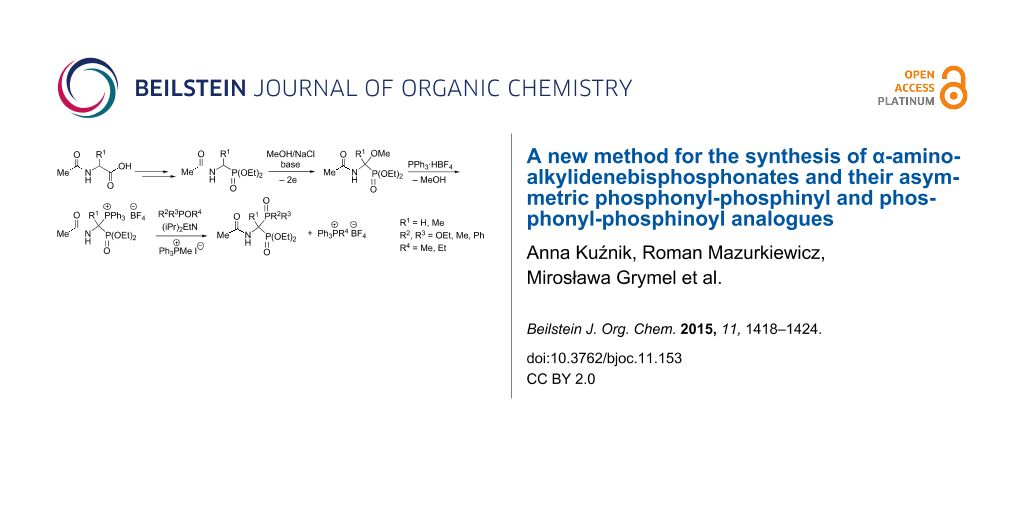
Abstract
A convenient approach has been developed to α-aminoalkylidenebisphosphonates and their asymmetric phosphonyl-phosphinyl and phosphonyl-phosphinoyl analogues by α-phosphonylation, α-phosphinylation or α-phosphinoylation of 1-(N-acylamino)alkylphosphonates, that, in turn, are easily accessible from N-acyl-α-amino acids. Effective electrophilic activation of the α-position of 1-(N-acetylamino)alkylphosphonates was achieved by electrochemical α-methoxylation of these compounds in methanol, mediated with NaCl, followed by displacement of the methoxy group with triphenylphosphonium tetrafluoroborate to give hitherto unknown 1-(N-acetylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborates. The latter compounds react smoothly with trialkyl phosphites, dialkyl phosphonites or alkyl phosphinites in the presence of Hünig’s base and methyltriphenylphosphonium iodide in a Michaelis–Arbuzov-like reaction to give the expected alkylidenebisphosphonates, 1-phosphinylalkylphosphonates or 1-phosphinoylalkylphosphonates, respectively, in good yields.

Graphical Abstract
Introduction
α-Aminophosphonic and α-aminophosphinic acids, as phosphorus analogues and bioisosters of α-amino acids, exhibit a variety of important biological properties [1-3]. α-Aminobisphosphonates of general formula 1 (Figure 1, X = NHR), that can be considered as one of the most interesting subclass of α-aminophosphonic acids [1,3], are an important class of drugs that are currently successfully being used to treat osteoporosis and similar diseases, including Paget’s disease (osteitis deformans), bone metastasis (with or without hypercalcaemia), Kahler’s disease (multiple myeloma), primary hyperparathyroidism, brittle bone disease (osteogenesis imperfecta), fibrous dysplasia and others [1,4].α‑Aminobisphosphonates (Figure 1, X = NHR) and α‑hydroxybisphosphonates (Figure 1, X = OH) that can easily be obtained from α‑amino derivatives [5,6] belong to two of the most important subclasses of bisphosphonates with many successful medical applications (Table 1) [1,3,4,7-9].
![[1860-5397-11-153-1]](/bjoc/content/figures/1860-5397-11-153-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: General structure of bisphosphonates.
Figure 1: General structure of bisphosphonates.
Table 1: Antiresorptive potency of selected α-amino and α-hydroxy derivatives of bisphosphonates [3,7,8].
| Comp. | Generic name | X | R1 | R2 | R3 | R4 | ED50 [µg/kg] |
|---|---|---|---|---|---|---|---|
| 1a | Incadronate | NHR2 | H |
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-153-i3.svg?max-width=637&scale=1.0)
|
H | Na | 7.0 |
| 1b | NE 58025 | NHR2 | H |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-153-i4.svg?max-width=637&scale=1.0)
|
H | H | |
| 1c | NE 97220 | NHR2 | H |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-153-i5.svg?max-width=637&scale=1.0)
|
H | H | |
| 1d | Risedronate | OH |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-153-i6.svg?max-width=637&scale=1.0)
|
– | H | H | 12.0 |
| 1e | Ibandronate | OH |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-153-i7.svg?max-width=637&scale=1.0)
|
– | H | H | 1.1 |
| 1f | Zoledronate | OH |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-153-i8.svg?max-width=637&scale=1.0)
|
– | H | H | 0.07 |
The most common methods for the synthesis of 1-aminoalkylidenebisphosphonic acid derivatives consist in the consecutive formation of two Cα–P bonds between a carbon electrophile (most often an electrophilic imine intermediate) and two identical molecules of the proper phosphorus nucleophile. Another group of methods requires the formation of a Cα–N bond between an easily accessible, symmetrical, nucleophilic methylenebisphosphonic acid derivative and a nitrogen electrophile. Both groups of methods result in symmetrical products with the same phosphonyl or dialkoxyphosphonyl groups [10-18].
