Abstract
Phosphonodepsipeptides are phosphorus analogues of depsipeptides and phosphonate-linked analogues of naturally occurring peptides. They are more stable than phosphonopeptides and have been widely applied as enzyme inhibitors, haptens for the production of antibodies, biological agents, and prodrugs. The synthetic strategies towards phosphonodepsipeptides are reviewed, including the phosphonylation of hydroxy esters with phosphonochloridates, the condensation of phosphonic monoesters and hydroxy esters, the alkylation of phosphonic monoesters with 1-(alkoxycarbonyl)alkyl halides or sulfonates, multicomponent condensation of amides, aldehydes, and dichlorophosphites followed by alcoholysis with hydroxy esters, the phosphinylation of hydroxy esters with phosphonochloridites followed by oxidation, and the carbene insertion of N-protected amino acids with 1-diazoalkylphosphonates. This review includes the synthesis of α-, β-, and γ-phosphonodepsipeptides and phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acids.

Graphical Abstract
Introduction
Both, phosphonopeptides and phosphonodepsipeptides are phosphorus analogues of peptides [1-5]. The phosphonopeptides are peptides with a phosphonamidate bond instead of an amide bond whereas the phosphonodepsipeptides are peptides with a phosphonate linkage instead of an amide. Phosphonodepsipeptides are structurally close analogues of depsipeptides (Figure 1). In general, phosphonodepsipeptides are more stable than the corresponding phosphonopeptides because the phosphonate bond is more inert than a phopshonamidate bond. Phosphonodepsipeptides are widely used as enzyme inhibitors [6-10], haptens for inducing catalytic antibodies [11,12], and produgs [8,9,13]. They have potential applications as antibiotics [14], antimicrobials [15], antimalarials [16], antitumor agents [17], and medicinal agents [18]. Thus, much attention has been paid to the synthesis of phosphonodepsipeptides.
![[1860-5397-17-41-1]](/bjoc/content/figures/1860-5397-17-41-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Phosphonopeptides, phosphonodepsipeptides, peptides, and depsipeptides.
Figure 1: Phosphonopeptides, phosphonodepsipeptides, peptides, and depsipeptides.
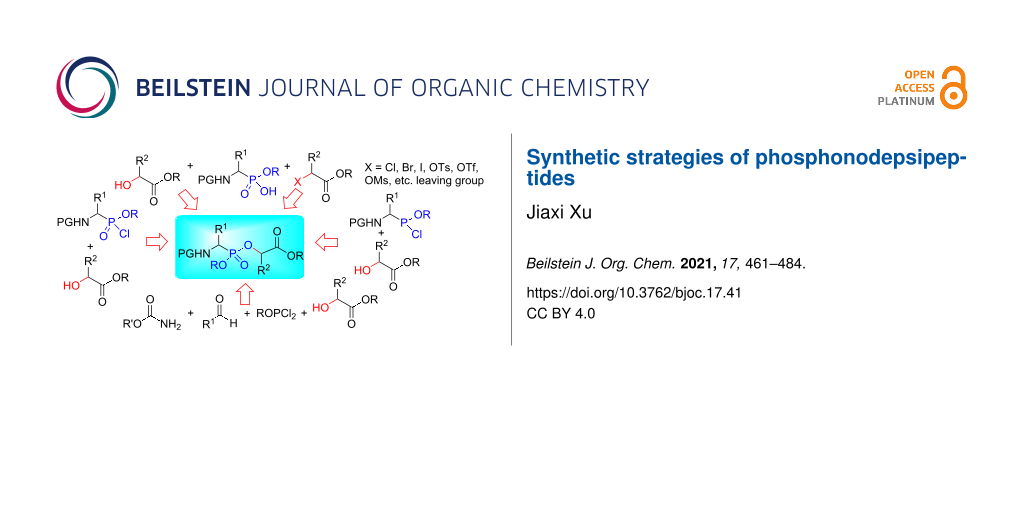
To date, diverse synthetic strategies of phosphonodepsipeptides 1 have been developed. The strategies comprise the phosphonylation of hydroxy esters 2 with N-protected aminoalkylphosphonochloridates 3 (method I), reactions of N-protected aminoalkylphosphonic monoesters 4 with hydroxy esters 2 (method II) or with 1-(alkoxycarbonyl)alkyl halides or sulfonates 5 (method III), pseudo-four-component condensations (method IV), and the phosphinylation of hydroxy esters 2 with N-protected aminoalkylphosphonochloridites 9 followed by oxidation (method V) (Figure 2). This review focuses on the synthetic methods of phosphonodepsipeptides and phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acids, excluding peptides with side-chain phosphonic acids.
![[1860-5397-17-41-2]](/bjoc/content/figures/1860-5397-17-41-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The diverse strategies for phosphonodepsipeptide synthesis.
Figure 2: The diverse strategies for phosphonodepsipeptide synthesis.
Review
Synthesis of phosphonodepsipeptides via the phosphonylation of hydroxy esters with phosphonochloridates
The phosphonylation of hydroxy esters 2 with alkyl N-protected aminoalkylphosphonochloridates 3 is a general and widely applied method for the synthesis of α-, β-, and γ-phosphonodepsipeptides 1. The N-protected aminoalkylphosphonic monomethyl/ethyl esters are very useful materials for the preparation of N-protected aminoalkylphosphonochloridates 3. N-Protected aminoalkylphosphonic diaryl esters should be converted into the corresponding monomethyl/ethyl esters via transesterification and selective hydrolysis.
Synthesis of α-phosphonodepsipeptides
In 1987, a series of phosphonodepsidipeptides 10 was synthesized as phosphorus analogues of peptides and evaluated as inhibitors of leucine aminopeptidase from porcine kidney and two compounds, i.e., 10e and 10h (R = isobutyl, benzyl) were modest inhibitors. The synthesis started from diphenyl N-Cbz-1-aminoalkylphosphonates 11 (Scheme 1). They were transformed to dimethyl esters via transesterification and further to monomethyl esters 12 via basic hydrolysis. After chlorination with thionyl chloride, the monomethyl esters 12 were converted into N-Cbz-1-aminoalkylphosphonochloridates 13, which were further coupled with methyl (S)-2-hydroxy-4-methylpentanoate (14), affording the protected phosphonodepsidipeptides 15. Finally, after deprotection the free phosphonodepsidipeptides 10 were obtained [19].
![[1860-5397-17-41-i1]](/bjoc/content/inline/1860-5397-17-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of α-phosphonodepsidipeptides as inhibitors of leucine aminopeptidase.
Scheme 1: Synthesis of α-phosphonodepsidipeptides as inhibitors of leucine aminopeptidase.
The disodium salts of the trans and cis isomers of 2-hydroxy-2-oxo-3-[(phenoxyacetyl)amino]-1,2-oxaphosphorinane-6-carboxylic acid (16, Figure 3) were prepared and evaluated as inhibitors of the zinc-containing β-lactamase II from B. cereus. However, neither stereoisomer had any significant activity [6].
![[1860-5397-17-41-3]](/bjoc/content/figures/1860-5397-17-41-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Structure of 2-hydroxy-2-oxo-3-[(phenoxyacetyl)amino]-1,2-oxaphosphorinane-6-carboxylic acid (16).
Figure 3: Structure of 2-hydroxy-2-oxo-3-[(phenoxyacetyl)amino]-1,2-oxaphosphorinane-6-carboxylic acid (16).
The phosphonodepsidipeptide 17 was synthesized via the coupling of methyl N-Cbz-1-aminoethylphosphonochloridate (13b) generated from phosphonic monomethyl ester 12b and methyl (S)-2-hydroxy-3-phenylpropanoate (18) followed by hydrogenolysis. It was further coupled to cyclen to afford various cyclen-containing phosphonodepsipeptides as inhibitors of carboxypeptidase A (Scheme 2) [20].
![[1860-5397-17-41-i2]](/bjoc/content/inline/1860-5397-17-41-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of α-phosphonodepsidipeptide 17 as coupling partner for cyclen-containing phosphonodepsipeptides as inhibitors of carboxypeptidase A.
Scheme 2: Synthesis of α-phosphonodepsidipeptide 17 as coupling partner for cyclen-containing phosphonodepsip...
VanX is a Zn(II)-dependent ᴅ-Ala-ᴅ-Ala dipeptidase. To prepare novel inhibitors of VanX, N-[(1-aminoethyl)hydroxyphosphinyl]-ᴅ-lactate (20a), and {S-[(aminoethyl)hydroxyphosphinyl]thio}acetic acid (20b) were synthesized via coupling of the methyl N-Cbz-protected 1-aminoethylphosphonochloridate 13b with methyl ᴅ-lactate (22a) and methyl mercaptoacetate (22b), respectively, followed by a basic hydrolysis and hydrogenolysis. The bioassay results indicated that the phosphonothiodepsidipeptide 20b did not inhibit VanX (Scheme 3) [21].
![[1860-5397-17-41-i3]](/bjoc/content/inline/1860-5397-17-41-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of α-phosphonodepsidipeptides containing enantiopure hydroxy ester as VanX inhibitors.
Scheme 3: Synthesis of α-phosphonodepsidipeptides containing enantiopure hydroxy ester as VanX inhibitors.
