
Abstract
The present paper describes the synthesis of new palladium–oxazoline complexes in one step with good to high yields (68–95%). The oxazolines were prepared from enantiomerically pure α-aminoalcohols. The structures of the synthesized palladium complexes were confirmed by NMR, FTIR, TOFMS, UV–visible spectroscopic analysis and X–ray diffraction. The optical properties of the complexes were evaluated by the determination of the gap energy values (Eg) ranging between 2.34 and 3.21 eV. Their catalytic activities were tested for the degradation of Eriochrome Blue Black B (a model of azo dyes) in the presence of an ecological oxidant (H2O2). The efficiency of the decolorization has been confirmed via UV–visible spectroscopic analysis and the factors affecting the degradation phenomenon have been studied. The removal of the Eriochrome reached high yields. We have found that the complex 9 promoted 84% of color elimination within 5 min (C0 = 30 mg/L, T = 22 °C, pH 7, H2O2 = 0.5 mL) and the energetic parameters have been also determined.
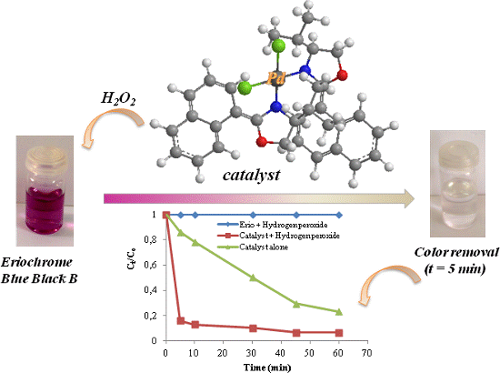
Graphical Abstract
Introduction
Palladium complexes have been used as starting materials to prepare polymers [1], agrochemicals [2], pharmaceuticals [3], flavors and fragrances [4]. They have also been used for the total synthesis of natural products and nanocompounds [5]. It is only since 1986 that oxazoline-based ligands have been utilized in asymmetric catalysis. This initiated considerable research activity in this field and triggered the synthesis of numerous chiral ligands containing at least one oxazoline ring [6]. Oxazoline units are expected to readily coordinate to a metal center and have been shown to bind a wide range of transition metals [7]. The wide variety of ligands with one or more oxazoline rings incorporating different heteroatoms, additional chiral groups, and specific structural features have been used in diverse reactions [8]. The importance of these ligands lies in the fact that the stereogenic centers are kept in close proximity to the metal and thereby having a strong and direct influence on the stereochemical course of the metal-catalyzed process.
Oxazolines possess several interesting advantages: versatility of the ligand design, their straightforward synthesis from readily available starting materials and variability of the chiral center, which are located near the donor atoms. The oxazoline unit is anticipated to increase the stability of a metal complex giving rise to more air- and possibly water-stable catalysts [9].
Commonly, difficulties arise when trying to treat waste waters containing dyes because the dyes are recalcitrant molecules, often resistant to aerobic digestion, and stable to light, heat, and oxidizing agents [10,11]. Recently, the catalytic oxidation was recognized as an effective method to treat colored waters [12].
In fact, the treatment of colored waters remains a serious environmental topic. Many industries such as textile, leather and paper discharge various dyes during their processing operations [13,14]. These dyes are toxic, mutagenic, and carcinogenic [15,16]. That’s why many unconventional methods and techniques have been investigated and a number of studies have been developed [17-19]. In particular, attention has been focused on the synthesis of supports having metal complexes in their structures due to their capacities and efficiencies to treat colored waters [20-22].
Owing to the easy formation of palladium–oxazoline complexes, it was very interesting to investigate the ability of some synthetic dyes to coordinate to palladium complexes by developing binary systems. This was done by complexing oxazoline with PdII ions, leading to the adsorption of dyes. Moreover, the decolorization of the solution by Pd and Pd complexes has also been applied [23].
In this paper, we report the synthesis and characterization of some new palladacycles. Optical properties were determined and the catalytic activity of these complexes was evaluated for the first time during the degradation of organic dyes such as Eriochrome Blue Black B referred to as Erio which was chosen as an example of azo dyes (Figure 1).
![[1860-5397-11-132-1]](/bjoc/content/figures/1860-5397-11-132-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of Eriochrome Blue Black B.
Figure 1: Structure of Eriochrome Blue Black B.
Results and Discussion
The use of oxazolines as ligands during the preparation of optically active cyclopalladated compounds can give rise to different applications [24-28].
The compilation of the literature shows that the presence of substituents on the ligand promotes the cyclopalladation. Smoliakova and co-workers performed the cyclopalladation of tert-butyl-substituted phenyloxazoline by stirring the reaction mixture at room temperature [29].
In the light of these results, we also attempted to carry out this process under milder conditions. When (S)-4-isopropyl-2-(naphthalen-1-yl)oxazoline (2) was added to an acetic acid solution of Pd(OAc)2, a yellowish precipitate was obtained and identified as (S,S)-di-μ-acetatobis[1-(4-isopropyloxazolin-2-yl)naphthalen-2-yl-C,N]dipalladium(II) (3) (Scheme 1). Unfortunately, complex dimer 3 was relatively unstable, so only its 1H NMR and FTIR data were performed. The metathesis of dimer 3 with lithium chloride in acetone afforded the more stable (S,S)-dimer 4 in which NMR analysis shows the presence of two dimeric forms. For better elucidation of their structures, dimeric complexes 4 were transformed into their mononuclear phosphane derivatives 5a and 5b using PPh3 in toluene. According to 1HNMR data, the mixture contains 5a and 5b in the ratio of 4:1. Essays to separate the later regioisomers by column chromatography or preparative TLC on silica gel were unsuccessful.
![[1860-5397-11-132-i1]](/bjoc/content/inline/1860-5397-11-132-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cyclopalladation reactions of (S)-4-isopropyl-2-(naphthalen-1-yl)oxazoline.
Scheme 1: Cyclopalladation reactions of (S)-4-isopropyl-2-(naphthalen-1-yl)oxazoline.