Up until 1989, very little was known about the preparative feasibility and biological activity of 1-phosphinylalkylphosphonates, including their most interesting 1-hydroxy and 1-amino derivatives (Figure 2, 2 and 3, respectively; R = H, Alk, Ar). In 1990, Ebetino et al. described both a contiguous and stepwise strategy for the synthesis of the pyridylaminomethane-based class of phosphinylalkylphosphonates 3 (Figure 2, R = Me or n-butyl, R1 = H, R2 = Py and its derivatives, R3, R4 = H), and discovered the bone resorption-inhibition ability of these compounds [19-21]. They also registered a number of patents relating to methods for treating or preventing diseases characterised by abnormal calcium and phosphate metabolism by utilising 1-phosphinylalkylphosphonic acid derivatives, especially their 1-amino and 1-hydroxy derivatives, and their pharmaceutical compositions [22-27].
![[1860-5397-11-153-2]](/bjoc/content/figures/1860-5397-11-153-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: General structures of 1-hydroxy- and 1-amino-1-phosphinylalkylphosphonates (2 and 3, respectively) and 1-amino-1-phosphinoylalkylphosphonates 4.
Figure 2: General structures of 1-hydroxy- and 1-amino-1-phosphinylalkylphosphonates (2 and 3, respectively) ...
In the contiguous method, the condensation of 2-amino-3-picoline with triethyl orthoformate, diethyl phosphite and monoethyl methylphosphonite, followed by chromatographic separation of the three possible condensation products and the final hydrolysis, gave the expected phosphinylalkylphosphonic acid in only 17% yield [20]. The stepwise method consisted in the condensation of diethyl chlorophosphonite with triethyl orthoformate to diethyl 1,1-diethoxymethylphosphonate [(EtO)2CHPO(OEt)2], followed by condensation of the latter compound with 2-amino-3-picoline and monoethyl methylphosphonite. The final hydrolysis gave the expected phosphinylalkylphosphonic acid in 35% yield based on 2-amino-3-picoline [20].
In 1989, Schrader and Steglich described an example of synthesis of diethyl 1-(N-acylamino)-1-[ethoxy(methylphosphinyl)]methylphosphonate using a Michaelis–Arbuzov-like condensation of the corresponding ethyl [1-(N-acylamino)-1-bromomethyl]methylphosphinate with triethyl phosphite in 79% yield [28].
The first described synthesis of 1-amino-1-phosphinoylalkylphosphonates 4 (Figure 2), reported by Kostka and Kotyński in 1990–1993 was the synthesis of diethyl 1-(N-salicyloylamino)-1-diphenylphosphinoylmethylphosphonate (Figure 2, 4: R = Ph, R1 = H, R2 = o-HOC6H4CO, R3 = Et) by condensation of diethyl 2,3-dihydro-4H-1,3-benzoxazin-4-one-2-phosphonate with diphenylphosphine oxide [29] or diphenyltrimethylsilyloxyphosphine [Ph2POSiMe3] [30]. In 2004–2007, Onys’ko et al. reported the synthesis of diethyl or diphenyl 1-(N-phenylsulfonylamino)-1-diphenylphosphinoylmethylphosphonate derivatives (Figure 2, 4: R = Ph, R1 = Ar or CCl3, R2 = SO2Ph, R3 = Et or Ph) and diethyl 1-amino-1-diphenylphosphinoyl-2,2,2-trifluoroethylphosphonate (Figure 2, 4: R = Ph, R1 = CF3, R2 = H, R3 = Et) by the addition of diphenylphosphine oxide to the corresponding arylimidoyl-, trichloroacetimidoyl- or trifluoroacetimidoylphosphonates [R1(C=NR2)PO(OR3)2; R1 = Ar, CCl3 or CF3, R2 = SO2Ph or H, R3 = Et or Ph] [31-34]. In 2009, Prishchenko et al. reported an example of synthesis of diethyl 1-dimethylamino-1-(dipropylphosphinoyl)methylphosphonate (Figure 2, 4: R = Pr, R1 = H, R3 = Et) by condensation of [1-ethoxy-1-(dimethylamino)methyl]dipropylphosphine oxide [(Pr)2POCH(NMe2)OEt] with diethyl phosphite in 74% yield [35].
In the present paper we report a novel method for α-phosphonylation, α-phosphinylation or α-phosphinoylation of 1-(N-acetylamino)alkylphosphonates that enables effective transformation of the starting compounds into 1-aminoalkylidenebisphosphonates 1, 1-amino-1-phosphinylalkylphosphonates 3, or 1-amino-1-phosphinoylalkylphosphonates 4, respectively. The two latter groups of compounds are not available by common methods used for the synthesis of 1-aminobisphosphonic acid derivatives.
Results and Discussion
Recently we developed a simple and effective two-step transformation of N-acyl-α-amino acids into their phosphonic analogues that allows for easy access to a variety of structurally diversified N-acyl-α-aminophosphonates 5 (Scheme 1) [36,37].
![[1860-5397-11-153-i1]](/bjoc/content/inline/1860-5397-11-153-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Electrochemical α-methoxylation of 1-(N-acylamino)alkylphosphonates.
Scheme 1: Electrochemical α-methoxylation of 1-(N-acylamino)alkylphosphonates.
Despite the electron-withdrawing inductive effect of N-acylamino- and dialkoxyphosphonyl groups of 1-aminophosphonates 5, the electrophilicity of these compounds’ α-carbon is displayed only after functionalisation of the α-position with a nucleofuge group, e.g. by α-bromination of 1-aminophosphonates with N-bromosuccinimide [28,38-41] or by alkoxylation or aryloxylation of this position [20,29,30].
