), 57 (14), 56 (17), 55 (28), 43 (18), 41 (58); MS (ESI, m/z, int [%]): 404 (15, M + Na), 249 (100), 242 (15); HRMS (ESI): calcd for C18H22N2O5ClNa: 404.1115, found, 404.1131; 1H NMR (200 MHz, δ, CDCl3, TMS, PhNHNH2) 1.25, 1.27, 1.29 (3s, 12H, 4×CH3), 1.75 (m, 2H, HHC-CH(O-)-CHH), 2.01 (m, 2H, HHC-CH(O-)-CHH), 5.22 (tt, J = 11.3 Hz, J = 4.3 Hz, 1H, CH-OC(=O)), 6.51 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO), 8.03 (d, J = 16.0 Hz, 1H, Ar-CH=CH-COO); 13C NMR (50 MHz, δ, CDCl3, TMS, PhNHNH2) 20.88 (2×CH3), 31.67 (2×CH3), 43.75 (2×CH2), 60.33 (2×C(CH3)2), 67.72 (CH-O-), 122.22 (CH), 125.02 (CH), 125.58 (CH), 128.53 (C), 128.87 (C), 128.90 (CH), 138.45 (CH), 139.10 (C), 165.31 (C=O); IR (cm−1, KBr) 2976, 1712, 1523, 1346, 1330, 1194, 1180, 990.
Supporting Information
| Supporting Information File 1: EIMS, ESIMS, 1H NMR, 13C NMR, and IR spectra of 3 and 5a–5i. | ||
| Format: PDF | Size: 3.1 MB | Download |
References
-
Zakrzewski, J.; Krawczyk, M. Heterocycl. Commun. 2014, 20, 89–91. doi:10.1515/hc-2013-0169
-
Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
De, P.; Bedos-Belval, F.; Vanucci-Bacqué, C.; Baltas, M. Curr. Org. Chem. 2012, 16, 747–768. doi:10.2174/138527212799958020
Return to citation in text: [1] [2] [3] -
De, P.; Baltas, M.; Bedos-Belval, F. Curr. Med. Chem. 2011, 18, 1672–1703. doi:10.2174/092986711795471347
Return to citation in text: [1] [2] [3] [4] -
Clifford, M. N. J. Sci. Food Agric. 2000, 80, 1033–1043. doi:10.1002/(SICI)1097-0010(20000515)80:7<1033::AID-JSFA595>3.0.CO;2-T
Return to citation in text: [1] -
Clifford, M. N. J. Sci. Food Agric. 1999, 79, 362–372. doi:10.1002/(SICI)1097-0010(19990301)79:3<362::AID-JSFA256>3.0.CO;2-D
Return to citation in text: [1] -
Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Narasimhan, B.; Belsare, D.; Pharande, D.; Mourya, V.; Dhake, A. Eur. J. Med. Chem. 2004, 39, 827–834. doi:10.1016/j.ejmech.2004.06.013
And references cited therein.
Return to citation in text: [1] [2] -
Sambandan, D. R.; Ratner, D. J. Am. Acad. Dermatol. 2011, 64, 748–758. doi:10.1016/j.jaad.2010.01.005
Return to citation in text: [1] -
Bhatia, S. P.; Cocchiara, J.; Wellington, G. A.; Lalko, J.; Letizia, C. S.; Api, A. M. Food Chem. Toxicol. 2011, 49 (Suppl. 2), S252–S255. doi:10.1016/j.fct.2011.07.051
Return to citation in text: [1] -
Belsito, D.; Bickers, D.; Bruze, M.; Calow, P.; Dagli, M.; Fryer, A. D.; Greim, H.; Miyachi, Y.; Saurat, J. H.; Sipes, I. G. Food Chem. Toxicol. 2011, 49 (Suppl. 2), S256–S267. doi:10.1016/j.fct.2011.07.053
Return to citation in text: [1] -
Waghmare, S. R.; Gaikwad, H. K. J. Chem. Pharm. Res. 2012, 4, 2415–2421.
Return to citation in text: [1] -
Wall, V. M.; Eisenstadt, A.; Ager, D. J.; Laneman, S. A. Platinum Met. Rev. 1999, 43, 138–145.