According to the literature, the PPh3 adducts of cyclopalladated oxazolines have the trans (N,P) geometry [30,31]. In addition, the palladacycles with oxazoline ligands exists in endo and exo regioisomers. Results obtained in the present study show that naphthyl-oxazoline has a tendency to form exclusively endo-cyclic complexes with the C=N bond within a palladacycle. Furthermore, NMR data analyses demonstrate that the five-membered palladacycle 5a was preferentially formed upon the six-membered 5b. Indeed, palladium insertion in the peri C–H bond of the naphthyl-oxazoline will take place at high temperature reactions [32].
In order to investigate the effect of solvents on the isomeric ratio, cyclopalladation of oxazoline 2 was also carried out with Pd(OAc)2 in refluxing MeCN. After 3.5 h the reaction yielded 89% of the expected dimer. However, a significant change on the isomer ratio was observed.
In this work, we report one example related to the direct palladation of enantiopure bis-oxazoline 7 having a chiral center at the 4-position and the substituent at the 2-position of the heterocycles. The addition of one equivalent of 7 to a methanolic solution of Na2PdCl4 gave the palladium complex 8 in 75% yield. The coordination sphere has two chlorine atoms bonding in a cis configuration to the Pd center and the oxazoline ligand chelating to the Pd center via the two nitrogen atoms to form a planar palladium ring system (Scheme 2). Crystallization by slow diffusion of pentane into a solution of 8 in dichloromethane gave suitable crystals for an X-ray diffraction study. The molecular structure is presented in Figure 2 (see Supporting Information File 1 for the cif of complex 8). The coordination geometry of 8 is pseudo-square planar with the four coordination sites occupied by the two nitrogen donors and the two chlorine atoms. The molecule possesses approximate C2 symmetry and its chirality has been confirmed to be the (S,S)-isomer. The Pd–N and the Pd–Cl distances (Pd–N16 distance (1.963 A°), Pd–N1 distance (2.031 A°)) are almost the same (Pd–Cl1 distance (2.281 A°), the Pd–Cl2 distance (2.259 A°)).
![[1860-5397-11-132-i2]](/bjoc/content/inline/1860-5397-11-132-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of cyclopalladated complex from bis-oxazoline.
Scheme 2: Synthesis of cyclopalladated complex from bis-oxazoline.
![[1860-5397-11-132-2]](/bjoc/content/figures/1860-5397-11-132-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP drawing of the complex 8.
Figure 2: ORTEP drawing of the complex 8.
On the other hand, the addition of (S)-4-isopropyl-2-(naphthalen-1-yl)oxazoline (2) and 3-[(4S)-4,5-dihydro-4-isopropyl-1,3-oxazol-2-yl]propanenitrile (11) to a methanolic solution of Na2PdCl4 at room temperature gave the palladium complexes 9 and 12 in 85% and 68% yields, respectively (Scheme 3). The two complexes are stable when exposed to air and water. The single crystal X-ray model of complex 9 is depicted in Figure 3 (see Supporting Information File 2 for the cif of complex 9). This structure confirms the expected monomeric nature of the complex and coordination of the oxazoline nitrogen to the palladium atom. The Pd(II) unit is coordinated to two monodentate ligands with the two nitrogen and two chlorine atoms in equatorial positions to complete the distorted tetragonal coordination sphere. The two ligands coordinate to the palladium center in a trans geometry with respect to each other.
![[1860-5397-11-132-i3]](/bjoc/content/inline/1860-5397-11-132-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the bis(oxazoline) coordinated complexes.
Scheme 3: Synthesis of the bis(oxazoline) coordinated complexes.
![[1860-5397-11-132-3]](/bjoc/content/figures/1860-5397-11-132-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP drawing of the complex 9.
Figure 3: ORTEP drawing of the complex 9.
All the complexes prepared in this work were characterized by IR, 1H, 13C, 2D NMR, X-ray diffraction (for 9 and 8) and UV–visible spectroscopy. The characterized bands in the IR spectra of all complexes are given in Table 1.
Table 1: The FTIR analysis of the complexes.
| vibration | bands ν (cm−1) | |||||
|---|---|---|---|---|---|---|
| attribution | 3 | 4 | 5 | 8 | 9 | 12 |
| ν(C–H) | 2956.4 | 2954.8 | 2956.7 | 2961.7 | 2957.8 | 2963.9 |
| ν(nitrile) | – | – | – | – | – | 2250.8 |
| ν(C=N) | 1645.4 | 1641.6 | 1637.4 | 1638.2 | 1642.2 | 1635.4 |
| ν(C–N) | 1350.2 | 1355.3 | 1436.7 | 1345.7 | 1377.8 | 1545.2 |
| δ(CH3) | 1205.2 | 1210.0 | 1201.0 | – | 1200.4 | 1260.3 |
| ν(P–C) | – | – | 1094.9 | – | – | – |
| ν(C–O) | 1016.1 | 1012.9 | 1011.0 | 1023.2 | 1030.8 | 1018.8 |
| δ(C–H) | 760 | 766.2 | 692.3 | 773.6 | 776.0 | – |
| δ(Pd–N) | – | 510.9 | 513.4 | 525.2 | 573 | – |
Optical properties
The gap energy values (Eg) of the complexes determined from the absorption edge of the solution spectra are given in Table 2. The gap energy was evaluated by the extrapolation of the tangent to the first inflexion point of the UV curve.
Finally, we have studied the catalytic activities of the new complexes during the decolorization of azo dyes in solutions which are discharged in textile industry.
Oxidative degradation of dyes
Six complexes were checked for the oxidative degradation of Eriochrome Blue Black B. The experimental results indicated that the complexes have potential activities during the degradation of the azo dyes in the aqueous medium and in the presence of hydrogen peroxide. From the preliminary data, it was found that all the prepared complexes have demonstrated a promising catalytic activity at the same conditions (t = 10 min, C0 = 30 mg/L, 10 mg of the catalyst, amount of H2O2 = 0.5 mL). Among the six compounds, catalyst 9 was found to be the most active during this study because the corresponding solution became almost colorless within five minutes (Figure 4). As also clearly depicted in Figure 5, the complete removal of Eriochrome was reached in 10 min for all samples tested.
![[1860-5397-11-132-4]](/bjoc/content/figures/1860-5397-11-132-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Change in color removal in the presence of different catalysts within 10 min (before filtration). T: sample which contain only the solution of dye and H2O2.