As we demonstrated, electrophilic activation of the α-carbon of 1-aminophosphonates can easily be achieved by electrochemical α-methoxylation of these compounds in methanol, mediated with NaCl (Scheme 1, Table 2). α-Methoxylations were carried out in the presence of sodium methoxide or 3-(1-piperidino)propyl-functionalised silica gel (SiO2-Pip) or without any base. The composition of the reaction mixture was controlled using 1H NMR to determine the optimal electric charge consumption. The best result of α-methoxylation of diethyl N-acetylaminomethylphosphonate (5a) was achieved without using any base, whereas the best result of the same reaction of the analogous ethylphosphonic acid derivative 5b was obtained in the presence of sodium methoxide. Attempts to carry out the analogous α-methoxylation for phosphonic derivatives of valine and phenylalanine failed, probably due to the steric hindrance exerted by a bulky substituent at the α-position.
Table 2: Electrochemical α-methoxylation of diethyl 1-(N-acetylamino)alkylphosphonates 5 (R = Me).
| substrate 5 | procedure | base | charge [F/mol] | product 6 | ||
|---|---|---|---|---|---|---|
| no. | R1 | no. | yielda [%] | |||
| 5a | H | A | – | 4.5 | 6a | 88 |
| 5a | H | B | MeONa | 4.5 | 6a | 41 |
| 5b | Me | A | – | 10 | 6b | 66 |
| 5b | Me | B | SiO2-Pip | 10 | 6b | 63 |
| 5b | Me | B | MeONa | 10 | 6b | 72 |
aThe yield was estimated based on the 1H NMR spectrum of the reaction mixture relative to dimethyldiphenylsilane as the internal standard.
Electrochemical α-alkoxylation of N-acyl-α-amino acid esters mediated by NaCl, NaBr, LiCl, KCl or KI is a well-known reaction [38,42-46], whereas the analogous reaction of N-acyl-α-aminophosphonic acid esters was mentioned in the literature only once, in relation to α-methoxylation of diethyl N-benzoylaminomethylphosphonate [42].
Attempts to carry out a Michaelis–Arbuzov-like reaction of the obtained diethyl 1-(N-acetylamino)-1-methoxyalkylphosphonates with triethyl phosphite failed due to still too low electrophilic activity of the α-carbon atom of the removed 1-methoxyalkylphosphonic acid derivatives. However, the electrophilic activity of the latter compounds was enough for their reaction with triphenylphosphine. Therefore, to enhance the electrophilic activity of the discussed 1-methoxyalkylphosphonic acid derivatives, their methoxy group was successfully displaced by the triphenylphosphonium group. Thus, heating the homogeneous mixture of diethyl 1-(N-acetylamino)-1-methoxymethylphosphonate (6a) with triphenylphosphonium tetrafluoroborate at 50 °C gave hitherto unknown phosphonium salt 7a as a resin-like compound stable at room temperature (Scheme 2). In the case of 1-(N-acetylamino)-1-methoxyethylphosphonate (6b), the analogous reaction with triphenylphosphonium tetrafluoroborate proceeded very quickly, without heating, but the resulting phosphonium salt 7b underwent slow decomposition even at low temperature (Scheme 2). The most characteristic feature of the 13C NMR spectra of these compounds was a doublet of doublets in the range of 45–58 ppm (J1 = 42–49 Hz, J2 = 151–160 Hz) assigned to the α-carbon atom coupled with two nonequivalent phosphorus atoms as well as the presence of two doublets of phosphorus atoms in the ranges of 11.7–16.5 and 28.5–37.0 ppm (J = 33–48 Hz) in the 31P NMR spectra.
![[1860-5397-11-153-i2]](/bjoc/content/inline/1860-5397-11-153-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Transformation of diethyl 1-(N-acetylamino)-1-methoxyalkylphosphonates into bisphosphoric acid esters via the corresponding phosphonium salts.
Scheme 2: Transformation of diethyl 1-(N-acetylamino)-1-methoxyalkylphosphonates into bisphosphoric acid este...
Finally, it was demonstrated that diethyl 1-(N-acetylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborates 7 react smoothly with trialkyl phosphites, dialkyl phosphonites or alkyl phosphinites in the presence of Hünig’s base and methyltriphenylphosphonium iodide as catalysts to give bisphosphonates 8a,b, 1-phosphinylalkylphosphonates 8c–e or 1-phosphinoylalkylphosphonates 8f,g, respectively, in good yields (Table 3). The nature of the catalytic activity of Hünig’s base and methyltriphenylphosphonium iodide in similar Michaelis–Arbuzov-like reactions of 1-(N-acylamino)alkylphosphonium salts was explained in our previous paper [47].
Table 3: Reaction of diethyl 1-(N-acetylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborates 7 with phosphorus nucleophiles.
| substrate | nucleophile | temp. [°C] | time [h] | product | |||||
|---|---|---|---|---|---|---|---|---|---|
| no. | R1 | R2 | R3 | R4 | no. | yield [%] | |||
| 7a | H | EtO | EtO | Et | 60 | 5 | 8a | 75 | |
| 7b | Me | EtO | EtO | Et | 20 | 2.5 | 8b | 70 | |
| 7a | H | EtO | Me | Et | 20 | 22 | 8ca | 50b | |
| 7a | H | EtO | Ph | Et | 60 | 6 | 8d | 51c | |
| 7b | Me | EtO | Ph | Et | 20 | 3 | 8e | 53c | |
| 7a | H | Ph | Ph | Me | 60 | 2 | 8f | 68 | |
| 7b | Me | Ph | Ph | Me | 20 | 2 | 8g | 76 | |
aSynthesis was performed under argon due to the sensitivity of the nucleophile to oxidation. bMixture of diastereomers in a ratio of 1:1. cMixture of diastereomers in a ratio of 1.4:1.