It was discovered that the enzymatic activity of VanX was inhibited competitively by phosphonodepsidipeptide 2-{[(1-aminoethyl)(hydroxy)phosphoryl]oxy}propanoic acid (25b). Seven phosphonodepsidipeptides 25 as analogues of ᴅ-Ala-ᴅ-Ala with various substituents were prepared through the reaction of methyl N-Cbz 1-aminoethylphosphonochloridate (13b) with different benzyl 1-hydroxyalkanoates 26 followed by hydrogenolysis and hydrolysis. The bioassay results indicated that six out of the seven synthetic phosphonodepsipeptides 25 inhibited VanX with IC50 values ranging from 0.48 to 8.21 mM (Scheme 4) [7].
![[1860-5397-17-41-i4]](/bjoc/content/inline/1860-5397-17-41-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of α-phosphonodepsidipeptides as VanX inhibitors.
Scheme 4: Synthesis of α-phosphonodepsidipeptides as VanX inhibitors.
Two optically active phosphonodepsidipeptides 28 were prepared and investigated as VanX inhibitors. The racemic N-Cbz 1-aminoethylphosphonic acid (29) was separated by chemical resolution with quinine, affording the (S)-N-Cbz-1-aminoethylphosphonic acid ((S)-29), which was transformed to the corresponding phosphonochloridate and further reacted with benzyl (R)-lactate ((R)-26b) or benzyl (R)-2-phenyllactate ((R)-26f). The optically active phosphonodepsidipeptides 28 were obtained after hydrogenolysis and tested for their biological activity (Scheme 5) [22].
![[1860-5397-17-41-i5]](/bjoc/content/inline/1860-5397-17-41-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of optically active α-phosphonodepsidipeptides as VanX inhibitors.
Scheme 5: Synthesis of optically active α-phosphonodepsidipeptides as VanX inhibitors.
The phosphonodepsidipeptides 31 were synthesized by the esterification of N-Cbz-aminoalkylphosphonic acids 32 and 4-nitrobenyl 2-hydroxyalkanoates 33 in the presence of SOCl2 in DMF. This was an efficient coupling reaction for the synthesis of phosphonodepsipeptides from N-protected phosphonic acids and hydroxy esters. The phosphonodepsidipeptide 31 (R1 = R2 = H, Scheme 6) was converted to the free phosphonodepsidipeptide 34 in 80% yield and the N-Cbz-phosphonodepsidipeptide 35 in 85% yield, respectively, via hydrogenolysis and basic hydrolysis, respectively (Scheme 6) [23]. The method actually is a convenient and direct method to synthesize phosphonodepsipeptides from N-protected phosphonic acids and hydroxy esters through phosphonochloridates as in situ-generated intermediates.
![[1860-5397-17-41-i6]](/bjoc/content/inline/1860-5397-17-41-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: The synthesis of phosphonodepsipeptides through a thionyl chloride-catalyzed esterification of N-Cbz aminoalkylphosphonic acids 32 and 4-nitrobenyl 2-hydroxyalkanoates 33.
Scheme 6: The synthesis of phosphonodepsipeptides through a thionyl chloride-catalyzed esterification of N-Cb...
To prepare an antigen to induce monoclonal catalytic antibodies capable of catalyzing peptide-bond formation reactions, the phosphonodepsidipeptide 39 was synthesized via the coupling of the hydroxy analog of tryptophan amide with 4-nitrobenzyl (R)-N-Fmoc 1-amino(cyclohexyl)methylphosphonochloridate (38), which was prepared from diethyl (R)-N-Fmoc 1-amino(cyclohexyl)methylphosphonate (36) via a selective basic hydrolysis, chlorination, esterification with 4-nitrobenzyl alcohol, selective basic hydrolysis, and chlorination. After the treatment of compound 39 with piperidine, the N-terminal free dipeptide was obtained and acylated with hexanedioic anhydride to afford the designed hapten 40 (Scheme 7) [11].
![[1860-5397-17-41-i7]](/bjoc/content/inline/1860-5397-17-41-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of α-phosphinodipeptidamide as a hapten.
Scheme 7: Synthesis of α-phosphinodipeptidamide as a hapten.
Phosphonodepsioctapeptide 41 was prepared as a variation of the partial sequence of a gene product of erb B-2. Two different hydroxypeptide esters 46 were first prepared and successfully coupled with methyl N-Cbz-1-aminoethylphosphonochloridate ((R)-13b) by use of their magnesium salts because the direct coupling of the hydroxypeptide esters 46 with the phosphonochloridate (R)-13b failed. The magnesium salts were in situ generated from the hydroxypeptide esters 46 with a Grignard reagent (Scheme 8) [24].
![[1860-5397-17-41-i8]](/bjoc/content/inline/1860-5397-17-41-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of α-phosphonodepsioctapeptide 41.
Scheme 8: Synthesis of α-phosphonodepsioctapeptide 41.
To minimize the side reactions of 1-aminoalkylphosphonochloridates, a convenient method for the synthesis of phosphonodepsipeptides was described. The N-Cbz-protected 2-aminoalkylphosphinates 50 were converted to their trimethylsilyl phosphonites 52 through the treatment with bis(trimethylsilyl)acetamide (51). The phosphonites 52 were then oxidized with CCl4 to generate the corresponding phosphonochloridates 53 as intermediates, which reacted with hydroxy esters 54 and 55 to give rise to the desired phosphonodepsipeptides 56 and 57, respectively. The oxidative activation was carried out in the presence of alcohols as nucleophiles so that stoichiometric formation of the phosphonochloridate was avoided and side reactions were minimized (Scheme 9) [25]. The current chlorination is a mild and neutral method for the preparation of phosphonochloridates.
![[1860-5397-17-41-i9]](/bjoc/content/inline/1860-5397-17-41-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of phosphonodepsipeptides via an in situ-generated phosphonochloridate.
Scheme 9: Synthesis of phosphonodepsipeptides via an in situ-generated phosphonochloridate.
Eleven phosphonodepsitetrapeptides 58 were synthesized and evaluated as inhibitors of the aspartic peptidase pepsin. The synthesis started from methyl (R)-N-Cbz-1-amino-2-phenylethylphosphinate (59). In wet acetonitrile compound 59 was oxidized into the corresponding phosphonic monomethyl ester (R)-12h with carbon tetrachloride due to hydrolysis of the phophonochloridate intermediate. The phosphonic monomethyl ester (R)-12h was further chlorinated to the phosphonochloridate (R)-13h, which was coupled with 3-(pyridin-4-yl)propyl (S)-2-hydroxy-3-phenylpropanoate (60) to give rise to the phosphonodepsidipeptide 61. The phosphonodepsidipeptide 61 was also obtained directly by the oxidation of the phosphinate 59 with carbon tetrachloride in the presence of the hydroxy ester 60. After hydrogenolysis of 61 and coupling with various N-protected dipeptides 62, the N-protected phosphonodepsitetrapeptides 63 were obtained and further transformed to the N-protected phosphonodepsipeptide ester lithium salts 58 after aminolysis with tertiary butylamine and treatment with Dowex-Li+ (Scheme 10) [26].
![[1860-5397-17-41-i10]](/bjoc/content/inline/1860-5397-17-41-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of α-phosphonodepsitetrapeptides 58 as inhibitors of the aspartic peptidase pepsin.
Scheme 10: Synthesis of α-phosphonodepsitetrapeptides 58 as inhibitors of the aspartic peptidase pepsin.
Synthesis of β-phosphonodepsipeptides
To develop iminocyclitol-based small molecule libraries against a bacterial TGase, an iminocyclitol was conjugated with a pyrophosphate mimic. After in situ screening, the first potent iminocyclitol-based inhibitor against bacterial TGases was efficiently developed [27].
The synthesis of N-pyrrolidine-derived β-phosphonodepsipeptides 64 is shown in Scheme 11. First, dibenzyl allylphosphonate (65) was converted to benzyl allylphosphonochloridate (66), which was then coupled with benzyl 2-azido-3-hydroxy-2-methylpropanoate (67) producing benzyl [allyl(benzyloxy)phosphoryl)oxy]propanoate (68). After the dihydroxylation with osmium tetroxide and oxidation with sodium periodate, benzyl 2-azido-3-(((benzyloxy)(2-oxoethyl)phosphoryl)oxy)-2-methylpropanoate (69) was obtained. The latter was further transformed to the final phosphonodepsipeptide library 64 after the reductive amination with pyrrolidine derivatives 70 and acylation with a library of carboxylic acids 72 in the presence of coupling reagents (Scheme 11) [27].
![[1860-5397-17-41-i11]](/bjoc/content/inline/1860-5397-17-41-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthesis of a β-phosphonodepsidipeptide library 64.
Scheme 11: Synthesis of a β-phosphonodepsidipeptide library 64.
Alternatively, the 1,3-protected glycerol 73 was first converted into various 2,3-protected glycerols 74, which were further transformed to methyl 2-alkoxy-3-hydroxypropanoates 75. Following a similar strategy as above, another library of phosphonodepsipeptides 78 was prepared (Scheme 12) [27].
![[1860-5397-17-41-i12]](/bjoc/content/inline/1860-5397-17-41-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of another β-phosphonodepsidipeptide library.
Scheme 12: Synthesis of another β-phosphonodepsidipeptide library.
Synthesis of γ-phosphonodepsipeptides
γ-Phosphonodepsipeptides 79 have been prepared from N-Cbz-ʟ-glutamic acid (80) and diethyl 2-hydroxyglutarate (84). To prepare phosphorus analogues of γ-glutamyl peptide, the starting N-Cbz-ʟ-glutamic acid (80) was first transformed to the corresponding dimethyl phosphonate 81. After aminolysis and chlorination the corresponding phosphonochloridate 83 was obtained. The latter was further reacted with diethyl 2-hydroxyglutarate (84), affording γ-phosphonodepsidipeptide 79 in only 6.7% yield, indicating that this strategy was not suitable for the synthesis of γ-phosphonodepsipeptides (Scheme 13) [28].