Figure 4: Change in color removal in the presence of different catalysts within 10 min (before filtration). T...
![[1860-5397-11-132-5]](/bjoc/content/figures/1860-5397-11-132-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Change in color removal in the presence of different catalysts (after filtration over 10 min).
Figure 5: Change in color removal in the presence of different catalysts (after filtration over 10 min).
In attempts to check the efficiency of the prepared complex 9 concerning the degradation of Eriochrome Blue Black B, we have discussed the results by varying the experimental conditions (contact time, initial dye concentration, temperature and H2O2 dose).
Figure 6 represents the evolution of the rate of the degradation of Eriochrome against time using H2O2, the complex 9 plus H2O2 and the complex alone [18]. First, it can be registered that the color solution remains persistent and stable in the presence of H2O2 without the addition of any catalyst. However, it was observed that the concentration of the dye declined sharply, in the presence of the system catalyst/H2O2. Indeed, 84% of the target was achieved in the presence of the prepared catalyst after only 5 min of reaction time at 22 °C whereas the dye removal does not exceed 14% using the complex alone. The efficiency of the combination of catalysts/H2O2 for the degradation of the studied azo dye is so confirmed and catalyst 9 is able to decompose the reaction products completely by the cleavage of the azo linkage (chromophore structure: –N=N–, responsible for the color) [33,34].
![[1860-5397-11-132-6]](/bjoc/content/figures/1860-5397-11-132-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Evolution of the color degradation against time using Eriochrome plus H2O2, the complex plus H2O2 or the complex alone.
Figure 6: Evolution of the color degradation against time using Eriochrome plus H2O2, the complex plus H2O2 o...
Effect of the hydrogen peroxide concentration
As proved in the previous section, the action of H2O2 alone did not show any degradation capacity for the studied dye solution, although this agent is considered a relatively powerful oxidant. In this section, we examine the effect of H2O2 dose on the rate of dye removal for an initial dye concentration of 30 mg/L using 10 mg of the catalyst. Data given by Figure 6 revealed that the presence of the catalyst, with the increment in H2O2 concentration, significantly reduced the time necessary to decolorize the solution with high target removal. As an example, we observe that after only a 5 min reaction and at the optimum concentration of H2O2 (0.5 mL), the decolorization of the Eriochrome solution reached 84% whereas the target removal is about 18% for the studied catalyst in the same condition and using 0.2 mL of H2O2. This means that the increase of the oxidant concentration generates more free-hydroxyl radicals, causing the dye decolorization. On the contrary, a slight decrease of the decolorization process from 84% to 79% has occurred for a highest amount of H2O2 (0.7 mL) because the solution undergoes self-quenching (scavenger) of OH· radicals by adding amounts of H2O2 to produce HO2· radicals, according to the following equations:
H2O2 + HO· → H2O + HO2·
HO2·+ HO· → H2O2 + O2
We observe that all curves depicted in Figure 7 have a pseudo-first-order kinetics shape, suggesting, therefore, a first-order kinetics law.
![[1860-5397-11-132-7]](/bjoc/content/figures/1860-5397-11-132-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Change of the concentration of the Erio solution with the variation of H2O2 dose.
Figure 7: Change of the concentration of the Erio solution with the variation of H2O2 dose.
Effect of the initial dye concentration on the decolorization process
The effect of the initial dye concentration on the decolorization process was studied at a constant dose of H2O2 and at a temperature of 22 °C. Data given in Figure 8 exhibited that the percentage of color removal decreased with the increase of the initial dye concentration. As an example, it decreased from 84% (C = 30 mg/L) to 64% (C = 70 mg/L) for a reaction time of 5 min in the presence of the studied catalyst. This confirms the fact that the decolorization process depends, on the concentration of H2O2 and the initial dye concentration. These trends were in agreement with those observed in our laboratory during the study of the degradation of azo dyes [18-20] and also with literature [35,36].
![[1860-5397-11-132-8]](/bjoc/content/figures/1860-5397-11-132-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Evolution of the color removal against initial dye concentration.
Figure 8: Evolution of the color removal against initial dye concentration.
Effect of the temperature on the decolorization process of dyes
In this section, the effect of the temperature was studied at a constant dose of H2O2 and an initial dye concentration of 30 mg/L. The results shown in Figure 9 indicated that the removal of the color was enhanced when the temperature increased. The target removal after 5 min of reaction at 22 °C and 60 °C were, respectively, 84% and 94%, in the presence of the studied catalyst.
![[1860-5397-11-132-9]](/bjoc/content/figures/1860-5397-11-132-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: Change of the color removal versus temperature.
Figure 9: Change of the color removal versus temperature.
Thermodynamic parameters
To better understand the degradation process, the pseudo first-order kinetic equation was used to determine the kinetic parameters. The Arrhenius law was used to calculate the activation energy (Ea). The determination of the entropy and the enthalpy of activations (ΔS* and ΔH*) were performed using the Eyring equation [37]. The results are summarized in Table 3.
The activation energy (Ea) has been found to be low (16.669 kJ mol−1) confirming that the complex 9 was very efficient for the degradation of azo dyes using H2O2.
Reuse of the catalysts
In this section, the reusability of the catalysts was considered. To do this, for example, the catalyst 9 was separated by filtration, washed with distilled water after each run, then dried and further subjected to subsequent runs under the same conditions. The data given in Figure 10 indicate that the regeneration process could be repeated for six cycles, without appreciable activity loss.
![[1860-5397-11-132-10]](/bjoc/content/figures/1860-5397-11-132-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Recycling experiments for Erio removal (C0 = 30 ppm, 20 mL) in the presence of catalyst 9 at pH 7 and T = 22 °C.
Figure 10: Recycling experiments for Erio removal (C0 = 30 ppm, 20 mL) in the presence of catalyst 9 at pH 7 a...
The reuse of the prepared catalysts is found to be possible after separation and washing. The regenerated catalysts were also characterized by FTIR analyses after each cycle, and no change was observed.