Conclusion
It was demonstrated that effective electrophilic activation of the α-position of 1-(N-acetylamino)alkylphosphonates can be achieved by electrochemical α-methoxylation of these compounds in methanol, mediated with NaCl, followed by displacement of the methoxy group with triphenylphosphonium tetrafluoroborate to give hitherto unknown 1-(N-acetylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborates. The Michaelis–Arbuzov-like reaction of the latter compounds with trialkyl phosphites, dialkyl phosphonites or alkyl phosphinites in the presence of Hünig’s base and methyltriphenylphosphonium iodide gave the expected alkylidenebisphosphonates, 1-phosphinylalkylphosphonates or 1-phosphinoylalkylphosphonates, respectively, in good yields. The reported set of reactions enables α-phosphonylation, α-phosphinylation or α-phosphinoylation of 1-(N-acylamino)alkylphosphonates.
Experimental
General methods: IR spectra were measured on a FTIR spectrophotometer (ATR method). 1H and 13C NMR spectra were recorded at operating frequencies of 400 or 600 MHz and 100 MHz, respectively, using TMS as the resonance shift standard. 31P NMR spectra were recorded at an operating frequency of 162 MHz, with 80% orthophosphoric acid as an external resonance shift standard. All chemical shifts (δ) are reported in ppm, and coupling constants (J) are in Hz. Spectroscopic properties of all synthesized compounds as well as 1H NMR, 13C NMR and 31P NMR spectra of selected bisphosphoric acid esters are given in Supporting Information File 1.
Electrochemical α-methoxylation of diethyl 1-(N-acetylamino)alkylphosphonates 5. Procedure A: MeOH (10 mL) and diethyl 1-(N-acetylamino)alkylphosphonate 5 (0.3 mmol) were added to a glass beaker (25 mL) equipped with a cylindrical Pt mesh anode (20 cm2) and a rotating flat cathode (platinised titanium, 1.2 cm2). In the case of 1-(N-acetylamino)methylphosphonate (5a), NaCl (70 mg, 1.2 mmol) was placed in the beaker in its entirety at the beginning of oxidation, as opposed to 1-(N-acetylamino)ethylphosphonate (5b), where the mediator was added in three portions during the process (0.6 mmol at the start and 2 × 0.3 mmol – each portion after the charge of 3.0 F/mol had been passed). The beaker was placed in an ice-water bath and its content was stirred for 5 min by means of a rotating cathode before commencement of the oxidation. Electrolysis was carried out while stirring at 0.1 A until a 4.5–10 F/mol charge had passed (Table 2). After evaporation of the methanol in vacuo, the residue was extracted with CH2Cl2. Subsequent evaporation of the solvent from the extracts under reduced pressure yielded 1-(N-acetylamino)-1-methoxyalkylphosphonates 6, which were used in the next step without further purification.
Procedure B: MeOH (10 mL), diethyl 1-(N-acetylamino)alkylphosphonate 5 (0.3 mmol), and NaCl (70 mg, 1.2 mmol), in one portion for 1-(N-acetylamino)methylphosphonate (5a) or in three portions for derivative 5b (as described above for Procedure A), and SiO2-Pip (150 mg, 0.15 mmol) or MeONa (0.6 mL methanolic solution, 0.3 mmol), were added to a glass beaker (25 mL) equipped with a cylindrical Pt mesh anode (20 cm2) and a rotating flat cathode (platinised titanium, 1.2 cm2). Electrolysis was carried out while stirring at 0.1 A until a 4.5–10 F/mol charge had passed (Table 2). After evaporation of MeOH under reduced pressure, the residue was extracted with CH2Cl2 followed by further evaporation of the solvent from the extracts in vacuo to obtain diethyl 1-(N-acetylamino)-1-methoxyalkylphosphonates 6.
Synthesis of 1-(N-acetylamino)-1-triphenylphosphoniummethylphosphonate tetrafluoroborate 7a. Triphenylphosphonium tetrafluoroborate (52.5 mg, 0.15 mmol) was added to a solution of diethyl 1-(N-acetylamino)-1-methoxymethylphosphonate (6a, 36 mg, 0.15 mmol) in CH2Cl2 (1 mL). After obtaining a homogeneous solution, the solvent was evaporated to dryness and the residue was heated in a flask equipped with a septum with a needle for 20 h at 50 °C to obtain compound 7a, which was used in the subsequent synthesis without further purification.
Synthesis of 1-(N-acetylamino)-1-triphenylphosphoniumethylphosphonate tetrafluoroborate 7b. Triphenylphosphonium tetrafluoroborate (52.5 mg, 0.15 mmol) was added to a solution of diethyl 1-(N-acetylamino)-1-methoxyethylphosphonate (6b, 38 mg, 0.15 mmol) in dichloromethane (1 mL). The homogeneous mixture was stirred at room temperature for 0.1 h, and then the solvent was evaporated to dryness to give product 7b, which was used in the subsequent synthesis without further purification.
Synthesis of bisphosphonates 8a,b, 1-phosphinylalkylphosphonates 8c–e and 1-phosphinoylalkylphosphonates 8f,g. General procedure: Methyltriphenylphosphonium iodide (36 mg, 0.088 mmol), phosphorus nucleophile (0.52 mmol) and (iPr)2EtN (6.0 μL, 0.035 mmol) were added to a solution of 1-(N-acetylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborate 7 (0.35 mmol) in CH2Cl2 (1 mL) in a glass vial with a screw-cap. For syntheses started from 1-(N-acetylamino)-1-triphenylphosphoniummethylphosphonate tetrafluoroborate (7a), the mixture was heated at 60 °C for the time shown in Table 3. For the syntheses using 1-(N-acetylamino)-1-triphenylphosphoniumethylphosphonate tetrafluoroborate (7b), the mixture was left at room temperature for 2–3 h (Table 3). The solvent was evaporated under reduced pressure, the residue was extracted with toluene, and the toluene was subsequently evaporated. The crude products were purified by column chromatography (silica gel, CH2Cl2/MeOH for 8a–f, and AcOEt/MeOH for 8g).