Return to citation in text: [1] -
Sheng, S.-R.; Luo, Q.-Y.; Huang, P.-G.; Guo, L.; Wang, Q.-Y.; Pei, X.-L. J. Chem. Res. 2006, 24–26. doi:10.3184/030823406776331070
Return to citation in text: [1] -
Liu, S.; Fukuyama, T.; Sato, M.; Ryu, I. Org. Process Res. Dev. 2004, 8, 477–481. doi:10.1021/op034200h
Return to citation in text: [1] -
Mingji, D.; Liang, B.; Wang, C.; You, Z.; Xiang, J.; Dong, G.; Chen, J.; Yang, Z. Adv. Synth. Catal. 2004, 346, 1669–1673. doi:10.1002/adsc.200404165
Return to citation in text: [1] -
Bumagin, N. A.; More, P. G.; Beletskaya, I. P. J. Organomet. Chem. 1989, 371, 397–401. doi:10.1016/0022-328X(89)85235-0
Return to citation in text: [1] -
Smith, M. R.; Kim, J. Y.; Ciufolini, M. A. Tetrahedron Lett. 2013, 54, 2042–2045. doi:10.1016/j.tetlet.2013.01.077
Return to citation in text: [1] -
Pastre, J. C.; Correia, C. R. D. Adv. Synth. Catal. 2009, 351, 1217–1223. doi:10.1002/adsc.200900032
Return to citation in text: [1] -
Sakakibara, T.; Nishimura, S.; Kimura, K.; Minato, I.; Odaira, Y. J. Org. Chem. 1970, 35, 3884–3887. doi:10.1021/jo00836a066
Return to citation in text: [1] -
Zakrzewski, J. Beilstein J. Org. Chem. 2012, 8, 1515–1522. doi:10.3762/bjoc.8.171
Return to citation in text: [1] [2] [3] -
Kálai, T.; Borza, E.; Antus, C.; Radnai, B.; Gulyás-Fekete, G.; Fehér, A.; Sümegi, B.; Hideg, K. Bioorg. Med. Chem. 2011, 19, 7311–7317. doi:10.1016/j.bmc.2011.10.066
Return to citation in text: [1] -
Kálai, T.; Bognár, B.; Jekő, J.; Hideg, K. Synthesis 2006, 2573–2579. doi:10.1055/s-2006-942439
Return to citation in text: [1] -
Keddie, D. J.; Johnson, T. E.; Arnold, D. P.; Bottle, S. E. Org. Biomol. Chem. 2005, 3, 2593–2598. doi:10.1039/b504354a
Return to citation in text: [1] -
Keddie, D. J. The Synthesis of Novel Profluorescent Nitroxide Probes. Ph.D. Thesis, Queensland University of Technology, Austraöia, 2008.
Return to citation in text: [1] -
Fairfull-Smith, K. E.; Bottle, S. E. Eur. J. Org. Chem. 2008, 5391–5400. doi:10.1002/ejoc.200800597
Return to citation in text: [1] -
Kálai, T.; Balog, M.; Jekő, J.; Hubbell, W. L.; Hideg, K. Synthesis 2002, 2365–2372. doi:10.1055/s-2002-35240
Return to citation in text: [1] -
Stroh, C.; Mayor, M.; von Hänisch, C. Eur. J. Org. Chem. 2005, 3697–3703. doi:10.1002/ejoc.200500116
Return to citation in text: [1] -
Kálai, T.; Jekő, J.; Berente, Z.; Hideg, K. Synthesis 2006, 439–446. doi:10.1055/s-2006-926279
Return to citation in text: [1] -
Keddie, D. J.; Fairfull-Smith, K. E.; Bottle, S. E. Org. Biomol. Chem. 2008, 6, 3135–3143. doi:10.1039/B806963H
Return to citation in text: [1] -
Miura, Y.; Ushitani, Y. Macromolecules 1993, 26, 7079–7082. doi:10.1021/ma00077a056
Return to citation in text: [1] -
Romero, F. M.; Ziessel, R. Tetrahedron Lett. 1999, 40, 1895–1898. doi:10.1016/S0040-4039(99)00069-6
Return to citation in text: [1] -
Neiman, M. B.; Rozantsev, E. G.; Mamedova, Yu. G. Nature 1962, 196, 472–474. doi:10.1038/196472a0
Return to citation in text: [1] -
Hyslop, D. K.; Parent, J.; Scott, J. S. Macromolecules 2012, 45, 8147–8154. doi:10.1021/ma3016135
Return to citation in text: [1] -
Appelt, M.; Schmidt-Naake, G. Macromol. Mater. Eng. 2004, 289, 245–253. doi:10.1002/mame.200300255
Return to citation in text: [1] [2] -
Rozantsev, E. G.; Suskina, V. J. Russ. Chem. Bull. 1968, 17, 1997–1999.