![[1860-5397-17-41-i13]](/bjoc/content/inline/1860-5397-17-41-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthesis of γ-phosphonodepsidipeptides.
Scheme 13: Synthesis of γ-phosphonodepsidipeptides.
Folylpolyglutamate synthetase catalyzes an ATP-dependent ligation reaction. The reaction results in the synthesis of poly(γ-glutamate) metabolites of folates and some antifolates. Three γ-phosphonodepsidipeptide derivatives 85 were designed as prototypes and mechanism-based folylpolyglutamate synthetase inhibitors. The synthesis started with dimethyl N-phthaloyl-protected γ-aminophosphonate 86 that was selectively hydrolyzed with thiophenol, affording the corresponding phosphonic monomethyl ester 87. The ester 87 was then coupled with dibenzyl (S)-2-hydroxypentanedioate (88) using BOP as the activating agent to generate the γ-phosphonodepsidipeptide 89. After hydrazinolysis, acylation with arenecarbonyl chlorides or arenecarboxylic acids, and further modification, the γ-phosphonodepsidipeptide derivatives 85 were obtained. The phosphonate moiety in these analogues represented an important new lead in the development of folylpolyglutamate synthetase inhibitors (Scheme 14) [29].
![[1860-5397-17-41-i14]](/bjoc/content/inline/1860-5397-17-41-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of phosphonodepsipeptides 85 as folylpolyglutamate synthetase inhibitors.
Scheme 14: Synthesis of phosphonodepsipeptides 85 as folylpolyglutamate synthetase inhibitors.
γ-Phosphonodepsipeptides were also designed and synthesized as potent inhibitors and active site probes of γ-glutamyl transpeptidase, which catalyzes the transfer of the γ-glutamyl group of glutathione and related γ-glutamyl amides to amino acids and peptides (transpeptidation) or to water (hydrolysis). For this purpose, N-Cbz-aminophosphonic acid 91 was first transformed to the corresponding dichloride 92, which underwent a sequential alcoholysis with phenol and benzyl (4-hydroxybutanoyl)glycinate (93), respectively, to give the protected phosphonodepsitripeptide 94. After hydrogenolysis, the free γ-phosphonodepsitripeptide 95 was obtained (Scheme 15) [30].
![[1860-5397-17-41-i15]](/bjoc/content/inline/1860-5397-17-41-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of the γ-phosphonodepsitripeptide 95 as an inhibitor of γ-gutamyl transpeptidase.
Scheme 15: Synthesis of the γ-phosphonodepsitripeptide 95 as an inhibitor of γ-gutamyl transpeptidase.
Seven years later, various enantiopure 2-hydroxyalkanoic acids 96 were prepared from optically pure amino acids and converted to the benzyl or methyl [2-hydroxyalkanoyl]glycinates 97. Following the similar strategy, phosphonodepsitripeptides 99 and 102 were synthesized. The phosphonodepsidipeptide 104 was prepared through the sequential coupling of N-Cbz-γ-aminophosphonodichloride 92 with phenol and methyl (S)-2-hydroxypentanoate (18). All synthetic phosphonodepsipeptides 99, 102, and 104 were considered as glutathione-analogue phosphonopeptides as mechanism-based inhibitors of γ-glutamyl transpeptidase for probing the cysteinyl-glycine binding site (Scheme 16) [31].
![[1860-5397-17-41-i16]](/bjoc/content/inline/1860-5397-17-41-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Synthesis of phosphonodepsipeptides as inhibitors and probes of γ-glutamyl transpeptidase.
Scheme 16: Synthesis of phosphonodepsipeptides as inhibitors and probes of γ-glutamyl transpeptidase.
Synthesis of phosphonodepsipeptides via the condensation of phosphonic monoesters and hydroxy esters
The condensation of N-protected aminoalkylphosphonic monoesters and hydroxy esters is an alternative general strategy that has been widely used for the synthesis of phosphonodepsipeptides with various coupling reagents including the Mitsunobu reagent.
Synthesis of α-phosphonodepsipeptides
N,N’-Dicyclohexylcarbodiimide (DCC) was the first attempted coupling reagent in the synthesis of phosphonyl depsipeptides 108 from hydrogen 4-phenylbutylphosphinic acid (105) and 2-hydroxypropanoic acid derivatives 106 followed by oxidation. The corresponding phosphinyl depsipeptides 107 were generated and further oxidized into phosphonyl depsipeptides 108 (Scheme 17) [32].
![[1860-5397-17-41-i17]](/bjoc/content/inline/1860-5397-17-41-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Synthesis of phosphonyl depsipeptides 108 via DCC-mediated condensation and oxidation.
Scheme 17: Synthesis of phosphonyl depsipeptides 108 via DCC-mediated condensation and oxidation.
The protected phosphonodepsidipeptides 111 were prepared without racemization from N-Cbz-protected α-aminoalkylphosphonic monomethyl (12h) or benzyl esters 109 and hydroxy esters 110 by using (1H-benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) or (1H-benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) as activating agents as well (Scheme 18) [33].
![[1860-5397-17-41-i18]](/bjoc/content/inline/1860-5397-17-41-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of phosphonodepsipeptides 111 with BOP and PyBOP as coupling reagents.
Scheme 18: Synthesis of phosphonodepsipeptides 111 with BOP and PyBOP as coupling reagents.
The synthetic method was further investigated and compared with alternative coupling reagents. Various optically active phosphonodepsidipeptides 113–118 were synthesized. The reaction mechanism was further studied, revealing that the reaction proceeds through benzotriazolyl esters as was shown by the comparison with other coupling reagents, including DCC, DCC/DMAP, DCC/1-hydroxybenzotriazole (HOBt), bromotris(dimethylamino)phosphonium hexafluorophosphate (BroP), or O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate (HBTU) and by 31P NMR analysis. The results indicated that the intermediates, benzotriazolyl phosphonates, were more reactive toward alcohols (hydroxy esters) than toward amines (amino esters), contrary to their carboxylic partners (Scheme 19) [34].
![[1860-5397-17-41-i19]](/bjoc/content/inline/1860-5397-17-41-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of optically active phosphonodepsipeptides with BOP and PyBOP as coupling reagents.
Scheme 19: Synthesis of optically active phosphonodepsipeptides with BOP and PyBOP as coupling reagents.
The coupling reagents BroP and N,N,N',N'-bis(tetramethylene)chlorouronium tetrafluoroborate (TPyCIU) were applied as activating agents in the synthesis of phosphonodepsidipeptides 121 from N-Cbz-1-amino-2-phenylethylphosphonic acid (119) and hydroxy esters 120 in the presence of diisopropylethylamine (DIPEA) in CH2Cl2 (Scheme 20) [35].
![[1860-5397-17-41-i20]](/bjoc/content/inline/1860-5397-17-41-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Synthesis of phosphonodepsipeptides with BroP and TPyCIU as coupling reagents.
Scheme 20: Synthesis of phosphonodepsipeptides with BroP and TPyCIU as coupling reagents.
Vancomycin resistance became a serious health care problem currently. The development of a catalytic monoclonal antibody to hydrolyze the depsidipeptide ᴅ-Ala-ᴅ-Lac was a new antibiotic strategy. In this regard, the phosphonodepsipeptide hapten 127 was designed to induce the antibody. First, N-Boc-(S)-1-aminoethylphosphinic acid (122) was coupled with benzyl alcohol in the presence of DCC as the coupling reagent, affording the benzyl N-Boc-1-aminoethylphosphinate 123. Oxidation with sodium periodate and neutralization with adamantan-1-amine (124), afforded the adamantan-1-ammonium benzyl phosphonate 125. After treated with hydrochloride and coupled with benzyl ᴅ-lactate ((R)-26b) with BOP as activating reagent, the ammonium compound 125 was transformed to the protected phosphonodepsidipeptide 126 (Scheme 21) [12], that was further converted to the designed hapten 127.
![[1860-5397-17-41-i21]](/bjoc/content/inline/1860-5397-17-41-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of a phosphonodepsipeptide hapten with BOP as coupling reagent.
Scheme 21: Synthesis of a phosphonodepsipeptide hapten with BOP as coupling reagent.
The new muramyl dipeptide (MDP) phosphorus analogue 131 related to LK 423 as potential immunomodulator was prepared by the coupling of methyl 1-(N-benzyloxycarbonyl)aminoethylphosphonate (12b) and (R)-2-hydroxyglutaric acid dimethyl ester (128) with BOP as coupling reagent, followed by hydrogenolysis and coupling with 2-(2-phthalimidoethoxy)acetic acid (130). The amide bond between ʟ-Ala and ᴅ-Glu was replaced by a phosphonate isostere, giving the phosphonodepsipeptide 131 (Scheme 22) [36].
![[1860-5397-17-41-i22]](/bjoc/content/inline/1860-5397-17-41-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of phosphonodepsitripeptide with BOP as coupling reagent.
Scheme 22: Synthesis of phosphonodepsitripeptide with BOP as coupling reagent.
The synthesis of the norleucine-derived phosphonodepsipeptides 135 and 138 was realized by a BOP-activated coupling of N-Cbz-1-aminopentylphosphonic monobenzyl ester (132), a phosphorus analogue of norleucine, with derivatives 133 and 136 of (S)-lactic or glycolic acids followed by hydrogenolysis (Scheme 23) [37].