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Kafarski, P.; Lejczak, B. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 301–312. doi:10.2174/1568011013354543
Return to citation in text: [1] [2] [3] [4] -
Orsini, F.; Sello, G.; Sisti, M. Curr. Med. Chem. 2010, 17, 264–289. doi:10.2174/092986710790149729
Return to citation in text: [1] -
Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, iv, 127–166. doi:10.3998/ark.5550190.0013.411
Return to citation in text: [1] [2] [3] [4] -
Chmielewska, E. The new bisphosphonates as potential drugs for osteoporosis. Ph.D. Thesis, Wrocław University of Technology, Poland, 2010.
Return to citation in text: [1] [2] -
Worms, K.-H.; Blum, H. Z. Anorg. Allg. Chem. 1979, 457, 209–213. doi:10.1002/zaac.19794570125
Return to citation in text: [1] -
Blum, H. Z. Naturforsch., B: J. Chem. Sci. 1988, 43, 75–81. doi:10.1515/znb-1988-0114
Return to citation in text: [1] -
Kamel, A. A.; Khidre, M. D. ARKIVOC 2008, xvii, 273–286. doi:10.3998/ark.5550190.0009.h26
Return to citation in text: [1] [2] -
Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmidt, J.; Schreiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i
Return to citation in text: [1] [2] -
Lejczak, B.; Kafarski, P. Biological activity of aminophosphonic acids and their short peptides. In Phosphorous Heterocycles I; Bansal, R. K., Ed.; Topics in Heterocyclic Chemistry, Vol. 20; Springer: Berlin, 2009; pp 31–63. doi:10.1007/7081_2008_14
Return to citation in text: [1] -
Kaboudin, B.; Alipour, S. Tetrahedron Lett. 2009, 50, 4243–4245. doi:10.1016/j.tetlet.2009.05.016
Return to citation in text: [1] -
Kubiček, V.; Rudovský, J.; Kotek, J.; Hermann, P.; Vander Elst, L.; Muller, R. N.; Kolar, Z. I.; Wolterbeek, H. T.; Peters, J. A.; Lukeš, I. J. Am. Chem. Soc. 2005, 127, 16477–16485. doi:10.1021/ja054905u
Return to citation in text: [1] -
Jiang, Q.; Yang, L.; Hai, L.; Wu, Y. Lett. Org. Chem. 2008, 5, 229–233. doi:10.2174/157017808783955862
Return to citation in text: [1] -
Balakrishna, A.; Reddy, M. V. N.; Rao, P. V.; Kumar, M. A.; Kumar, B. S.; Nayak, S. K.; Reddy, C. S. Eur. J. Med. Chem. 2011, 46, 1798–1802. doi:10.1016/j.ejmech.2011.02.038
Return to citation in text: [1] -
Lin, Y.-S.; Park, J.; De Schutter, J. W.; Huang, X. F.; Berghuis, A. M.; Sebag, M.; Tsantrizos, Y. S. J. Med. Chem. 2012, 55, 3201–3215. doi:10.1021/jm201657x
Return to citation in text: [1] -
Wu, M.; Chen, R.; Huang, Y. Synth. Commun. 2004, 34, 1393–1398. doi:10.1081/SCC-120030688
Return to citation in text: [1] -
McNulty, J.; Das, P. Tetrahedron 2009, 65, 7794–7800. doi:10.1016/j.tet.2009.07.032
Return to citation in text: [1] -
Tanaka, K. S. E.; Dietrich, E.; Ciblat, S.; Métayer, C.; Arhin, F. F.; Sarmiento, I.; Moeck, G.; Parr, T. R., Jr.; Far, A. R. Bioorg. Med. Chem. Lett. 2010, 20, 1355–1359. doi:10.1016/j.bmcl.2010.01.006
Return to citation in text: [1] -
Lecerclé, D.; Gabillet, S.; Gomis, J.-M.; Taran, F. Tetrahedron Lett. 2008, 49, 2083–2087. doi:10.1016/j.tetlet.2008.01.127
Return to citation in text: [1] -
Ebetino, F. H.; Jamieson, L. A. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 51, 23–26. doi:10.1080/10426509008040673
Return to citation in text: [1] -
Ebetino, F. H.; Berk, J. D. J. Organomet. Chem. 1997, 529, 135–142. doi:10.1016/S0022-328X(96)06846-5
Return to citation in text: [1] [2] [3] [4] -
Ebetino, F. H. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 144, 9–12. doi:10.1080/10426509908546169
Return to citation in text: [1] -
Ebetino, F. H. Methylene phosphonoalkylphosphinates, pharmaceutical compositions, and methods for treating abnormal calcium and phosphate metabolism. Eur. Patent EP0298553 A1, Jan 11, 1989.
Return to citation in text: [1] -
Ebetino, F. H. Process for synthesis of aminomethylene phosphonoalkylphosphinates. Eur. Patent EP0298555 A1, Jan 11, 1989.
Return to citation in text: [1] -
Ebetino, F. H.; Dansereau, S. M.; Francis, M. D.; Nelson, D. G. A.; Janusz, J. M. Quaternary nitrogen-containing phosphonate compounds for treating abnormal calcium and phosphate metabolism as well as dental calculus and plaque. Eur. Patent EP0646119 B1, July 15, 1998.