Suggested mechanism of decolorization
On the evidence of the kinetic studies and the literature data, we propose the mechanistic pathway depicted in Scheme 4. The first step involves the complexation of the azo dye to palladium(II) hydroperoxide 13, followed by a peroxymetalation of the azo moiety. This then affords the pseudocyclic five membered hydroperoxometallic intermediaite 15 which decomposes to azoxy product 16 and palladium hydroxide complex 17. The degradation of the azoxy product 16 can afford quinolinones and diazonium salts as it established by many authors [38-40].
![[1860-5397-11-132-i4]](/bjoc/content/inline/1860-5397-11-132-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism of decolorization.
Scheme 4: Proposed mechanism of decolorization.
Conclusion
In summary, new palladium-complexes using oxazolines as ligands were synthesized, characterized and tested for the catalytic activity during the degradation of the dye Eriochrome Blue Black B. The variation of the experimental conditions exhibited that the degradation process is contact time, initial dye concentration, temperature and H2O2 dose-dependent. 84% of target removal of Eriochrome Blue Black B was reached within minutes, under some experimental conditions. These new complexes prove to be active and also to be a reusable catalyst for the decolorization of Erio solutions in the presence of hydrogen peroxide. Further work is ongoing to apply the same strategy for the degradation of other organic pollutants.
Experimental
Analytical methods
Unless otherwise noted, all starting materials were obtained from commercial suppliers and used without purification. NMR spectra were recorded on a 300 MHz and 200 MHz Bruker spectrometer. Chemical shifts were reported in ppm relative to the residual solvent peak (7.27 ppm for CHCl3) for 1H spectra and (77.00 ppm for CDCl3) for 13C spectra. All chemical shifts were reported as δ values (ppm) relative to internal tetramethylsilane. High resolution mass spectrometry data were recorded on an Autospec Ultima (Waters/Micromass) device with a resolution of 5000 RP at 5%. Thin-layer chromatography (TLC) was carried out on aluminium sheets precoated with silica gel 60 F254. Microwave irradiations were realized using an Anton Paar Monowave 300 apparatus. Microwave heating was performed with a single mode cavity Discover Microwave Synthesizer, producing continuous irradiation with IR temperature control. An ultraviolet–visible spectrophotometer (U-2000 Hitachi), wavelengths of range 200–800 nm and a quartz cell were employed for the absorbance measurements.
Synthesis of the ligands
The amino acids and (R)-2-aminobutan-1-ol were purchased from Sigma-Aldrich. The other α-aminoalcohols were obtained by the reduction of the corresponding amino acids using the method developed by Meyers [41]. The ligands bis- and mono-oxazolines were prepared from the available optically pure α-aminoalcohols (derived from the corresponding amino acids). (S)-4-Isopropyl-2-(naphthalen-1-yl)oxazoline (2) was isolated in a moderate yield from the condensation of the L-valinol with naphthonitrile under microwave irradiation, while the second ligand 1,2-bis[(S)-4-phenyloxazoline]benzene (7) was synthesized from L-(α)-(+)-phenylglycinol under the same conditions of the reaction as described by B. Ben Hassine et al. [42]. The third ligand 3-[(4S)-4,5-dihydro-4-isopropyl-1,3-oxazol-2-yl]propanenitrile (11) was obtained using the reaction of 4-ethoxy-4-iminobutanenitrile monohydrochloride with L-valinol in high yield [43].
Synthesis of the cyclopalladated complexes
Synthesis of (S)-chloro-[(4-isopropyloxazolinyl)-2-naphthyl](triphenylphosphine)palladium(II) (5): The complex (3) was synthesized using two methods:
Method A: A mixture of Pd(OAc)2 (50 mg, 0.22 mmol, 1 equiv), AcONa (18.3 mg, 0.22 mmol, 1 equiv) and (S)-4-isopropyl-2-(naphthalen-1-yl)oxazoline (2, 59 mg, 0.24 mmol, 1.1 equiv) in acetic acid (3.0 mL) was heated in an oil bath at 80 °C for 3.5 h. Complex 3 was isolated in 90% yield.
Method B: Pd(OAc)2 (50 mg, 0.22 mmol) was added to an acetonitrile solution (3 mL) of oxazoline (2) and refluxed for 3 h at 78 °C. The mixture was allowed to cool to rt and filtered through celite. The solvent was evaporated, and the crude product was recrystallized from ether/petroleum ether to obtain 3 (89%).
The metathesis of dimer 3 (0.196 mmol, 1 equiv) with LiCl (18.5 mg, 0.43 mmol, 2.2 equiv) in acetone (7.0 mL) at room temperature for 24 h afforded dimer 4 in 91% yield. PPh3 (94.4 mg, 0.36 mmol, 2 equiv) was added to a stirred solution of the dimer 4 (138 mg, 0.18 mmol, 1 equiv) in toluene (10.0 mL). After 12 h, the solvent was evaporated to obtain a pale-yellow solid, which was purified by trituration with petroleum ether or recrystallization from pentane/CH2Cl2 to afford pure 5 as a yellow powder in 78% yield. [α]D −350 ± 36.9 (c 0.02, MeCN); 1H NMR (CDCl3, 300 MHz) δ 8.03 (dd, J = 7.2 Hz, J = 1.5 Hz, 1H), 7.80 (dd, J = 9.6 Hz, J = 1.2 Hz, 1H), 7.73–7.66 (m, 2H), 7.48–7.42, 7.20–7.16 (m, 15H(PPh3)), 7.09 (d, 1H), 6.97 (t, 1H), 5.46 (dt, J = 9.6 Hz, J = 4.8 Hz, CH-N, 1H), 4.58 (m, J = 9.6 Hz, J = 8.7 Hz, 1H), 4.37 (m, J = 8.7 Hz, J = 5.4 Hz, 1H), 2.28–2.19 (m, 1H), 0.92 (d, J = 6.9 Hz, 3H), 0.71 (d, J 6.9 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.1, 143.2–123.2 (C(PPh3)), 134.8, 132.9, 131.7, 131.1, 129.9, 128.5, 128.0, 127.8, 124.8, 123.9, 70.2, 68.0, 31.3, 18.5, 15.9; 31P NMR (MeOD, 75 MHz) δ 36.0 ppm; TOF–MS (ES+) for (C34H31NOPPd): theoretical [M − Cl]+: 602.1199; measured [M − Cl]+: 602.1201; FTIR (KBr pellets, cm−1): 2956.7, 1637.4, 1436.7, 1201.0, 1094.9, 1011.0, 692.3, 513.4.