Izv.. Akad.. Nauk. SSSR, S. Kh. 1968, 2106-2109.
Return to citation in text: [1] -
Dieck, H. A.; Heck, R. F. J. Am. Chem. Soc. 1974, 96, 1133–1136. doi:10.1021/ja00811a029
Return to citation in text: [1] -
Coulson, D. R.; Satek, L. C.; Grim, S. O. Inorg. Synth. 1972, 13, 121–124. doi:10.1002/9780470132449.ch23
Return to citation in text: [1] [2] -
Shezad, N.; Oakes, R. S.; Clifford, A. A.; Rayner, C. M. Tetrahedron Lett. 1999, 40, 2221–2224. doi:10.1016/S0040-4039(99)00176-8
Return to citation in text: [1] -
Reaxa Ltd, Aldrich; Pd EnCat™, Experimental Guide.
Return to citation in text: [1] -
Reaxa Ltd; Pd(II) EnCatTM Experimental User Guide. May 2006.
Return to citation in text: [1] -
Mendes Da Silva, J. F.; Yepes Perez, A. F.; Pinto de Almeida, N. RSC Adv. 2014, 4, 28148–28155. doi:10.1039/C4RA03586K
Return to citation in text: [1] -
Barros, J. C.; Yaunner, R. S.; de Souza, A. L. F.; da Silva, J. F. M.; Antunes, O. A. C. Appl. Organomet. Chem. 2011, 25, 820–823. doi:10.1002/aoc.1845
Return to citation in text: [1] -
Broadwater, S. J.; McQuade, D. T. J. Org. Chem. 2006, 71, 2131–2134. doi:10.1021/jo0517904
Return to citation in text: [1] -
Lee, C. K. Y.; Holmes, A. B.; Ley, S. V.; McConvey, I. F.; Al-Duri, B.; Leeke, G. A.; Santos, R. C. D.; Seville, J. P. K. Chem. Commun. 2005, 2175–2177. doi:10.1039/b418669a
Return to citation in text: [1] -
Tandukar, S.; Sen, A. J. Mol. Catal. A: Chem. 2007, 268, 112–119. doi:10.1016/j.molcata.2006.12.003
Return to citation in text: [1] -
Leikoski, T.; Wrigstedt, P.; Helminen, J.; Matikainen, J.; Sipilä, J.; Yli-Kauhaluoma, J. Tetrahedron 2013, 69, 839–843. doi:10.1016/j.tet.2012.10.092
Return to citation in text: [1] -
Smith, M. R.; Jang, Y. I.; Kim, J. Y.; Ciufolini, M. A. Tetrahedron 2013, 69, 10139–10151. doi:10.1016/j.tet.2013.09.019
Return to citation in text: [1] -
Lee, T. D.; Keana, J. F. W. J. Org. Chem. 1975, 40, 3145–3147. doi:10.1021/jo00909a033
Return to citation in text: [1] [2] -
López-Peña, H. A.; Hernández-Muñoz, L. S.; Frontana-Uribe, B. A.; González, F. J.; González, I.; Frontana, C.; Cardoso, J. J. Phys. Chem. B 2012, 116, 5542–5550. doi:10.1021/jp301207v
Return to citation in text: [1] -
Li, Y.; Lei, X.; Li, X.; Lawler, R. G.; Murata, Y.; Komatsu, K.; Turro, N. J. Chem. Commun. 2011, 47, 12527–12529. doi:10.1039/c1cc15149e
Return to citation in text: [1] -
Li, Y.; Lei, X.; Lawler, R. G.; Murata, Y.; Komatsu, K.; Turro, N. J. Org. Lett. 2012, 14, 3822–3825. doi:10.1021/ol3013155
Return to citation in text: [1] -
Morrison, A.; Davies, A. P. Org. Mass Spectrom. 1970, 3, 353–366. doi:10.1002/oms.1210030310
Return to citation in text: [1] -
Kaliská, V.; Toma, Š.; Leško, J. Collect. Czech. Chem. Commun. 1987, 52, 2266–2273. doi:10.1135/cccc19872266
Return to citation in text: [1]
| 39. | Shezad, N.; Oakes, R. S.; Clifford, A. A.; Rayner, C. M. Tetrahedron Lett. 1999, 40, 2221–2224. doi:10.1016/S0040-4039(99)00176-8 |
| 40. | Reaxa Ltd, Aldrich; Pd EnCat™, Experimental Guide. |
| 41. | Reaxa Ltd; Pd(II) EnCatTM Experimental User Guide. May 2006. |