Return to citation in text: [1] -
Ebetino, F. H.; Kaas, S. M.; Francis, M. D.; Nelson, D. G. A.; Janusz, J. M. Quaternary nitrogen-containing phosphonate compounds, pharmaceutical compositions, and methods of treating abnormal calcium and phosphate metabolism and methods of treating and preventing dental calculus and plaque. U.S. Patent US5,770,586, June 23, 1998.
Return to citation in text: [1] -
Francis, M. D.; Kaas, S. M.; Ebetino, F. H. Sulfur-containing phosphonate compounds, pharmaceutical compositions, and methods of treating abnormal calcium and phosphate metabolism. U.S. Patent US5,824,661, Oct 20, 1998.
Return to citation in text: [1] -
Kaas, S. M.; Ebetino, F. H.; Francis, M. D. Sulfur-containing phosphonate compounds for treating abnormal calcium and phosphate metabolism. Eur. Patent EP0642519 B1, Aug 9, 2000.
Return to citation in text: [1] -
Schrader, T.; Steglich, W. Synthesis 1989, 97–101. doi:10.1055/s-1989-27162
Return to citation in text: [1] [2] -
Kostka, K.; Kotyński, A. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 47, 261–265. doi:10.1080/10426509008037977
Return to citation in text: [1] [2] -
Kostka, K.; Kotyński, A. Phosphorus, Sulfur Silicon Relat. Elem. 1993, 83, 215–221. doi:10.1080/10426509308034365
Return to citation in text: [1] [2] -
Rassukana, Yu. V.; Sinitsa, A. A.; Onys’ko, P. P. Russ. Chem. Bull. 2005, 54, 2652–2655. doi:10.1007/s11172-006-0171-9
Return to citation in text: [1] -
Kolotilo, N. V.; Sinitsa, A. A.; Rassukanaya, Yu. V.; Onys’ko, P. P. Russ. J. Gen. Chem. 2006, 76, 1210–1218. doi:10.1134/S1070363206080068
Return to citation in text: [1] -
Rassukana, Yu. V.; Onys’ ko, P. P.; Davydova, K. O.; Sinitsa, A. D. Tetrahedron Lett. 2004, 45, 3899–3902. doi:10.1016/j.tetlet.2004.03.114
Return to citation in text: [1] -
Rassukana, Yu. V.; Kolotylo, M. V.; Sinitsa, O. A.; Pirozhenko, V. V.; Onys’ko, P. P. Synthesis 2007, 2627–2630. doi:10.1055/s-2007-983838
Return to citation in text: [1] -
Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Petrosyan, V. S. Heteroat. Chem. 2009, 20, 319–324. doi:10.1002/hc.20552
Return to citation in text: [1] -
Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. J. Org. Chem. 2012, 77, 1952–1960. doi:10.1021/jo202534u
Return to citation in text: [1] -
Mazurkiewicz, R.; Październiok-Holewa, A.; Kononienko, A. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 1986–1992. doi:10.1080/10426500903436735
Return to citation in text: [1] -
Schrader, T.; Kober, R.; Steglich, W. Synthesis 1986, 372–375. doi:10.1055/s-1986-31638
Return to citation in text: [1] [2] -
Schrader, T.; Steglich, W. Synthesis 1990, 1153–1156. doi:10.1055/s-1990-27122
Return to citation in text: [1] -
Elachqar, A.; El Hallaouiq, A.; Roumestant, M. L.; Viallefont, P. Synth. Commun. 1994, 24, 1279–1286. doi:10.1080/00397919408011729
Return to citation in text: [1] -
Boukallaba, K.; Elachqar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Labriti, B.; Atmani, A.; El Bali, B.; Lachkar, M.; Allouchi, H.; Martinez, J.; Roland, V. Phosphorus, Sulfur Silicon Relat. Elem. 2007, 182, 1045–1052. doi:10.1080/10426500601093739
Return to citation in text: [1] -
Ginzel, K.-D.; Brungs, P.; Steckhan, E. Tetrahedron 1989, 45, 1691–1701. doi:10.1016/S0040-4020(01)80034-X
Return to citation in text: [1] [2] -
Shono, T.; Matsumura, Y.; Inoue, K. J. Org. Chem. 1983, 48, 1388–1389. doi:10.1021/jo00156a060
Return to citation in text: [1] -
Papadopoulos, A.; Lewall, B.; Steckhan, E.; Ginzel, K.-D.; Knoch, F.; Nieger, M. Tetrahedron 1991, 47, 563–572. doi:10.1016/S0040-4020(01)87046-0
Return to citation in text: [1] -
Papadopoulos, A.; Heyer, J.; Ginzel, K.-D.; Steckhan, E. Chem. Ber. 1989, 122, 2159–2164. doi:10.1002/cber.19891221117
Return to citation in text: [1] -
Shono, T. Tetrahedron 1984, 40, 811–850. doi:10.1016/S0040-4020(01)91472-3
Return to citation in text: [1] -
Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 967–980. doi:10.1080/10426507.2012.729237
Return to citation in text: [1]
| 20. | Ebetino, F. H.; Berk, J. D. J. Organomet. Chem. 1997, 529, 135–142. doi:10.1016/S0022-328X(96)06846-5 |
| 29. | Kostka, K.; Kotyński, A. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 47, 261–265. doi:10.1080/10426509008037977 |
| 30. | Kostka, K.; Kotyński, A. Phosphorus, Sulfur Silicon Relat. Elem. 1993, 83, 215–221. doi:10.1080/10426509308034365 |