Synthesis of dichloro-[1,2-bis((S)-4-phenyl-4,5-dihydrooxazol-2-yl)benzene]palladium(II) (8): Complex 8 was synthesized from 1,2-bis((S)-4-phenyl-4,5-dihydrooxazol-2-yl)benzene (7) (170 mg, 0.46 mmol, 1.01 equiv) and sodium tetrachloropalladate(II) (134 mg, 0.45 mmol, 1 equiv) in freshly distilled and thoroughly degassed methanol (5 mL). The red solution was allowed to stand for 1 h at room temperature. After filtration, the solid was washed with methanol to afford the expected palladium(II) complex 8 (0.34 mmol) in 75% yield. 1H NMR (CDCl3, 300 MHz) δ 7.99–7.96 (m, 4H), 7.65–7.58 (m, 5H, Haromat), 7.31–7.04 (m, 5H, Haromat), 5.88 (dd, J = 10.2 Hz, J = 5.7 Hz, 1H), 5.06 (t, J = 9.3 Hz, 1H), 4.89 (dd, J = 9.3 Hz, J = 5.7 Hz, 1H), 4.62 (t, J = 9.3 Hz, 1H), 4.51 (t, J = 9 Hz, 1H), 4.10 (t, J = 9 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 162.5, 142.9–124.4, 73.2, 70.5; TOF–MS (ES+) for (C24H20ClN2O2Pd): theoretical [M − Cl]+: 511.0202; measured [M − Cl]+: 511.0201.
Synthesis of the bis(oxazoline) coordinated complexes
Synthesis of dichlorobis[(4-isopropyl-2-naphthalen-1-yl)oxazoline)]palladium(II) (9) and dichlorobis(4-isopropyl-2-(2-cyanoethyl)oxazoline)palladium(II) (12): Complexes 9 and 12 were synthesized using the same procedure. A solution of sodium tetrachloropalladate(II) Na2PdCl4 (0.34 mmol, 1 equiv) in absolute MeOH (3 mL) was added to (0.75 mmol, 2.2 equiv) of the ligand. A yellow precipitate was formed immediately. The mixture was stirred for 24 h at room temperature. After removal of the solvent under reduced pressure, the yellow solid was washed with methanol, and recrystallized from CHCl3/hexane. Yields of dichloro[bis(4-isopropyl-2-naphthalen-1-yl)oxazoline]palladium(II) (9) and dichloro[bis(4-isopropyl-2-(2-cyanoethyl)oxazoline)]palladium(II) (12) are 85% and 68%, respectively.
Complex 9: [α]D −113 ± 28 (c 0.1, MeCN); 1H NMR (CDCl3, 300 MHz) δ 8.49 (d, J = 6.9 Hz, 1H, H1), 8.00–7.97 (m, 2H, H3,7), 7.90–7.87 (m, 1H, H5), 7.55–7.49 (m, 2H, H6,7), 7.43 (bs, 1H, H2), 4.58–4.50 (m, 1H, CH-N), 4.46–4.38 (m, 2H, CH2-O), 2.62 (m, 1H, CH(CH3)2), 0.99 (d, J = 6.9 Hz, 3H, CH3), 0.92 (d, J = 6 Hz, 3H, CH3); 13C NMR (CDCl3, 75 MHz) δ 168.9, 133.2, 132.0, 130.7, 130.0, 128.2, 127.1, 126.4, 125.8, 124.6, 124.2, 70.8, 69.3, 30.0, 19.1, 15.3; TOF–MS (ES+) for (C18H29ClN4O2Pd): theoretical [M − Cl]+: 615.1365; measured [M − Cl]+: 615.1361; FTIR (KBr pellets, cm−1): 2957.8, 1642.2, 1377.8, 1200.4, 1030.8, 776.0, 573.
Complex 12: [α]D −5.7 ± 0.5 (c 0.94, CHCl3); 1H NMR (MeOD, 300 MHz) δ 3.64–3.56 (m, 1H), 3.50–3.39 (m, 2H), 2.66–2.45 (m, 1H), 1.83–1.73 (m, 1H), 0.88–0.80 (m, 6H); 13C NMR (MeOD, 75 MHz) δ 173.0, 119.4, 64.0, 58.9, 33.1, 30.8, 20.8, 19.6, 14.7; TOF–MS (ES+) for (C18H29Cl2N4O2Pd): theoretical [M + H]+: 509.0594; measured [M + H]+: 509.0598; FTIR (KBr pellets, cm−1): 2963.9, 2250.8, 1635.4, 1545.2, 1260.3, 1018.8.
Oxidative degradation procedures of dyes
All experiments were carried out in a batch system for a period of time (sufficient to achieve equilibrium) and were uniformly agitated at a speed of 150 rpm. The pH of the solution was adjusted to 7.0 (buffered aqueous solution). 0.2 mg of each catalyst were added to 20 mL of the dye solution (C0 = 30 mg/L) followed by the addition of a calculated dose of H2O2. At the end of each contact time, the content of the flasks was filtered using a Whatman No. 41 filter paper. The concentration of dye in each filtrate was determined at the maximum wavelength (531 nm). Factors affecting the degradation of the dyes such as H2O2 dose, initial concentration and temperature were studied in the ranges 0–0.7 mL/L, 30–70 mg/L and 22–60 °C, respectively.
Supporting Information
The electronic Supporting Information includes the X-ray diffraction of the structures of complexes 8 and 9. The crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data Center, CCDC 1052659-1052660 for 8 and 9, respectively. Crystallographic data associated with this article can be obtained, free of charge, on application to CCDC, at http://www.ccdc.cam.ac.uk/deposit/.
| Supporting Information File 1: CIF file for complex 8. | ||
| Format: CIF | Size: 1.7 MB | Download |
| Supporting Information File 2: CIF file for complex 9. | ||
| Format: CIF | Size: 681.7 KB | Download |
| Supporting Information File 3: Experimental procedures, spectroscopic and analytical data, and copies of spectra of the products. | ||
| Format: PDF | Size: 1.0 MB | Download |
Acknowledgements
The authors are grateful to the DGRS “Direction Generale de la Recherche Scientifique” (Directorate General for Scientific Research) of the Tunisian Ministry of Higher Education and Scientific Research, the CNRS, the University of Versailles Saint Quentin-en-Yvelines and ANR (ANR-11-BS07-030-01) France for the financial support.