| 42. | Mendes Da Silva, J. F.; Yepes Perez, A. F.; Pinto de Almeida, N. RSC Adv. 2014, 4, 28148–28155. doi:10.1039/C4RA03586K |
| 43. | Barros, J. C.; Yaunner, R. S.; de Souza, A. L. F.; da Silva, J. F. M.; Antunes, O. A. C. Appl. Organomet. Chem. 2011, 25, 820–823. doi:10.1002/aoc.1845 |
| 44. | Broadwater, S. J.; McQuade, D. T. J. Org. Chem. 2006, 71, 2131–2134. doi:10.1021/jo0517904 |
| 45. | Lee, C. K. Y.; Holmes, A. B.; Ley, S. V.; McConvey, I. F.; Al-Duri, B.; Leeke, G. A.; Santos, R. C. D.; Seville, J. P. K. Chem. Commun. 2005, 2175–2177. doi:10.1039/b418669a |
| 46. | Tandukar, S.; Sen, A. J. Mol. Catal. A: Chem. 2007, 268, 112–119. doi:10.1016/j.molcata.2006.12.003 |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 3. | De, P.; Bedos-Belval, F.; Vanucci-Bacqué, C.; Baltas, M. Curr. Org. Chem. 2012, 16, 747–768. doi:10.2174/138527212799958020 |
| 4. | De, P.; Baltas, M.; Bedos-Belval, F. Curr. Med. Chem. 2011, 18, 1672–1703. doi:10.2174/092986711795471347 |
| 5. | Clifford, M. N. J. Sci. Food Agric. 2000, 80, 1033–1043. doi:10.1002/(SICI)1097-0010(20000515)80:7<1033::AID-JSFA595>3.0.CO;2-T |
| 6. | Clifford, M. N. J. Sci. Food Agric. 1999, 79, 362–372. doi:10.1002/(SICI)1097-0010(19990301)79:3<362::AID-JSFA256>3.0.CO;2-D |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 8. |
Narasimhan, B.; Belsare, D.; Pharande, D.; Mourya, V.; Dhake, A. Eur. J. Med. Chem. 2004, 39, 827–834. doi:10.1016/j.ejmech.2004.06.013
And references cited therein. |
| 21. | Zakrzewski, J. Beilstein J. Org. Chem. 2012, 8, 1515–1522. doi:10.3762/bjoc.8.171 |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 10. | Bhatia, S. P.; Cocchiara, J.; Wellington, G. A.; Lalko, J.; Letizia, C. S.; Api, A. M. Food Chem. Toxicol. 2011, 49 (Suppl. 2), S252–S255. doi:10.1016/j.fct.2011.07.051 |
| 11. | Belsito, D.; Bickers, D.; Bruze, M.; Calow, P.; Dagli, M.; Fryer, A. D.; Greim, H.; Miyachi, Y.; Saurat, J. H.; Sipes, I. G. Food Chem. Toxicol. 2011, 49 (Suppl. 2), S256–S267. doi:10.1016/j.fct.2011.07.053 |
| 38. | Coulson, D. R.; Satek, L. C.; Grim, S. O. Inorg. Synth. 1972, 13, 121–124. doi:10.1002/9780470132449.ch23 |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 35. | Appelt, M.; Schmidt-Naake, G. Macromol. Mater. Eng. 2004, 289, 245–253. doi:10.1002/mame.200300255 |
| 4. | De, P.; Baltas, M.; Bedos-Belval, F. Curr. Med. Chem. 2011, 18, 1672–1703. doi:10.2174/092986711795471347 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 9. | Sambandan, D. R.; Ratner, D. J. Am. Acad. Dermatol. 2011, 64, 748–758. doi:10.1016/j.jaad.2010.01.005 |
| 53. | Morrison, A.; Davies, A. P. Org. Mass Spectrom. 1970, 3, 353–366. doi:10.1002/oms.1210030310 |
| 54. | Kaliská, V.; Toma, Š.; Leško, J. Collect. Czech. Chem. Commun. 1987, 52, 2266–2273. doi:10.1135/cccc19872266 |
| 3. | De, P.; Bedos-Belval, F.; Vanucci-Bacqué, C.; Baltas, M. Curr. Org. Chem. 2012, 16, 747–768. doi:10.2174/138527212799958020 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 4. | De, P.; Baltas, M.; Bedos-Belval, F. Curr. Med. Chem. 2011, 18, 1672–1703. doi:10.2174/092986711795471347 |