| 36. | Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. J. Org. Chem. 2012, 77, 1952–1960. doi:10.1021/jo202534u |
| 37. | Mazurkiewicz, R.; Październiok-Holewa, A.; Kononienko, A. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 1986–1992. doi:10.1080/10426500903436735 |
| 28. | Schrader, T.; Steglich, W. Synthesis 1989, 97–101. doi:10.1055/s-1989-27162 |
| 38. | Schrader, T.; Kober, R.; Steglich, W. Synthesis 1986, 372–375. doi:10.1055/s-1986-31638 |
| 39. | Schrader, T.; Steglich, W. Synthesis 1990, 1153–1156. doi:10.1055/s-1990-27122 |
| 40. | Elachqar, A.; El Hallaouiq, A.; Roumestant, M. L.; Viallefont, P. Synth. Commun. 1994, 24, 1279–1286. doi:10.1080/00397919408011729 |
| 41. | Boukallaba, K.; Elachqar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Labriti, B.; Atmani, A.; El Bali, B.; Lachkar, M.; Allouchi, H.; Martinez, J.; Roland, V. Phosphorus, Sulfur Silicon Relat. Elem. 2007, 182, 1045–1052. doi:10.1080/10426500601093739 |
| 1. | Kafarski, P.; Lejczak, B. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 301–312. doi:10.2174/1568011013354543 |
| 2. | Orsini, F.; Sello, G.; Sisti, M. Curr. Med. Chem. 2010, 17, 264–289. doi:10.2174/092986710790149729 |
| 3. | Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, iv, 127–166. doi:10.3998/ark.5550190.0013.411 |
| 1. | Kafarski, P.; Lejczak, B. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 301–312. doi:10.2174/1568011013354543 |
| 3. | Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, iv, 127–166. doi:10.3998/ark.5550190.0013.411 |
| 4. | Chmielewska, E. The new bisphosphonates as potential drugs for osteoporosis. Ph.D. Thesis, Wrocław University of Technology, Poland, 2010. |
| 7. | Kamel, A. A.; Khidre, M. D. ARKIVOC 2008, xvii, 273–286. doi:10.3998/ark.5550190.0009.h26 |
| 8. | Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmidt, J.; Schreiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i |
| 9. | Lejczak, B.; Kafarski, P. Biological activity of aminophosphonic acids and their short peptides. In Phosphorous Heterocycles I; Bansal, R. K., Ed.; Topics in Heterocyclic Chemistry, Vol. 20; Springer: Berlin, 2009; pp 31–63. doi:10.1007/7081_2008_14 |
| 31. | Rassukana, Yu. V.; Sinitsa, A. A.; Onys’ko, P. P. Russ. Chem. Bull. 2005, 54, 2652–2655. doi:10.1007/s11172-006-0171-9 |
| 32. | Kolotilo, N. V.; Sinitsa, A. A.; Rassukanaya, Yu. V.; Onys’ko, P. P. Russ. J. Gen. Chem. 2006, 76, 1210–1218. doi:10.1134/S1070363206080068 |
| 33. | Rassukana, Yu. V.; Onys’ ko, P. P.; Davydova, K. O.; Sinitsa, A. D. Tetrahedron Lett. 2004, 45, 3899–3902. doi:10.1016/j.tetlet.2004.03.114 |
| 34. | Rassukana, Yu. V.; Kolotylo, M. V.; Sinitsa, O. A.; Pirozhenko, V. V.; Onys’ko, P. P. Synthesis 2007, 2627–2630. doi:10.1055/s-2007-983838 |
| 5. | Worms, K.-H.; Blum, H. Z. Anorg. Allg. Chem. 1979, 457, 209–213. doi:10.1002/zaac.19794570125 |
| 6. | Blum, H. Z. Naturforsch., B: J. Chem. Sci. 1988, 43, 75–81. doi:10.1515/znb-1988-0114 |
| 35. | Prishchenko, A. A.; Livantsov, M. V.; Novikova, O. P.; Livantsova, L. I.; Petrosyan, V. S. Heteroat. Chem. 2009, 20, 319–324. doi:10.1002/hc.20552 |
| 1. | Kafarski, P.; Lejczak, B. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 301–312. doi:10.2174/1568011013354543 |
| 4. | Chmielewska, E. The new bisphosphonates as potential drugs for osteoporosis. Ph.D. Thesis, Wrocław University of Technology, Poland, 2010. |
| 29. | Kostka, K.; Kotyński, A. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 47, 261–265. doi:10.1080/10426509008037977 |
| 1. | Kafarski, P.; Lejczak, B. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 301–312. doi:10.2174/1568011013354543 |
| 3. | Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, iv, 127–166. doi:10.3998/ark.5550190.0013.411 |
| 30. | Kostka, K.; Kotyński, A. Phosphorus, Sulfur Silicon Relat. Elem. 1993, 83, 215–221. doi:10.1080/10426509308034365 |
| 22. | Ebetino, F. H. Methylene phosphonoalkylphosphinates, pharmaceutical compositions, and methods for treating abnormal calcium and phosphate metabolism. Eur. Patent EP0298553 A1, Jan 11, 1989. |
| 23. | Ebetino, F. H. Process for synthesis of aminomethylene phosphonoalkylphosphinates. Eur. Patent EP0298555 A1, Jan 11, 1989. |