References
-
Laine, T. V.; Piironen, U.; Lappalainen, K.; Klinga, M.; Aitola, E.; Leskelä, M. J. Organomet. Chem. 2000, 606, 112–124. doi:10.1016/S0022-328X(00)00291-6
Return to citation in text: [1] -
Karch, R.; Fazla, D. Novel nickel-palladium and platinum–carbene complexes, their preparation and use in catalytic reactions. Canadian Patent CA2,494,685 C, Sept 25, 2012.
Return to citation in text: [1] -
Abu-Surrah, A. S.; Al-Sadoni, H. H.; Abdalla, M. Y. Cancer Ther. 2008, 6, 1–10.
Return to citation in text: [1] -
de Vries, J. G. Top. Organomet. Chem. 2012, 42, 1–34. doi:10.1007/3418_2012_32
Return to citation in text: [1] -
Modha, S. G.; Mehta, V. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2013, 42, 5042–5055. doi:10.1039/c3cs60041f
Return to citation in text: [1] -
Bolm, C. Angew. Chem., Int. Ed. Engl. 1991, 30, 542–543. doi:10.1002/anie.199105421
Return to citation in text: [1] -
Gómez-Simón, M.; Jansat, S.; Muller, G.; Panyella, D.; Font-Bardia, M.; Solans, X. J. Chem. Soc., Dalton Trans. 1997, 3755–3764. doi:10.1039/A703951D
Return to citation in text: [1] -
McManus, H. A.; Guiry, P. J. Chem. Rev. 2004, 104, 4151–4202. doi:10.1021/cr040642v
Return to citation in text: [1] -
Hussain, S. M. S.; Ibrahim, M. B.; Fazal, A.; Suleiman, R.; Fettouhi, M.; El Ali, B. Polyhedron 2014, 70, 39–46. doi:10.1016/j.poly.2013.12.023
Return to citation in text: [1] -
Robinson, T.; Chandran, B.; Nigam, P. Water Res. 2002, 36, 2824–2830. doi:10.1016/S0043-1354(01)00521-8
Return to citation in text: [1] -
Han, M. H.; Yun, Y.-S. Biochem. Eng. J. 2007, 36, 2–7. doi:10.1016/j.bej.2006.06.010
Return to citation in text: [1] -
Santos, V. P.; Pereira, M. F. R.; Faria, P. C. C.; Órfão, J. J. M. J. Hazard. Mater. 2009, 162, 736–742. doi:10.1016/j.jhazmat.2008.05.090
Return to citation in text: [1] -
Vaidya, A. A.; Date, K. V. Colourage 1982, 14, 3.
Return to citation in text: [1] -
Boer, C. G.; Obici, L.; Marques de Souza, C. G.; Peralta, R. M. Bioresour. Technol. 2004, 94, 107–112. doi:10.1016/j.biortech.2003.12.015
Return to citation in text: [1] -
Anliker, R. Ecotoxicol. Environ. Saf. 1979, 3, 59–74. doi:10.1016/0147-6513(79)90060-5
Return to citation in text: [1] -
Chung, K.-T.; Stevens, S. E., Jr. Environ. Toxicol. Chem. 1993, 12, 2121–2132. doi:10.1002/etc.5620121120
Return to citation in text: [1] -
Aloui, F.; Jabli, M.; Ben Hassine, B. Synth. Commun. 2013, 43, 277–291. doi:10.1080/00397911.2011.598254
Return to citation in text: [1] -
Jabli, M.; Touati, R.; Kacem, Y.; Ben Hassine, B. J. Text. Inst. 2012, 103, 434–450. doi:10.1080/00405000.2011.581797
Return to citation in text: [1] [2] [3] -
Haji, S.; Benstaali, B.; Al-Bastaki, N. Chem. Eng. J. 2011, 168, 134–139. doi:10.1016/j.cej.2010.12.050
Return to citation in text: [1] [2] -
Kuo, C.-Y. J. Hazard. Mater. 2009, 163, 239–244. doi:10.1016/j.jhazmat.2008.06.083
Return to citation in text: [1] [2] -
Huang, Y.-H.; Huang, Y.-F.; Chang, P.-S.; Chen, C.-Y. J. Hazard. Mater. 2008, 154, 655–662. doi:10.1016/j.jhazmat.2007.10.077
Return to citation in text: [1] -
Jabli, M.; Aloui, F.; Ben Hassine, B. J. Eng. Fibers Fabr. 2013, 8 (3), 19–34.