| 49. | Lee, T. D.; Keana, J. F. W. J. Org. Chem. 1975, 40, 3145–3147. doi:10.1021/jo00909a033 |
| 50. | López-Peña, H. A.; Hernández-Muñoz, L. S.; Frontana-Uribe, B. A.; González, F. J.; González, I.; Frontana, C.; Cardoso, J. J. Phys. Chem. B 2012, 116, 5542–5550. doi:10.1021/jp301207v |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 8. |
Narasimhan, B.; Belsare, D.; Pharande, D.; Mourya, V.; Dhake, A. Eur. J. Med. Chem. 2004, 39, 827–834. doi:10.1016/j.ejmech.2004.06.013
And references cited therein. |
| 4. | De, P.; Baltas, M.; Bedos-Belval, F. Curr. Med. Chem. 2011, 18, 1672–1703. doi:10.2174/092986711795471347 |
| 51. | Li, Y.; Lei, X.; Li, X.; Lawler, R. G.; Murata, Y.; Komatsu, K.; Turro, N. J. Chem. Commun. 2011, 47, 12527–12529. doi:10.1039/c1cc15149e |
| 52. | Li, Y.; Lei, X.; Lawler, R. G.; Murata, Y.; Komatsu, K.; Turro, N. J. Org. Lett. 2012, 14, 3822–3825. doi:10.1021/ol3013155 |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 47. | Leikoski, T.; Wrigstedt, P.; Helminen, J.; Matikainen, J.; Sipilä, J.; Yli-Kauhaluoma, J. Tetrahedron 2013, 69, 839–843. doi:10.1016/j.tet.2012.10.092 |
| 2. | Sova, M. Mini-Rev. Med. Chem. 2012, 12, 749–767. doi:10.2174/138955712801264792 |
| 3. | De, P.; Bedos-Belval, F.; Vanucci-Bacqué, C.; Baltas, M. Curr. Org. Chem. 2012, 16, 747–768. doi:10.2174/138527212799958020 |
| 48. | Smith, M. R.; Jang, Y. I.; Kim, J. Y.; Ciufolini, M. A. Tetrahedron 2013, 69, 10139–10151. doi:10.1016/j.tet.2013.09.019 |
| 7. | Sharma, P. J. Chem. Pharm. Res. 2011, 3, 403–423. |
| 13. | Wall, V. M.; Eisenstadt, A.; Ager, D. J.; Laneman, S. A. Platinum Met. Rev. 1999, 43, 138–145. |
| 14. | Sheng, S.-R.; Luo, Q.-Y.; Huang, P.-G.; Guo, L.; Wang, Q.-Y.; Pei, X.-L. J. Chem. Res. 2006, 24–26. doi:10.3184/030823406776331070 |
| 15. | Liu, S.; Fukuyama, T.; Sato, M.; Ryu, I. Org. Process Res. Dev. 2004, 8, 477–481. doi:10.1021/op034200h |
| 16. | Mingji, D.; Liang, B.; Wang, C.; You, Z.; Xiang, J.; Dong, G.; Chen, J.; Yang, Z. Adv. Synth. Catal. 2004, 346, 1669–1673. doi:10.1002/adsc.200404165 |
| 17. | Bumagin, N. A.; More, P. G.; Beletskaya, I. P. J. Organomet. Chem. 1989, 371, 397–401. doi:10.1016/0022-328X(89)85235-0 |
| 49. | Lee, T. D.; Keana, J. F. W. J. Org. Chem. 1975, 40, 3145–3147. doi:10.1021/jo00909a033 |
| 37. | Dieck, H. A.; Heck, R. F. J. Am. Chem. Soc. 1974, 96, 1133–1136. doi:10.1021/ja00811a029 |
| 38. | Coulson, D. R.; Satek, L. C.; Grim, S. O. Inorg. Synth. 1972, 13, 121–124. doi:10.1002/9780470132449.ch23 |
| 33. | Neiman, M. B.; Rozantsev, E. G.; Mamedova, Yu. G. Nature 1962, 196, 472–474. doi:10.1038/196472a0 |
| 21. | Zakrzewski, J. Beilstein J. Org. Chem. 2012, 8, 1515–1522. doi:10.3762/bjoc.8.171 |
| 34. | Hyslop, D. K.; Parent, J.; Scott, J. S. Macromolecules 2012, 45, 8147–8154. doi:10.1021/ma3016135 |
| 35. | Appelt, M.; Schmidt-Naake, G. Macromol. Mater. Eng. 2004, 289, 245–253. doi:10.1002/mame.200300255 |
| 36. |
Rozantsev, E. G.; Suskina, V. J. Russ. Chem. Bull. 1968, 17, 1997–1999.