![[1860-5397-17-41-i23]](/bjoc/content/inline/1860-5397-17-41-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Synthesis of norleucine-derived phosphonodepsipeptides 135 and 138.
Scheme 23: Synthesis of norleucine-derived phosphonodepsipeptides 135 and 138.
Following a similar strategy, phosphonodepsidipeptides 141 and phosphonodepsitripeptides 144 were synthesized as norleucine-derived phosphonopeptides (Scheme 24) [38].
![[1860-5397-17-41-i24]](/bjoc/content/inline/1860-5397-17-41-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of norleucine-derived phosphonodepsipeptides 141 and 144.
Scheme 24: Synthesis of norleucine-derived phosphonodepsipeptides 141 and 144.
The protected phosphonodepsipeptide 145 was applied in a solid-phase phosphonodepsipeptide synthesis. After hydrogenolysis and reaction with Fmoc-OSu, the N-protected phosphonodepsipeptide 145 was transformed to the N-Fmoc-protected phosphonodepsidipeptide 147, which was then coupled with the resin-loaded Cys(Trt)-NH-resin 148. The resin-loaded tripeptide 149 was deprotected with piperidine, coupled with Fmoc-Tyr(Ot-Bu) 150, deprotected again with piperidine, and cleaved with TFA and trapping agents. The free phosphonodepsipeptide 151 was obtained in 45% yield after HPLC purification (Scheme 25) [38].
![[1860-5397-17-41-i25]](/bjoc/content/inline/1860-5397-17-41-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Solid-phase synthesis of phosphonodepsipeptides.
Scheme 25: Solid-phase synthesis of phosphonodepsipeptides.
A general and high yielding synthesis of phosphonodepsidipeptides 152 was realized via a Mitsunobu reaction of the N-Cbz-1-aminophosphonic monomethyl esters 12b,d and hydroxy esters 106 followed by the selective demethylation with TMSBr in a one-pot reaction. The method provides a mild route to prepare phosphonodepsipeptides. The yields were insensitive to the steric encumbrance of both reactants being coupled (Scheme 26) [39].
![[1860-5397-17-41-i26]](/bjoc/content/inline/1860-5397-17-41-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Synthesis of phosphonodepsidipeptides via the Mitsunobu reaction.
Scheme 26: Synthesis of phosphonodepsidipeptides via the Mitsunobu reaction.
Synthesis of γ-phosphonodepsipeptides
To prepare phosphorus analogues of γ-glutamyl peptide, N-Cbz-ʟ-glutamic acid (80) was initially converted to the corresponding dimethyl phosphonate 81 and further to the methyl phosphonic monoester 82. The ester 82 was then coupled with diethyl 2-hydroxyglutarate (84) in a Mitsunobu reaction to generate the γ-phosphonodepsipeptide 79 in a high yield of 66% (Scheme 27) [28].
![[1860-5397-17-41-i27]](/bjoc/content/inline/1860-5397-17-41-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of γ-phosphonodepsipeptide via the Mitsunobu reaction.
Scheme 27: Synthesis of γ-phosphonodepsipeptide via the Mitsunobu reaction.
Synthesis of phosphonodepsipeptides via the multicomponent condensation of amides, aldehydes, and phosphites followed by alcoholysis with hydroxy esters
Previously, the Mannich-type reaction of benzyl carbamate, aldehydes, and trialkyl phosphites in acetyl chloride gave rise to N-Cbz-1-aminoalkylphosphonates [40]. When the reactions were conducted in benzene followed by an aminolysis or alcoholysis, phosphonamidates [41], phosphonopeptide [42], and mixed esters [43,44] were obtained directly. The multicomponent condensation reaction was applied as a direct synthetic method for phosphonodepsipeptides via the formation of 1-aminoalkylphosphonic acids and simultaneous construction of the phosphonate bond. A series of phosphonodepsipeptides 158 was prepared in good yields in a one-pot reaction directly from the simple and commercially available chemicals, benzyl carbamate (154), aldehydes 155, and methyl dichlorophosphite (156), followed by the alcoholysis with the hydroxy esters 157. The current strategy is a highly efficient and convergent synthesis of phosphonodepsipeptides that does not require the preparation of 1-aminoalkylphosphonic acid or 1-aminoalkylphosphonous acid derivatives first as starting materials (Scheme 28) [42].
![[1860-5397-17-41-i28]](/bjoc/content/inline/1860-5397-17-41-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Synthesis of phosphonodepsipeptides via a multicomponent condensation reaction.
Scheme 28: Synthesis of phosphonodepsipeptides via a multicomponent condensation reaction.
Similarly, phosphinopeptides [45,46], phosphinodepsipeptides [47], and hybrid sulfonophosphinopeptides [48,49] were prepared from amino amides and 2-aminoalkanesulfonamides by using this strategy.
Also side-chain functionalized phosphonodepsipeptides 160 were prepared in satisfactory yields directly through the one-pot reactions of benzyl carbamate (154), aldehydes 155, and diethyl (R,R)-2-chloro-1,3,2-dioxaphospholane-4,5-dicarboxylate (159), which was synthesized from diethyl ʟ-tartrate and phosphorus trichloride. A pair of diastereomeric products 160 was obtained in diasteromeric ratios of 1.7:1.0 to 2.5:1.0. The configuration of the major diastereomeric product was determined by hydrolysis of the product 160a and comparison of the obtained acid (R)-161a with the corresponding reported authentic sample (Scheme 29) [50].
![[1860-5397-17-41-i29]](/bjoc/content/inline/1860-5397-17-41-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Synthesis of phosphonodepsipeptides with a functionalized side-chain via a multicomponent condensation.
Scheme 29: Synthesis of phosphonodepsipeptides with a functionalized side-chain via a multicomponent condensat...
A straightforward method for the synthesis of phosphonodepsipeptides 163 was developed via the multicomponent condensation reaction of the simple starting materials, benzyl carbamate (154), aldehydes 155, and 1-ethoxycarbonylalkyl phosphorodichloridites 162, which were prepared from ethyl 2-hydroxyalkanoates 157 and phosphorus trichloride. Compared with the previous methods, the current strategy provides a more efficient, convenient, convergent, and practical synthetic route to phosphonodepsipeptides 163 under mild reaction conditions with good yields. Good diastereoselectivities were observed with diastereomeric ratios of 84:12 to 88:12 (Scheme 30) [51]. However, when using substrates with arylmethyl groups, the phosphorodichloridites favored an elimination reaction generating the corresponding ethyl cinnamate derivatives during their preparation.
![[1860-5397-17-41-i30]](/bjoc/content/inline/1860-5397-17-41-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: High yielding synthesis of phosphonodepsipeptides via a multicomponent condensation.
Scheme 30: High yielding synthesis of phosphonodepsipeptides via a multicomponent condensation.
To prepare optically active phosphonodepsipeptides, ethyl (R)-2-((dichlorophosphanyl)oxy)-2-phenylacetate ((R)-162c) was first prepared through the reaction of ethyl (R)-2-hydroxy-2-phenylacetate and phosphorus trichloride and further reacted with benzyl carbamate (154) and benzaldehyde (155a), affording a pair of optically active phosphonodepsipeptides 164 and 165 in an 86% yield and 85:15 diastereomeric ratio (Scheme 31) [51].
![[1860-5397-17-41-i31]](/bjoc/content/inline/1860-5397-17-41-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Synthesis of optically active phosphonodepsipeptides via a multicomponent condensation reaction.
Scheme 31: Synthesis of optically active phosphonodepsipeptides via a multicomponent condensation reaction.
Following a similar strategy, the three component condensation of diethyl phosphoramidate (166), aromatic aldehydes 167, and diisopropyl (4R,5R)-2-chloro-1,3,2-dioxaphospholane-4,5-dicarboxylate (168) gave the corresponding phosphonodepsipeptides 169 in 65–86% yields under mild conditions. Although the diisopropyl ester was applied instead of diethyl ʟ-tartrate, a low diastereoselectivity (ratios varied from 55:45 to 67:33) was observed as well. Acetophenone (170) produced the desired product 171 in 62% yield in the reaction. However, aliphatic aldehydes did not work (Scheme 32) [52].
![[1860-5397-17-41-i32]](/bjoc/content/inline/1860-5397-17-41-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Synthesis of N-phosphoryl phosphonodepsipeptides.
Scheme 32: Synthesis of N-phosphoryl phosphonodepsipeptides.
Synthesis of phosphonodepsipeptides via the alkylation of phosphonic monoesters with 1-(alkoxycarbonyl)alkyl halides or sulfonates
The alkylation of N-protected 1-aminoalkylphosphonic monoesters with 1-(alkoxycarbonyl)alkyl halides or sulfonates is also a general method for the synthesis of phosphonodepsipeptides. However, the strategy has not been utilized widely.
Synthesis of α-phosphonodepsipeptides
Skwarczynski and Kafarski synthesized various alkyl 1-aminoalkylphosphonates via the nucleophilic esterification of potassium 1-(N-benzyloxycarbonylamino)alkylphosphonates 172 with alkyl halides in the presence of 18-crown-6. They also prepared a phosphonodepsidipeptide 174 with ethyl chloroacetate (173) as an electrophile (Scheme 33) [53].
![[1860-5397-17-41-i33]](/bjoc/content/inline/1860-5397-17-41-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Synthesis of phosphonodepsipeptides via the alkylation of phosphonic monoesters.
Scheme 33: Synthesis of phosphonodepsipeptides via the alkylation of phosphonic monoesters.