Return to citation in text: [1] -
Vincent, T.; Guibal, E. Langmuir 2003, 20, 8475–8483. doi:10.1021/la034364r
Return to citation in text: [1] -
Gossage, R. A.; Jenkins, H. A.; Yadav, P. N. Tetrahedron Lett. 2004, 45, 7689–7691. doi:10.1016/j.tetlet.2004.08.082
Return to citation in text: [1] -
Overman, L. E.; Owen, C. E.; Pavan, M. M. Org. Lett. 2003, 5, 1809–1812. doi:10.1021/ol0271786
Return to citation in text: [1] -
von Matt, P.; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 566–568. doi:10.1002/anie.199305661
Return to citation in text: [1] -
Jin, Y.; Du, D.-M. Tetrahedron 2012, 68, 3633–3640. doi:10.1016/j.tet.2012.02.078
Return to citation in text: [1] -
Jansat, S.; Gómez, M.; Philippot, K.; Muller, G.; Guiu, E.; Claver, C.; Castillón, S.; Chaudret, B. J. Am. Chem. Soc. 2004, 126, 1592–1593. doi:10.1021/ja036132k
Return to citation in text: [1] -
Peterson, D. L.; Keuseman, K. J.; Kataeva, N. A.; Kuz’mina, L. G.; Howard, J. A. K.; Dunina, V. V.; Smoliakova, I. P. J. Organomet. Chem. 2002, 654, 66–73. doi:10.1016/S0022-328X(02)01376-1
Return to citation in text: [1] -
Mawo, R. Y.; Johnson, D. M.; Wood, J. L.; Smoliakova, I. P. J. Organomet. Chem. 2008, 693, 33–45. doi:10.1016/j.jorganchem.2007.10.011
Return to citation in text: [1] -
Mawo, R. Y.; Mustakim, S.; Young, V. G., Jr.; Hoffmann, M. R.; Smoliakova, I. P. Organometallics 2007, 26, 1801–1810. doi:10.1021/om061132p
Return to citation in text: [1] -
Kilian, P.; Knight, F. R.; Woollins, J. D. Chem. – Eur. J. 2011, 17, 2302–2328. doi:10.1002/chem.201001750
Return to citation in text: [1] -
Zille, A.; Górnacka, B.; Rehorek, A.; Cavaco-Paulo, A. Appl. Environ. Microbiol. 2005, 71, 6711–6718. doi:10.1128/AEM.71.11.6711-6718.2005
Return to citation in text: [1] -
Elmorsi, T. M.; Riyad, Y. M.; Mohamed, Z. H.; El Bary, H. M. H. A. J. Hazard. Mater. 2010, 174, 352–358. doi:10.1016/j.jhazmat.2009.09.057
Return to citation in text: [1] -
Gazi, S.; Rajakumar, A.; Singh, N. D. P. J. Hazard. Mater. 2010, 183, 894–901. doi:10.1016/j.jhazmat.2010.07.113
Return to citation in text: [1] -
Shen, C.; Song, S.; Zang, L.; Kang, X.; Wen, Y.; Liu, W.; Fu, L. J. Hazard. Mater. 2010, 177, 560–566. doi:10.1016/j.jhazmat.2009.12.069
Return to citation in text: [1] -
Sismanoglu, T.; Pura, S. Colloids Surf., A 2001, 180, 1–6. doi:10.1016/S0927-7757(00)00751-2
Return to citation in text: [1] -
Molnár, A.; Boros, S.; Simon, K.; Hermecz, I.; Gönczi, C. ARKIVOC 2010, No. x, 199–207.
Return to citation in text: [1] -
Bhirud, R. G.; Srisankar, E. V.; Narayan, K. S. Chem. Sci. 1991, 103, 83–93.
Return to citation in text: [1] -
Lin, J.-j.; Zhao, X.-s.; Liu, D.; Yu, Z.-g.; Zhang, Y.; Xu, H. J. Hazard. Mater. 2008, 157, 541–546. doi:10.1016/j.jhazmat.2008.01.050
Return to citation in text: [1] -
McKennon, M. J.; Meyers, A. I.; Drauz, K.; Schwarm, M. J. Org. Chem. 1993, 58, 3568–3571. doi:10.1021/jo00065a020
Return to citation in text: [1] -
Hassani, R.; Requet, A.; Marque, S.; Gaucher, A.; Prim, D.; Kacem, Y.; Ben Hassine, B. Tetrahedron: Asymmetry 2014, 25, 1275–1279. doi:10.1016/j.tetasy.2014.08.010
Return to citation in text: [1] -
Hassani, R.; Kacem, K.; Ben Mansour, H.; Ben Ammar, H.; Ben Hassine, B. Chem.-Biol. Interact. 2014, 217, 41–48. doi:10.1016/j.cbi.2014.04.003
Return to citation in text: [1]
| 42. | Hassani, R.; Requet, A.; Marque, S.; Gaucher, A.; Prim, D.; Kacem, Y.; Ben Hassine, B. Tetrahedron: Asymmetry 2014, 25, 1275–1279. doi:10.1016/j.tetasy.2014.08.010 |
| 43. | Hassani, R.; Kacem, K.; Ben Mansour, H.; Ben Ammar, H.; Ben Hassine, B. Chem.-Biol. Interact. 2014, 217, 41–48. doi:10.1016/j.cbi.2014.04.003 |
| 1. | Laine, T. V.; Piironen, U.; Lappalainen, K.; Klinga, M.; Aitola, E.; Leskelä, M. J. Organomet. Chem. 2000, 606, 112–124. doi:10.1016/S0022-328X(00)00291-6 |
| 5. | Modha, S. G.; Mehta, V. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2013, 42, 5042–5055. doi:10.1039/c3cs60041f |
| 20. | Kuo, C.-Y. J. Hazard. Mater. 2009, 163, 239–244. doi:10.1016/j.jhazmat.2008.06.083 |
| 21. | Huang, Y.-H.; Huang, Y.-F.; Chang, P.-S.; Chen, C.-Y. J. Hazard. Mater. 2008, 154, 655–662. doi:10.1016/j.jhazmat.2007.10.077 |
| 22. | Jabli, M.; Aloui, F.; Ben Hassine, B. J. Eng. Fibers Fabr. 2013, 8 (3), 19–34. |
| 3. | Abu-Surrah, A. S.; Al-Sadoni, H. H.; Abdalla, M. Y. Cancer Ther. 2008, 6, 1–10. |
| 15. | Anliker, R. Ecotoxicol. Environ. Saf. 1979, 3, 59–74. doi:10.1016/0147-6513(79)90060-5 |
| 16. | Chung, K.-T.; Stevens, S. E., Jr. Environ. Toxicol. Chem. 1993, 12, 2121–2132. doi:10.1002/etc.5620121120 |