Izv.. Akad.. Nauk. SSSR, S. Kh. 1968, 2106-2109. |
| 21. | Zakrzewski, J. Beilstein J. Org. Chem. 2012, 8, 1515–1522. doi:10.3762/bjoc.8.171 |
| 22. | Kálai, T.; Borza, E.; Antus, C.; Radnai, B.; Gulyás-Fekete, G.; Fehér, A.; Sümegi, B.; Hideg, K. Bioorg. Med. Chem. 2011, 19, 7311–7317. doi:10.1016/j.bmc.2011.10.066 |
| 23. | Kálai, T.; Bognár, B.; Jekő, J.; Hideg, K. Synthesis 2006, 2573–2579. doi:10.1055/s-2006-942439 |
| 24. | Keddie, D. J.; Johnson, T. E.; Arnold, D. P.; Bottle, S. E. Org. Biomol. Chem. 2005, 3, 2593–2598. doi:10.1039/b504354a |
| 25. | Keddie, D. J. The Synthesis of Novel Profluorescent Nitroxide Probes. Ph.D. Thesis, Queensland University of Technology, Austraöia, 2008. |
| 26. | Fairfull-Smith, K. E.; Bottle, S. E. Eur. J. Org. Chem. 2008, 5391–5400. doi:10.1002/ejoc.200800597 |
| 27. | Kálai, T.; Balog, M.; Jekő, J.; Hubbell, W. L.; Hideg, K. Synthesis 2002, 2365–2372. doi:10.1055/s-2002-35240 |
| 28. | Stroh, C.; Mayor, M.; von Hänisch, C. Eur. J. Org. Chem. 2005, 3697–3703. doi:10.1002/ejoc.200500116 |
| 29. | Kálai, T.; Jekő, J.; Berente, Z.; Hideg, K. Synthesis 2006, 439–446. doi:10.1055/s-2006-926279 |
| 30. | Keddie, D. J.; Fairfull-Smith, K. E.; Bottle, S. E. Org. Biomol. Chem. 2008, 6, 3135–3143. doi:10.1039/B806963H |
| 31. | Miura, Y.; Ushitani, Y. Macromolecules 1993, 26, 7079–7082. doi:10.1021/ma00077a056 |
| 32. | Romero, F. M.; Ziessel, R. Tetrahedron Lett. 1999, 40, 1895–1898. doi:10.1016/S0040-4039(99)00069-6 |
| 18. | Smith, M. R.; Kim, J. Y.; Ciufolini, M. A. Tetrahedron Lett. 2013, 54, 2042–2045. doi:10.1016/j.tetlet.2013.01.077 |
| 19. | Pastre, J. C.; Correia, C. R. D. Adv. Synth. Catal. 2009, 351, 1217–1223. doi:10.1002/adsc.200900032 |
| 20. | Sakakibara, T.; Nishimura, S.; Kimura, K.; Minato, I.; Odaira, Y. J. Org. Chem. 1970, 35, 3884–3887. doi:10.1021/jo00836a066 |
© 2015 Zakrzewski and Huras; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)