The macrocyclic peptidyl phosphonodepsipeptide 180 was designed on the basis of the acyclic conformational analog bound to the aspartic protease penicillopepsin. Dimethyl N-Cbz-1-amino-2-(naphthalen-2-yl)ethylphosphonate 176 was first prepared from 7-bromo-3,4-dihydronaphthalen-1(2H)-one (175) and further transformed to the macrocyclic peptidyl phosphonic monomethyl ester 177. The latter compound was alkylated with methyl 3-phenyl-2-trifluoromethanesulfonyloxypropanoate (178) to produce the macrocyclic peptidyl phosphonodepsipeptide 179, which was selectively hydrolyzed with TMSBr and treated with Dowex-Na+ to afford the macrocyclic peptidyl phosphonodepsipeptide sodium salt 180. By using a similar method, two acyclic analogues 183 and 186 were synthesized as well. The macrocyclic phosphonodepsipeptide 180 and the two acyclic analogues 183 and 186 were evaluated for their potential as inhibitors. The NMR analysis results indicated that the conformation of the macrocyclic phosphonodepsipeptide backbone closely approximated that of the lead inhibitor and showed the low-energy conformation accommodated in the active site of penicillopepsin without significant distortion (Scheme 34) [54].
![[1860-5397-17-41-i34]](/bjoc/content/inline/1860-5397-17-41-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Synthesis of phosphonodepsipeptides as inhibitors of aspartic protease penicillopepsin.
Scheme 34: Synthesis of phosphonodepsipeptides as inhibitors of aspartic protease penicillopepsin.
Synthesis of γ-phosphonodepsipeptides
The acyloxyalkyl esters 194 are derivatives of the new antimalarial drug fosmidomycin and inhibited the 1-deoxy-ᴅ-xylulose 5-phosphate reductoisomerase. The phosphonodepsipeptides 194 were synthesized as prodrugs with an increased activity after oral administration due to a chemical modification of the phosphonate moiety. For the synthesis, diethyl 3,3-diethoxypropylphosphonate (187) was hydrolyzed to 3-oxopropylphosphonate 188, which underwent a reductive amination with benzyloxyamine to give diethyl 3-benzyloxyaminopropylphosphonate (189). After the sequential treatment with acetyl chloride and TMSBr, alkylation with methyl or tert-butyl chloroacetate 192, and hydrogenolysis, the target phosphonodepsipeptides 194 were obtained (Scheme 35) [8,9,13].
![[1860-5397-17-41-i35]](/bjoc/content/inline/1860-5397-17-41-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Synthesis of phosphonodepsipeptides as prodrugs.
Scheme 35: Synthesis of phosphonodepsipeptides as prodrugs.
Synthesis of phosphonodepsipeptides via phosphinylation of hydroxy esters with phosphonochloridites followed by oxidation
Hammer’s group developed a new route to prepare phosphonodepsithioxopeptides 198 via the reaction of N-protected aminoalkylphosphonochloridites 196 with the hydroxy ester (S)-106b followed by sulfur oxidation. They first transformed the N-Boc-protected 1-aminoalkylphosphinate 195 to the corresponding phosphonochloridite 196 with dichlorotriphenylphosphorane. The phosphonochloridite 196 was then further reacted with methyl (S)-lactate ((S)-106b) followed by sulfurization with sulfur, affording the phosphonodepsithioxopeptide 198 in a one-pot activation–coupling–oxidation procedure (Scheme 36) [55]. Although it was mentioned that the phosphonochloridites were a more active species than the corresponding phosphonochloridates in the esterification, this synthetic strategy had not been applied by others. There is only one example reported till now possibly due to the inconvenient preparation of the phosphonochloridites.
![[1860-5397-17-41-i36]](/bjoc/content/inline/1860-5397-17-41-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Synthesis of phosphonodepsithioxopeptides 198.
Scheme 36: Synthesis of phosphonodepsithioxopeptides 198.
Synthesis of phosphonodepsipeptides via the addition of tetraoxyspirophosphoranes to imines
The addition reaction of the P–H bond of tetraoxyspirophosphoranes 199 to long-chain imines 200 of benzaldehyde, acetaldehyde, and dodecanal at room temperature generated the corresponding (α-aminoalkyl)spirophosphoranes 201 via the Pudovik reaction. The one-pot selective hydrolysis of the P–C bond of the spirophosphoranes 201 readily proceeded at room temperature in the presence of moist solvents to give the corresponding phosphonodepsipeptides 202 in high yields (Scheme 37) [56].
![[1860-5397-17-41-i37]](/bjoc/content/inline/1860-5397-17-41-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Synthesis of phosphonodepsipeptides.
Scheme 37: Synthesis of phosphonodepsipeptides.
An asymmetric synthesis of this class of phosphonodepsipeptides was realized with enantiopure (S)-α-hydroxyisovaleric acid-derived spirophosphoranes as the phosphorus reagents [57]. By using a similar strategy, linker-linked bisphosphonodepsidipeptides were synthesized [10]. The current method is also an interesting synthetic strategy of phosphonodepsipeptides. It can realize an asymmetric synthesis of phosphonodepsipeptides.
Synthesis of phosphonodepsipeptides with C-hydroxyalkylphosphonic acids
Phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acids can be considered as a class of important phosphorous analogues of depsipeptides. The coupling of N-protected amino acids and 1-hydroxyalkylphosphonates is a general method to prepare these compounds [58].
(R)-1-Amino-3-methylbutylphosphonic acid ((R)-204), a phosphonic ʟ-Leu analogue, is a potent inhibitor of the metalloenzyme leucine aminopeptidase. Racemic dibenzyl 1-hydroxy-3-methylbutylphosphonate (203), an oxyanalog of phosphonic leucine, was partially debenzylated by the treatment with NaI to generate the corresponding monobenzyl ester 204. The ester 204 was resolved with (−)-ephedrine and then O-benzylated with O-benzyl-N,N'-dicyclohexylurea (205) to give the (R)-1-hydroxy-3-methylbutylphosphonate ((R)-203). The latter was coupled with N-Boc-protected amino acids 206 to give the corresponding protected phosphonopeptides 207. After deprotection by hydrogenolysis and treatment with CF3CO2H, the phosphonodepsipeptides 208 were obtained (Scheme 38) [59].
![[1860-5397-17-41-i38]](/bjoc/content/inline/1860-5397-17-41-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Synthesis of phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acid.
Scheme 38: Synthesis of phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acid.
Phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acids are also accessible through carbene insertion reactions. The reaction of the N-protected amino acids 209 and 210 with diethyl 1-diazo-2,2,2-trifluoroethylphosphonate (211) gave rise to the trifluoromethyl-containing phosphonodepsipeptides 212 and 213 with C-1-hydroxyalkylphosphonic acids in good yields under the catalysis of dirhodium tetraacetate (Scheme 39) [60,61].
![[1860-5397-17-41-i39]](/bjoc/content/inline/1860-5397-17-41-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Synthesis of phosphonodepsipeptides with C-1-hydroxyalkylphosphonate via the rhodium-catalyzed carbene insertion.
Scheme 39: Synthesis of phosphonodepsipeptides with C-1-hydroxyalkylphosphonate via the rhodium-catalyzed carb...
To develop novel bone-targeting prodrugs, a copper-catalyzed carbene insertion of tetraethyl diazomethyldiphosphonate (216) with N-Boc-protected amino acids 214 and 215 provided a simple method to synthesize phosphonodepsipeptides 217 and 218 containing a C-1-hydroxyalkylphosphonate motif in good yields (Scheme 40) [62].
![[1860-5397-17-41-i40]](/bjoc/content/inline/1860-5397-17-41-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Synthesis of phosphonodepsipeptides with a C-1-hydroxyalkylphosphonate motif via a copper-catalyzed carbene insertion reaction.
Scheme 40: Synthesis of phosphonodepsipeptides with a C-1-hydroxyalkylphosphonate motif via a copper-catalyzed...
The transition metal-catalyzed carbene insertion of 1-diazoalkylphononates and N-protected amino acids is an efficient and convenient method for the synthesis of phosphonodepsipeptides with C-1-hydroxyalkylphosphonic acids because the required 1-diazoalkylphononates can be readily prepared from the corresponding 1-aminoalkylphosphonates via nitrosation with amyl nitrite.
Conclusion
Phosphonodepsipeptides are phosphorus analogues of depsipeptides. They are more stable than the corresponding phosphonopeptides and have been widely used as enzyme inhibitors, haptens for the production of antibodies, biological agents, and prodrugs. Various synthetic methods of phosphonodepsipeptides have been developed, including the phosphonylation of hydroxy esters with phosphonochloridates or with phosphonic monoesters in the presence of coupling reagents, the alkylation of phosphonic monoesters with 1-(alkoxylcarbonyl)alkyl halides or sulfonates, the phosphinylation of hydroxy esters with phosphonochloridites and subsequent oxidation, and the Mannich-type condensation of amides, aldehydes, and dichlorophosphites followed by alcoholysis with hydroxy esters. Among the synthetic methods, the multicomponent Mannich-type condensation strategy shows a high efficiency, convergent feature, and product diversity. It can be expected that the convergent multicomponent condensation synthetic strategy will show wide applications in the preparation of biologically active phosphonodepsipeptides in the future. However, highly stereoselective asymmetric synthetic methods of phosphonodepsipeptides are of high demand and need to be developed in the near future.