| 2. | Karch, R.; Fazla, D. Novel nickel-palladium and platinum–carbene complexes, their preparation and use in catalytic reactions. Canadian Patent CA2,494,685 C, Sept 25, 2012. |
| 17. | Aloui, F.; Jabli, M.; Ben Hassine, B. Synth. Commun. 2013, 43, 277–291. doi:10.1080/00397911.2011.598254 |
| 18. | Jabli, M.; Touati, R.; Kacem, Y.; Ben Hassine, B. J. Text. Inst. 2012, 103, 434–450. doi:10.1080/00405000.2011.581797 |
| 19. | Haji, S.; Benstaali, B.; Al-Bastaki, N. Chem. Eng. J. 2011, 168, 134–139. doi:10.1016/j.cej.2010.12.050 |
| 9. | Hussain, S. M. S.; Ibrahim, M. B.; Fazal, A.; Suleiman, R.; Fettouhi, M.; El Ali, B. Polyhedron 2014, 70, 39–46. doi:10.1016/j.poly.2013.12.023 |
| 12. | Santos, V. P.; Pereira, M. F. R.; Faria, P. C. C.; Órfão, J. J. M. J. Hazard. Mater. 2009, 162, 736–742. doi:10.1016/j.jhazmat.2008.05.090 |
| 8. | McManus, H. A.; Guiry, P. J. Chem. Rev. 2004, 104, 4151–4202. doi:10.1021/cr040642v |
| 13. | Vaidya, A. A.; Date, K. V. Colourage 1982, 14, 3. |
| 14. | Boer, C. G.; Obici, L.; Marques de Souza, C. G.; Peralta, R. M. Bioresour. Technol. 2004, 94, 107–112. doi:10.1016/j.biortech.2003.12.015 |
| 7. | Gómez-Simón, M.; Jansat, S.; Muller, G.; Panyella, D.; Font-Bardia, M.; Solans, X. J. Chem. Soc., Dalton Trans. 1997, 3755–3764. doi:10.1039/A703951D |
| 6. | Bolm, C. Angew. Chem., Int. Ed. Engl. 1991, 30, 542–543. doi:10.1002/anie.199105421 |
| 10. | Robinson, T.; Chandran, B.; Nigam, P. Water Res. 2002, 36, 2824–2830. doi:10.1016/S0043-1354(01)00521-8 |
| 11. | Han, M. H.; Yun, Y.-S. Biochem. Eng. J. 2007, 36, 2–7. doi:10.1016/j.bej.2006.06.010 |
| 30. | Mawo, R. Y.; Johnson, D. M.; Wood, J. L.; Smoliakova, I. P. J. Organomet. Chem. 2008, 693, 33–45. doi:10.1016/j.jorganchem.2007.10.011 |
| 31. | Mawo, R. Y.; Mustakim, S.; Young, V. G., Jr.; Hoffmann, M. R.; Smoliakova, I. P. Organometallics 2007, 26, 1801–1810. doi:10.1021/om061132p |
| 24. | Gossage, R. A.; Jenkins, H. A.; Yadav, P. N. Tetrahedron Lett. 2004, 45, 7689–7691. doi:10.1016/j.tetlet.2004.08.082 |
| 25. | Overman, L. E.; Owen, C. E.; Pavan, M. M. Org. Lett. 2003, 5, 1809–1812. doi:10.1021/ol0271786 |
| 26. | von Matt, P.; Pfaltz, A. Angew. Chem., Int. Ed. Engl. 1993, 32, 566–568. doi:10.1002/anie.199305661 |
| 27. | Jin, Y.; Du, D.-M. Tetrahedron 2012, 68, 3633–3640. doi:10.1016/j.tet.2012.02.078 |
| 28. | Jansat, S.; Gómez, M.; Philippot, K.; Muller, G.; Guiu, E.; Claver, C.; Castillón, S.; Chaudret, B. J. Am. Chem. Soc. 2004, 126, 1592–1593. doi:10.1021/ja036132k |
| 29. | Peterson, D. L.; Keuseman, K. J.; Kataeva, N. A.; Kuz’mina, L. G.; Howard, J. A. K.; Dunina, V. V.; Smoliakova, I. P. J. Organomet. Chem. 2002, 654, 66–73. doi:10.1016/S0022-328X(02)01376-1 |
| 38. | Molnár, A.; Boros, S.; Simon, K.; Hermecz, I.; Gönczi, C. ARKIVOC 2010, No. x, 199–207. |
| 39. | Bhirud, R. G.; Srisankar, E. V.; Narayan, K. S. Chem. Sci. 1991, 103, 83–93. |
| 40. | Lin, J.-j.; Zhao, X.-s.; Liu, D.; Yu, Z.-g.; Zhang, Y.; Xu, H. J. Hazard. Mater. 2008, 157, 541–546. doi:10.1016/j.jhazmat.2008.01.050 |
| 41. | McKennon, M. J.; Meyers, A. I.; Drauz, K.; Schwarm, M. J. Org. Chem. 1993, 58, 3568–3571. doi:10.1021/jo00065a020 |
| 35. | Gazi, S.; Rajakumar, A.; Singh, N. D. P. J. Hazard. Mater. 2010, 183, 894–901. doi:10.1016/j.jhazmat.2010.07.113 |
| 36. | Shen, C.; Song, S.; Zang, L.; Kang, X.; Wen, Y.; Liu, W.; Fu, L. J. Hazard. Mater. 2010, 177, 560–566. doi:10.1016/j.jhazmat.2009.12.069 |
| 37. | Sismanoglu, T.; Pura, S. Colloids Surf., A 2001, 180, 1–6. doi:10.1016/S0927-7757(00)00751-2 |
| 33. | Zille, A.; Górnacka, B.; Rehorek, A.; Cavaco-Paulo, A. Appl. Environ. Microbiol. 2005, 71, 6711–6718. doi:10.1128/AEM.71.11.6711-6718.2005 |
| 34. | Elmorsi, T. M.; Riyad, Y. M.; Mohamed, Z. H.; El Bary, H. M. H. A. J. Hazard. Mater. 2010, 174, 352–358. doi:10.1016/j.jhazmat.2009.09.057 |
| 18. | Jabli, M.; Touati, R.; Kacem, Y.; Ben Hassine, B. J. Text. Inst. 2012, 103, 434–450. doi:10.1080/00405000.2011.581797 |
| 19. | Haji, S.; Benstaali, B.; Al-Bastaki, N. Chem. Eng. J. 2011, 168, 134–139. doi:10.1016/j.cej.2010.12.050 |
| 20. | Kuo, C.-Y. J. Hazard. Mater. 2009, 163, 239–244. doi:10.1016/j.jhazmat.2008.06.083 |
| 32. | Kilian, P.; Knight, F. R.; Woollins, J. D. Chem. – Eur. J. 2011, 17, 2302–2328. doi:10.1002/chem.201001750 |
| 18. | Jabli, M.; Touati, R.; Kacem, Y.; Ben Hassine, B. J. Text. Inst. 2012, 103, 434–450. doi:10.1080/00405000.2011.581797 |
© 2015 Hassani et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