| 24. | Ebetino, F. H.; Dansereau, S. M.; Francis, M. D.; Nelson, D. G. A.; Janusz, J. M. Quaternary nitrogen-containing phosphonate compounds for treating abnormal calcium and phosphate metabolism as well as dental calculus and plaque. Eur. Patent EP0646119 B1, July 15, 1998. |
| 25. | Ebetino, F. H.; Kaas, S. M.; Francis, M. D.; Nelson, D. G. A.; Janusz, J. M. Quaternary nitrogen-containing phosphonate compounds, pharmaceutical compositions, and methods of treating abnormal calcium and phosphate metabolism and methods of treating and preventing dental calculus and plaque. U.S. Patent US5,770,586, June 23, 1998. |
| 26. | Francis, M. D.; Kaas, S. M.; Ebetino, F. H. Sulfur-containing phosphonate compounds, pharmaceutical compositions, and methods of treating abnormal calcium and phosphate metabolism. U.S. Patent US5,824,661, Oct 20, 1998. |
| 27. | Kaas, S. M.; Ebetino, F. H.; Francis, M. D. Sulfur-containing phosphonate compounds for treating abnormal calcium and phosphate metabolism. Eur. Patent EP0642519 B1, Aug 9, 2000. |
| 20. | Ebetino, F. H.; Berk, J. D. J. Organomet. Chem. 1997, 529, 135–142. doi:10.1016/S0022-328X(96)06846-5 |
| 47. | Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 967–980. doi:10.1080/10426507.2012.729237 |
| 19. | Ebetino, F. H.; Jamieson, L. A. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 51, 23–26. doi:10.1080/10426509008040673 |
| 20. | Ebetino, F. H.; Berk, J. D. J. Organomet. Chem. 1997, 529, 135–142. doi:10.1016/S0022-328X(96)06846-5 |
| 21. | Ebetino, F. H. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 144, 9–12. doi:10.1080/10426509908546169 |
| 10. | Kaboudin, B.; Alipour, S. Tetrahedron Lett. 2009, 50, 4243–4245. doi:10.1016/j.tetlet.2009.05.016 |
| 11. | Kubiček, V.; Rudovský, J.; Kotek, J.; Hermann, P.; Vander Elst, L.; Muller, R. N.; Kolar, Z. I.; Wolterbeek, H. T.; Peters, J. A.; Lukeš, I. J. Am. Chem. Soc. 2005, 127, 16477–16485. doi:10.1021/ja054905u |
| 12. | Jiang, Q.; Yang, L.; Hai, L.; Wu, Y. Lett. Org. Chem. 2008, 5, 229–233. doi:10.2174/157017808783955862 |
| 13. | Balakrishna, A.; Reddy, M. V. N.; Rao, P. V.; Kumar, M. A.; Kumar, B. S.; Nayak, S. K.; Reddy, C. S. Eur. J. Med. Chem. 2011, 46, 1798–1802. doi:10.1016/j.ejmech.2011.02.038 |
| 14. | Lin, Y.-S.; Park, J.; De Schutter, J. W.; Huang, X. F.; Berghuis, A. M.; Sebag, M.; Tsantrizos, Y. S. J. Med. Chem. 2012, 55, 3201–3215. doi:10.1021/jm201657x |
| 15. | Wu, M.; Chen, R.; Huang, Y. Synth. Commun. 2004, 34, 1393–1398. doi:10.1081/SCC-120030688 |
| 16. | McNulty, J.; Das, P. Tetrahedron 2009, 65, 7794–7800. doi:10.1016/j.tet.2009.07.032 |
| 17. | Tanaka, K. S. E.; Dietrich, E.; Ciblat, S.; Métayer, C.; Arhin, F. F.; Sarmiento, I.; Moeck, G.; Parr, T. R., Jr.; Far, A. R. Bioorg. Med. Chem. Lett. 2010, 20, 1355–1359. doi:10.1016/j.bmcl.2010.01.006 |
| 18. | Lecerclé, D.; Gabillet, S.; Gomis, J.-M.; Taran, F. Tetrahedron Lett. 2008, 49, 2083–2087. doi:10.1016/j.tetlet.2008.01.127 |
| 38. | Schrader, T.; Kober, R.; Steglich, W. Synthesis 1986, 372–375. doi:10.1055/s-1986-31638 |
| 42. | Ginzel, K.-D.; Brungs, P.; Steckhan, E. Tetrahedron 1989, 45, 1691–1701. doi:10.1016/S0040-4020(01)80034-X |
| 43. | Shono, T.; Matsumura, Y.; Inoue, K. J. Org. Chem. 1983, 48, 1388–1389. doi:10.1021/jo00156a060 |
| 44. | Papadopoulos, A.; Lewall, B.; Steckhan, E.; Ginzel, K.-D.; Knoch, F.; Nieger, M. Tetrahedron 1991, 47, 563–572. doi:10.1016/S0040-4020(01)87046-0 |
| 45. | Papadopoulos, A.; Heyer, J.; Ginzel, K.-D.; Steckhan, E. Chem. Ber. 1989, 122, 2159–2164. doi:10.1002/cber.19891221117 |
| 46. | Shono, T. Tetrahedron 1984, 40, 811–850. doi:10.1016/S0040-4020(01)91472-3 |
| 3. | Romanenko, V. D.; Kukhar, V. P. ARKIVOC 2012, iv, 127–166. doi:10.3998/ark.5550190.0013.411 |
| 7. | Kamel, A. A.; Khidre, M. D. ARKIVOC 2008, xvii, 273–286. doi:10.3998/ark.5550190.0009.h26 |
| 8. | Widler, L.; Jaeggi, K. A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; Klein, R.; Ramseier, U.; Schmidt, J.; Schreiber, G.; Seltenmeyer, Y.; Green, J. R. J. Med. Chem. 2002, 45, 3721–3738. doi:10.1021/jm020819i |
| 20. | Ebetino, F. H.; Berk, J. D. J. Organomet. Chem. 1997, 529, 135–142. doi:10.1016/S0022-328X(96)06846-5 |
| 42. | Ginzel, K.-D.; Brungs, P.; Steckhan, E. Tetrahedron 1989, 45, 1691–1701. doi:10.1016/S0040-4020(01)80034-X |
© 2015 Kuźnik et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)