References
-
Kafarski, P.; Lejczak, B. Synthesis of phosphono- and phosphinopeptides. In Aminophosphonic and Aminophosphinic Acids; Kukhar, V. P.; Hudson, H. R., Eds.; John Wiley & Sons: West Sussex, England, 2000; pp 173–203.
Return to citation in text: [1] -
Xu, J. Sci. Sin.: Chim. 2013, 43, 995–1004. doi:10.1360/032013-159
Return to citation in text: [1] -
Drabowicz, J.; Kiełbasiński, P.; Łyżwa, P.; Mikołajczyk, M.; Zając, A. Product Class 15: Alkylphosphonic Acids and Derivatives. In Science of Synthesis; Mathey, F., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany, 2009; Vol. 42, pp 679–778. doi:10.1055/sos-sd-042-00767
Return to citation in text: [1] -
Xu, J. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 487–492. doi:10.1080/10426507.2018.1540481
Return to citation in text: [1] -
Xu, J.; Xia, C.; Yu, L.; Zhou, Q. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 152, 35–44. doi:10.1080/10426509908031615
Return to citation in text: [1] -
Bartlett, P. A.; Vanmaele, L. J.; Kezer, W. B. Bull. Soc. Chim. Fr. 1986, 776–780.
Return to citation in text: [1] [2] -
Jia, C.; Yang, K.-W.; Liu, C.-C.; Feng, L.; Xiao, J.-M.; Zhou, L.-S.; Zhang, Y.-L. Bioorg. Med. Chem. Lett. 2012, 22, 482–484. doi:10.1016/j.bmcl.2011.10.094
Return to citation in text: [1] [2] -
Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Arch. Pharm. (Weinheim, Ger.) 2005, 338, 305–314. doi:10.1002/ardp.200500976
Return to citation in text: [1] [2] [3] -
Uh, E.; Jackson, E. R.; San Jose, G.; Maddox, M.; Lee, R. E.; Lee, R. E.; Boshoff, H. I.; Dowd, C. S. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. doi:10.1016/j.bmcl.2011.09.123
Return to citation in text: [1] [2] [3] -
Vercruysse-Moreira, K.; Déjugnat, C.; Etemad-Moghadam, G. Tetrahedron 2002, 58, 5651–5658. doi:10.1016/s0040-4020(02)00535-5
Return to citation in text: [1] [2] -
Smith, A. B., III; Taylor, C. M.; Benkovic, S. J.; Hirschmann, R. Tetrahedron Lett. 1994, 35, 6853–6856. doi:10.1016/0040-4039(94)85022-4
Return to citation in text: [1] [2] -
Isomura, S.; Ashley, J. A.; Wirsching, P.; Janda, K. D. Bioorg. Med. Chem. Lett. 2002, 12, 861–864. doi:10.1016/s0960-894x(02)00047-1
Return to citation in text: [1] [2] -
Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Bioorg. Med. Chem. Lett. 2003, 13, 2163–2166. doi:10.1016/s0960-894x(03)00354-8
Return to citation in text: [1] [2] -
Ntatsopoulos, V.; Macegoniuk, K.; Mucha, A.; Vassiliou, S.; Berlicki, Ł. Eur. J. Med. Chem. 2018, 159, 307–316. doi:10.1016/j.ejmech.2018.09.074
Return to citation in text: [1] -
Montenegro, I. P. F. M.; Mucha, A. P.; Reis, I.; Rodrigues, P.; Almeida, C. M. R. Int. J. Environ. Sci. Technol. 2017, 14, 943–955. doi:10.1007/s13762-016-1215-9
Return to citation in text: [1] -
Skinner-Adams, T. S.; Lowther, J.; Teuscher, F.; Stack, C. M.; Grembecka, J.; Mucha, A.; Kafarski, P.; Trenholme, K. R.; Dalton, J. P.; Gardiner, D. L. J. Med. Chem. 2007, 50, 6024–6031. doi:10.1021/jm070733v
Return to citation in text: [1] -
Carramiñana, V.; Ochoa de Retana, A. M.; Palacios, F.; de los Santos, J. M. Molecules 2020, 25, 3332. doi:10.3390/molecules25153332
Return to citation in text: [1] -
Mucha, A.; Kafarski, P.; Berlicki, Ł. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f
Return to citation in text: [1] -
Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014
Return to citation in text: [1] -
Song, J.-B.; Hah, S.-S.; Suh, J.-H. Bull. Korean Chem. Soc. 2004, 25, 1703–1706. doi:10.5012/bkcs.2004.25.11.1703
Return to citation in text: [1] -
Yang, K.-W.; Brandt, J. J.; Chatwood, L. L.; Crowder, M. W. Bioorg. Med. Chem. Lett. 2000, 10, 1085–1087. doi:10.1016/s0960-894x(00)00186-4
Return to citation in text: [1] -
Chang, Y.-P.; Tseng, M.-J.; Chu, Y.-H. Anal. Biochem. 2006, 359, 63–71. doi:10.1016/j.ab.2006.08.009
Return to citation in text: [1] -
Hoffmann, M. Aust. J. Chem. 1988, 41, 605–607. doi:10.1071/ch9880605
Return to citation in text: [1] -
Inami, K.; Teshima, T.; Miyashita, H.; Shiba, T. Bull. Chem. Soc. Jpn. 1995, 68, 942–949. doi:10.1246/bcsj.68.942
Return to citation in text: [1] -
Sampson, N. S.; Bartlett, P. A. J. Org. Chem. 1988, 53, 4500–4503. doi:10.1021/jo00254a015
Return to citation in text: [1] -
Bartlett, P. A.; Giangiordano, M. A. J. Org. Chem. 1996, 61, 3433–3438. doi:10.1021/jo952074c
Return to citation in text: [1] -
Shih, H.-W.; Chen, K.-T.; Chen, S.-K.; Huang, C.-Y.; Cheng, T.-J. R.; Ma, C.; Wong, C.-H.; Cheng, W.-C. Org. Biomol. Chem. 2010, 8, 2586–2593. doi:10.1039/c000622j
Return to citation in text: [1] [2] [3] -
Malachowski, W. P.; Coward, J. K. J. Org. Chem. 1994, 59, 7625–7634. doi:10.1021/jo00104a017
Return to citation in text: [1] [2] -
Tsukamoto, T.; Haile, W. H.; McGuire, J. J.; Coward, J. K. Arch. Biochem. Biophys. 1998, 355, 109–118. doi:10.1006/abbi.1998.0703
Return to citation in text: [1] -
Han, L.; Hiratake, J.; Kamiyama, A.; Sakata, K. Biochemistry 2007, 46, 1432–1447. doi:10.1021/bi061890j
Return to citation in text: [1] -
Nakajima, M.; Watanabe, B.; Han, L.; Shimizu, B.-i.; Wada, K.; Fukuyama, K.; Suzuki, H.; Hiratake, J. Bioorg. Med. Chem. 2014, 22, 1176–1194. doi:10.1016/j.bmc.2013.12.034
Return to citation in text: [1] -
Karanewsky, D. S.; Badia, M. C. Tetrahedron Lett. 1986, 27, 1751–1754. doi:10.1016/s0040-4039(00)84364-6
Return to citation in text: [1] -
Campagne, J.-M.; Coste, J.; Jouin, P. Tetrahedron Lett. 1993, 34, 6743–6744. doi:10.1016/s0040-4039(00)61690-8
Return to citation in text: [1] -
Campagne, J.-M.; Coste, J.; Jouin, P. J. Org. Chem. 1995, 60, 5214–5223. doi:10.1021/jo00121a045
Return to citation in text: [1] -
Galéotti, N.; Coste, J.; Bedos, P.; Jouin, P. Tetrahedron Lett. 1996, 37, 3997–3998. doi:10.1016/0040-4039(96)00742-3
Return to citation in text: [1] -
Gobec, S.; Urleb, U. Molecules 2002, 7, 394–404. doi:10.3390/70400394
Return to citation in text: [1] -
Pícha, J.; Buděšínský, M.; Šanda, M.; Jiráček, J. Tetrahedron Lett. 2008, 49, 4366–4368. doi:10.1016/j.tetlet.2008.05.028
Return to citation in text: [1] -
Pícha, J.; Buděšínský, M.; Hančlová, I.; Šanda, M.; Fiedler, P.; Vaněk, V.; Jiráček, J. Tetrahedron 2009, 65, 6090–6103. doi:10.1016/j.tet.2009.05.051
Return to citation in text: [1] [2] -
Campbell, D. A. J. Org. Chem. 1992, 57, 6331–6335. doi:10.1021/jo00049a051
Return to citation in text: [1] -
Yuan, C.; Wang, G. Phosphorus, Sulfur Silicon Relat. Elem. 1992, 71, 207–212. doi:10.1080/10426509208034513
Return to citation in text: [1] -
Xu, J.; Fu, N. Synth. Commun. 2000, 30, 4137–4145. doi:10.1080/00397910008087030
Return to citation in text: [1] -
Fu, N.; Zhang, Q.; Duan, L.; Xu, J. J. Pept. Sci. 2006, 12, 303–309. doi:10.1002/psc.727
Return to citation in text: [1] [2] -
Xu, J.; Fu, N. J. Chem. Soc., Perkin Trans. 1 2001, 1223–1226. doi:10.1039/b008340m
Return to citation in text: [1] -
Xu, J.; Wei, M. Synth. Commun. 2001, 31, 1489–1497. doi:10.1081/scc-100104060
Return to citation in text: [1] -
Li, B.; Cai, S.; Du, D.-M.; Xu, J. Org. Lett. 2007, 9, 2257–2260. doi:10.1021/ol070360s
Return to citation in text: [1] -
Meng, F.; Xu, J. Amino Acids 2010, 39, 533–538. doi:10.1007/s00726-009-0469-7
Return to citation in text: [1] -
Meng, F.; Xu, J. Tetrahedron 2013, 69, 4944–4952. doi:10.1016/j.tet.2013.04.032
Return to citation in text: [1] -
He, F.; Meng, F.; Song, X.; Hu, W.; Xu, J. Org. Lett. 2009, 11, 3922–3925. doi:10.1021/ol901543y
Return to citation in text: [1] -
Meng, F.; He, F.; Song, X.; Zhang, L.; Hu, W.; Liu, G.; Xu, J. Amino Acids 2012, 43, 423–429. doi:10.1007/s00726-011-1098-5
Return to citation in text: [1] -
Liu, H.; Cai, S.; Xu, J. J. Pept. Sci. 2006, 12, 337–340. doi:10.1002/psc.731
Return to citation in text: [1] -
Xu, J.; Gao, Y. Synthesis 2006, 783–788. doi:10.1055/s-2006-926324
Return to citation in text: [1] [2] -
Fang, Z.; Yang, H.; Miao, Z.; Chen, R. Helv. Chim. Acta 2011, 94, 1586–1593. doi:10.1002/hlca.201100025
Return to citation in text: [1] -
Skwarczyński, M.; Kafarski, P. Synth. Commun. 1995, 25, 3565–3571. doi:10.1080/00397919508015491
Return to citation in text: [1] -
Meyer, J. H.; Bartlett, P. A. J. Am. Chem. Soc. 1998, 120, 4600–4609. doi:10.1021/ja973715j
Return to citation in text: [1] -
de Fatima Fernandez, M.; Vlaar, C. P.; Fan, H.; Liu, Y.-H.; Fronczek, F. R.; Hammer, R. P. J. Org. Chem. 1995, 60, 7390–7391. doi:10.1021/jo00128a006
Return to citation in text: [1] -
Vercruysse, K.; Déjugnat, C.; Munoz, A.; Etemad-Moghadam, G. Eur. J. Org. Chem. 2000, 281–289. doi:10.1002/(sici)1099-0690(200001)2000:2<281::aid-ejoc281>3.0.co;2-2
Return to citation in text: [1] -
Déjugnat, C.; Etemad-Moghadam, G.; Rico-Lattes, I. Chem. Commun. 2003, 1858–1859. doi:10.1039/b304420c
Return to citation in text: [1] -
Yang, J. Q.; Yang, X.; Zeng, F. K.; Li, P. Preparation method of phosphonate derivative containing amino acid fragments and antineoplastic application. Chinese Pat. CN105503947A, April 20, 2016.
Return to citation in text: [1] -
Hoffmann, M. J. Prakt. Chem. 1990, 332, 251–255. doi:10.1002/prac.19903320217
Return to citation in text: [1] -
Titanyuk, I. D.; Vorob’eva, D. V.; Osipov, S. N.; Beletskaya, I. P. Synlett 2006, 1355–1358. doi:10.1055/s-2006-939704
Return to citation in text: [1] -
Titanyuk, I. D.; Vorob’eva, D. V.; Osipov, S. N.; Beletskaya, I. P. Russ. J. Org. Chem. 2010, 46, 619–623. doi:10.1134/s1070428010050015
Return to citation in text: [1] -
Wang, X.; Zhang, C.; Ma, Q.; Xiao, W.; Guo, L.; Wu, Y. Tetrahedron Lett. 2018, 59, 280–283. doi:10.1016/j.tetlet.2017.12.038
Return to citation in text: [1]
| 32. | Karanewsky, D. S.; Badia, M. C. Tetrahedron Lett. 1986, 27, 1751–1754. doi:10.1016/s0040-4039(00)84364-6 |
| 33. | Campagne, J.-M.; Coste, J.; Jouin, P. Tetrahedron Lett. 1993, 34, 6743–6744. doi:10.1016/s0040-4039(00)61690-8 |
| 34. | Campagne, J.-M.; Coste, J.; Jouin, P. J. Org. Chem. 1995, 60, 5214–5223. doi:10.1021/jo00121a045 |
| 28. | Malachowski, W. P.; Coward, J. K. J. Org. Chem. 1994, 59, 7625–7634. doi:10.1021/jo00104a017 |
| 38. | Pícha, J.; Buděšínský, M.; Hančlová, I.; Šanda, M.; Fiedler, P.; Vaněk, V.; Jiráček, J. Tetrahedron 2009, 65, 6090–6103. doi:10.1016/j.tet.2009.05.051 |
| 38. | Pícha, J.; Buděšínský, M.; Hančlová, I.; Šanda, M.; Fiedler, P.; Vaněk, V.; Jiráček, J. Tetrahedron 2009, 65, 6090–6103. doi:10.1016/j.tet.2009.05.051 |
| 37. | Pícha, J.; Buděšínský, M.; Šanda, M.; Jiráček, J. Tetrahedron Lett. 2008, 49, 4366–4368. doi:10.1016/j.tetlet.2008.05.028 |
| 35. | Galéotti, N.; Coste, J.; Bedos, P.; Jouin, P. Tetrahedron Lett. 1996, 37, 3997–3998. doi:10.1016/0040-4039(96)00742-3 |
| 12. | Isomura, S.; Ashley, J. A.; Wirsching, P.; Janda, K. D. Bioorg. Med. Chem. Lett. 2002, 12, 861–864. doi:10.1016/s0960-894x(02)00047-1 |
| 40. | Yuan, C.; Wang, G. Phosphorus, Sulfur Silicon Relat. Elem. 1992, 71, 207–212. doi:10.1080/10426509208034513 |
| 41. | Xu, J.; Fu, N. Synth. Commun. 2000, 30, 4137–4145. doi:10.1080/00397910008087030 |
| 42. | Fu, N.; Zhang, Q.; Duan, L.; Xu, J. J. Pept. Sci. 2006, 12, 303–309. doi:10.1002/psc.727 |
| 48. | He, F.; Meng, F.; Song, X.; Hu, W.; Xu, J. Org. Lett. 2009, 11, 3922–3925. doi:10.1021/ol901543y |
| 49. | Meng, F.; He, F.; Song, X.; Zhang, L.; Hu, W.; Liu, G.; Xu, J. Amino Acids 2012, 43, 423–429. doi:10.1007/s00726-011-1098-5 |
| 50. | Liu, H.; Cai, S.; Xu, J. J. Pept. Sci. 2006, 12, 337–340. doi:10.1002/psc.731 |
| 45. | Li, B.; Cai, S.; Du, D.-M.; Xu, J. Org. Lett. 2007, 9, 2257–2260. doi:10.1021/ol070360s |
| 46. | Meng, F.; Xu, J. Amino Acids 2010, 39, 533–538. doi:10.1007/s00726-009-0469-7 |
| 47. | Meng, F.; Xu, J. Tetrahedron 2013, 69, 4944–4952. doi:10.1016/j.tet.2013.04.032 |
| 43. | Xu, J.; Fu, N. J. Chem. Soc., Perkin Trans. 1 2001, 1223–1226. doi:10.1039/b008340m |
| 44. | Xu, J.; Wei, M. Synth. Commun. 2001, 31, 1489–1497. doi:10.1081/scc-100104060 |
| 42. | Fu, N.; Zhang, Q.; Duan, L.; Xu, J. J. Pept. Sci. 2006, 12, 303–309. doi:10.1002/psc.727 |
| 53. | Skwarczyński, M.; Kafarski, P. Synth. Commun. 1995, 25, 3565–3571. doi:10.1080/00397919508015491 |
| 54. | Meyer, J. H.; Bartlett, P. A. J. Am. Chem. Soc. 1998, 120, 4600–4609. doi:10.1021/ja973715j |
| 52. | Fang, Z.; Yang, H.; Miao, Z.; Chen, R. Helv. Chim. Acta 2011, 94, 1586–1593. doi:10.1002/hlca.201100025 |
| 1. | Kafarski, P.; Lejczak, B. Synthesis of phosphono- and phosphinopeptides. In Aminophosphonic and Aminophosphinic Acids; Kukhar, V. P.; Hudson, H. R., Eds.; John Wiley & Sons: West Sussex, England, 2000; pp 173–203. |
| 2. | Xu, J. Sci. Sin.: Chim. 2013, 43, 995–1004. doi:10.1360/032013-159 |
| 3. | Drabowicz, J.; Kiełbasiński, P.; Łyżwa, P.; Mikołajczyk, M.; Zając, A. Product Class 15: Alkylphosphonic Acids and Derivatives. In Science of Synthesis; Mathey, F., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany, 2009; Vol. 42, pp 679–778. doi:10.1055/sos-sd-042-00767 |
| 4. | Xu, J. Phosphorus, Sulfur Silicon Relat. Elem. 2019, 194, 487–492. doi:10.1080/10426507.2018.1540481 |
| 5. | Xu, J.; Xia, C.; Yu, L.; Zhou, Q. Phosphorus, Sulfur Silicon Relat. Elem. 1999, 152, 35–44. doi:10.1080/10426509908031615 |
| 14. | Ntatsopoulos, V.; Macegoniuk, K.; Mucha, A.; Vassiliou, S.; Berlicki, Ł. Eur. J. Med. Chem. 2018, 159, 307–316. doi:10.1016/j.ejmech.2018.09.074 |
| 22. | Chang, Y.-P.; Tseng, M.-J.; Chu, Y.-H. Anal. Biochem. 2006, 359, 63–71. doi:10.1016/j.ab.2006.08.009 |
| 59. | Hoffmann, M. J. Prakt. Chem. 1990, 332, 251–255. doi:10.1002/prac.19903320217 |
| 8. | Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Arch. Pharm. (Weinheim, Ger.) 2005, 338, 305–314. doi:10.1002/ardp.200500976 |
| 9. | Uh, E.; Jackson, E. R.; San Jose, G.; Maddox, M.; Lee, R. E.; Lee, R. E.; Boshoff, H. I.; Dowd, C. S. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. doi:10.1016/j.bmcl.2011.09.123 |
| 13. | Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Bioorg. Med. Chem. Lett. 2003, 13, 2163–2166. doi:10.1016/s0960-894x(03)00354-8 |
| 11. | Smith, A. B., III; Taylor, C. M.; Benkovic, S. J.; Hirschmann, R. Tetrahedron Lett. 1994, 35, 6853–6856. doi:10.1016/0040-4039(94)85022-4 |
| 12. | Isomura, S.; Ashley, J. A.; Wirsching, P.; Janda, K. D. Bioorg. Med. Chem. Lett. 2002, 12, 861–864. doi:10.1016/s0960-894x(02)00047-1 |
| 21. | Yang, K.-W.; Brandt, J. J.; Chatwood, L. L.; Crowder, M. W. Bioorg. Med. Chem. Lett. 2000, 10, 1085–1087. doi:10.1016/s0960-894x(00)00186-4 |
| 10. | Vercruysse-Moreira, K.; Déjugnat, C.; Etemad-Moghadam, G. Tetrahedron 2002, 58, 5651–5658. doi:10.1016/s0040-4020(02)00535-5 |
| 6. | Bartlett, P. A.; Vanmaele, L. J.; Kezer, W. B. Bull. Soc. Chim. Fr. 1986, 776–780. |
| 7. | Jia, C.; Yang, K.-W.; Liu, C.-C.; Feng, L.; Xiao, J.-M.; Zhou, L.-S.; Zhang, Y.-L. Bioorg. Med. Chem. Lett. 2012, 22, 482–484. doi:10.1016/j.bmcl.2011.10.094 |
| 8. | Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Arch. Pharm. (Weinheim, Ger.) 2005, 338, 305–314. doi:10.1002/ardp.200500976 |
| 9. | Uh, E.; Jackson, E. R.; San Jose, G.; Maddox, M.; Lee, R. E.; Lee, R. E.; Boshoff, H. I.; Dowd, C. S. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. doi:10.1016/j.bmcl.2011.09.123 |
| 10. | Vercruysse-Moreira, K.; Déjugnat, C.; Etemad-Moghadam, G. Tetrahedron 2002, 58, 5651–5658. doi:10.1016/s0040-4020(02)00535-5 |
| 7. | Jia, C.; Yang, K.-W.; Liu, C.-C.; Feng, L.; Xiao, J.-M.; Zhou, L.-S.; Zhang, Y.-L. Bioorg. Med. Chem. Lett. 2012, 22, 482–484. doi:10.1016/j.bmcl.2011.10.094 |
| 58. | Yang, J. Q.; Yang, X.; Zeng, F. K.; Li, P. Preparation method of phosphonate derivative containing amino acid fragments and antineoplastic application. Chinese Pat. CN105503947A, April 20, 2016. |
| 18. | Mucha, A.; Kafarski, P.; Berlicki, Ł. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 6. | Bartlett, P. A.; Vanmaele, L. J.; Kezer, W. B. Bull. Soc. Chim. Fr. 1986, 776–780. |
| 56. | Vercruysse, K.; Déjugnat, C.; Munoz, A.; Etemad-Moghadam, G. Eur. J. Org. Chem. 2000, 281–289. doi:10.1002/(sici)1099-0690(200001)2000:2<281::aid-ejoc281>3.0.co;2-2 |
| 17. | Carramiñana, V.; Ochoa de Retana, A. M.; Palacios, F.; de los Santos, J. M. Molecules 2020, 25, 3332. doi:10.3390/molecules25153332 |
| 20. | Song, J.-B.; Hah, S.-S.; Suh, J.-H. Bull. Korean Chem. Soc. 2004, 25, 1703–1706. doi:10.5012/bkcs.2004.25.11.1703 |
| 57. | Déjugnat, C.; Etemad-Moghadam, G.; Rico-Lattes, I. Chem. Commun. 2003, 1858–1859. doi:10.1039/b304420c |
| 16. | Skinner-Adams, T. S.; Lowther, J.; Teuscher, F.; Stack, C. M.; Grembecka, J.; Mucha, A.; Kafarski, P.; Trenholme, K. R.; Dalton, J. P.; Gardiner, D. L. J. Med. Chem. 2007, 50, 6024–6031. doi:10.1021/jm070733v |
| 8. | Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Arch. Pharm. (Weinheim, Ger.) 2005, 338, 305–314. doi:10.1002/ardp.200500976 |
| 9. | Uh, E.; Jackson, E. R.; San Jose, G.; Maddox, M.; Lee, R. E.; Lee, R. E.; Boshoff, H. I.; Dowd, C. S. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. doi:10.1016/j.bmcl.2011.09.123 |
| 13. | Ortmann, R.; Wiesner, J.; Reichenberg, A.; Henschker, D.; Beck, E.; Jomaa, H.; Schlitzer, M. Bioorg. Med. Chem. Lett. 2003, 13, 2163–2166. doi:10.1016/s0960-894x(03)00354-8 |
| 15. | Montenegro, I. P. F. M.; Mucha, A. P.; Reis, I.; Rodrigues, P.; Almeida, C. M. R. Int. J. Environ. Sci. Technol. 2017, 14, 943–955. doi:10.1007/s13762-016-1215-9 |
| 19. | Giannousis, P. P.; Bartlett, P. A. J. Med. Chem. 1987, 30, 1603–1609. doi:10.1021/jm00392a014 |
| 55. | de Fatima Fernandez, M.; Vlaar, C. P.; Fan, H.; Liu, Y.-H.; Fronczek, F. R.; Hammer, R. P. J. Org. Chem. 1995, 60, 7390–7391. doi:10.1021/jo00128a006 |
| 25. | Sampson, N. S.; Bartlett, P. A. J. Org. Chem. 1988, 53, 4500–4503. doi:10.1021/jo00254a015 |
| 11. | Smith, A. B., III; Taylor, C. M.; Benkovic, S. J.; Hirschmann, R. Tetrahedron Lett. 1994, 35, 6853–6856. doi:10.1016/0040-4039(94)85022-4 |
| 24. | Inami, K.; Teshima, T.; Miyashita, H.; Shiba, T. Bull. Chem. Soc. Jpn. 1995, 68, 942–949. doi:10.1246/bcsj.68.942 |
| 60. | Titanyuk, I. D.; Vorob’eva, D. V.; Osipov, S. N.; Beletskaya, I. P. Synlett 2006, 1355–1358. doi:10.1055/s-2006-939704 |
| 61. | Titanyuk, I. D.; Vorob’eva, D. V.; Osipov, S. N.; Beletskaya, I. P. Russ. J. Org. Chem. 2010, 46, 619–623. doi:10.1134/s1070428010050015 |
| 62. | Wang, X.; Zhang, C.; Ma, Q.; Xiao, W.; Guo, L.; Wu, Y. Tetrahedron Lett. 2018, 59, 280–283. doi:10.1016/j.tetlet.2017.12.038 |
| 30. | Han, L.; Hiratake, J.; Kamiyama, A.; Sakata, K. Biochemistry 2007, 46, 1432–1447. doi:10.1021/bi061890j |
| 31. | Nakajima, M.; Watanabe, B.; Han, L.; Shimizu, B.-i.; Wada, K.; Fukuyama, K.; Suzuki, H.; Hiratake, J. Bioorg. Med. Chem. 2014, 22, 1176–1194. doi:10.1016/j.bmc.2013.12.034 |
| 28. | Malachowski, W. P.; Coward, J. K. J. Org. Chem. 1994, 59, 7625–7634. doi:10.1021/jo00104a017 |
| 29. | Tsukamoto, T.; Haile, W. H.; McGuire, J. J.; Coward, J. K. Arch. Biochem. Biophys. 1998, 355, 109–118. doi:10.1006/abbi.1998.0703 |
| 27. | Shih, H.-W.; Chen, K.-T.; Chen, S.-K.; Huang, C.-Y.; Cheng, T.-J. R.; Ma, C.; Wong, C.-H.; Cheng, W.-C. Org. Biomol. Chem. 2010, 8, 2586–2593. doi:10.1039/c000622j |
| 27. | Shih, H.-W.; Chen, K.-T.; Chen, S.-K.; Huang, C.-Y.; Cheng, T.-J. R.; Ma, C.; Wong, C.-H.; Cheng, W.-C. Org. Biomol. Chem. 2010, 8, 2586–2593. doi:10.1039/c000622j |
| 26. | Bartlett, P. A.; Giangiordano, M. A. J. Org. Chem. 1996, 61, 3433–3438. doi:10.1021/jo952074c |
| 27. | Shih, H.-W.; Chen, K.-T.; Chen, S.-K.; Huang, C.-Y.; Cheng, T.-J. R.; Ma, C.; Wong, C.-H.; Cheng, W.-C. Org. Biomol. Chem. 2010, 8, 2586–2593. doi:10.1039/c000622j |
© 2021 Xu; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)