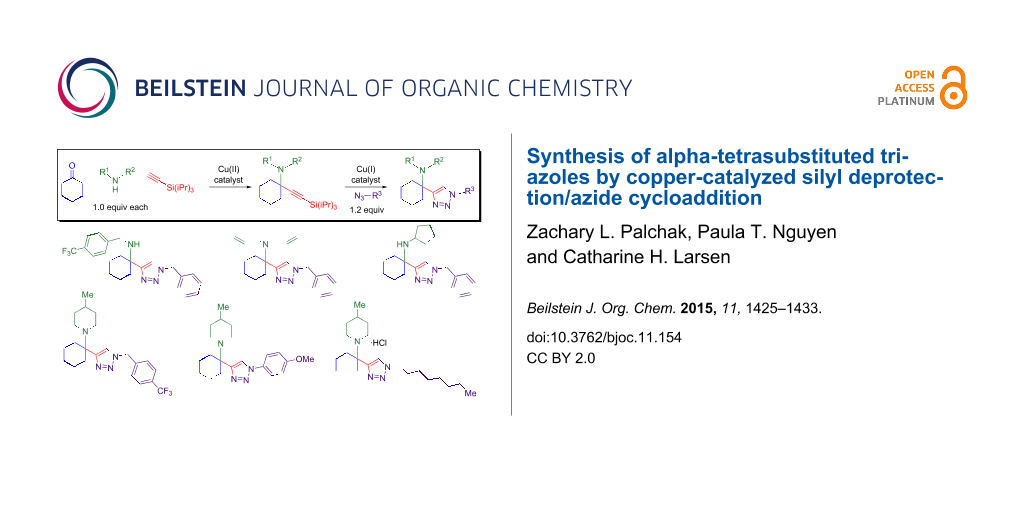
Abstract
Propargylamines are popular substrates for triazole formation, but tetrasubstituted variants have required multistep syntheses involving stoichiometric amounts of metal. A recent cyclohexanone–amine–silylacetylene coupling forms silyl-protected tetrasubstituted propargylamines in a single copper-catalyzed step. The development of the tandem silyl deprotection–triazole formation reported herein offers rapid access to alpha-tetrasubstituted triazoles. A streamlined two-step approach to this uncommon class of hindered triazoles will accelerate exploration of their therapeutic potential. The superior activity of copper(II) triflate in the formation of triazoles from sensitive alkyne substrates extends to simple terminal alkynes.

Graphical Abstract
Introduction
1,2,3-Triazoles demonstrate wide spread application in biological systems and drug development [1-12]. Copper-catalyzed azide–alkyne cycloadditions (CuAAC) regioselectively introduce a wide variety of substituents on 1,4-disubstituted 1,2,3-triazoles from the organic azide or terminal alkyne starting materials [1,2]. These Huisgen reactions [13] facilitate rapid drug screening by allowing for tracking in biological systems and the exploration of structure-activity relationships [10,14-19].
Propargylamines are a popular choice for the terminal alkyne component and form highly selective inhibitors (Figure 1) [2]. Due to the difficulty of forming tetrasubstituted propargylamines, the incorporation of deprotectable variants into triazoles is extremely rare. The Ellman group demonstrates the power of their chiral sulfinylimine protocol to synthesize propargylamine-derived alpha-tetrasubstituted triazoles (tetrasubstituted carbon bearing amine highlighted in red, Figure 1). One such triazole is a cruzain inhibitor with activity against parasite Trypanosoma cruzi, which causes Chagas’ disease [6]. An alpha-tetrasubstituted triazole that inhibits cathepsin S can potentially treat ailments ranging from inflammation to autoimmune disorders [7,8].
![[1860-5397-11-154-1]](/bjoc/content/figures/1860-5397-11-154-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: A sampling of propargylamine-derived triazoles with therapeutic effects includes alpha-tetrasubstituted triazoles as cruzain and cathepsin inhibitors.
Figure 1: A sampling of propargylamine-derived triazoles with therapeutic effects includes alpha-tetrasubstit...
The core of the cathepsin S inhibitor is synthesized in six steps. Synthesis and isolation of an N-sulfinyl ketimine is followed by stoichiometric alkynylation with a trimethylsilyl-protected alkynyllithium reagent. Removal of the silyl and sulfinyl protecting groups allows for CuAAC with a resin-bound azide. Acylation of the amine followed by dehydration yields the active alpha-tetrasubstituted triazole [7].
The lengthy synthesis of tetrasubstituted propargylamine precursors limits the exploration of such alpha-tetrasubstitued triazoles. The vast majority of three-component couplings produce trisubstituted propargylamines. Copper remains the most popular catalyst for these multicomponent reactions, abbreviated as A3 reactions to indicate the Aldehyde, Amine, and Alkyne reaction partners [20,21]. Methods for the corresponding KA2, Ketone–Amine–Alkyne, three-component coupling reaction are rare due to the lower electrophilicity and greater steric hindrance of ketones [22-26]. Due to the release of torsional strain when the sp2 center in the six-membered ring is attacked, cyclohexanone represents a special case as this cyclic ketone is nearly as reactive as an aldehyde [24].
The resultant tetrasubstituted (red) cyclohexylamine is found in natural alkaloids such as (–)-lycodine (Figure 2), [27] and this motif is also critical to the activity of drugs like ketamine and phencyclidine (1-(1-phenylcyclohexyl)piperidine, PCP) [28]. Tetrasubstituted carbons bearing amines can provide much higher levels of activity than the corresponding trisubstituted center. For example, fentanyl is an anesthetic that is 100 times as powerful as morphine (Figure 2) [29]. By creating a tetrasubstituted variant, the activity is increased two orders of magnitude: carfentanil is over 10,000 times as active as morphine.
![[1860-5397-11-154-2]](/bjoc/content/figures/1860-5397-11-154-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: A tetrasubstituted carbon bearing an amine (red) can provide 100-fold increase in activity compared to the trisubstituted carbon bearing an amine (blue).
Figure 2: A tetrasubstituted carbon bearing an amine (red) can provide 100-fold increase in activity compared...
Results and Discussion
Reaction optimization
The two-step/three-reaction sequence shown in Scheme 1 would streamline the synthesis of alpha-tetrasubstituted triazoles 6. In the first step, our solvent-free copper-catalyzed three-component coupling of cyclohexanone (1), amines 2, and alkynes 3 provides high yields of silyl-protected propargylic amines 4 [24,25]. Trimethylsilyl (TMS) acetylene was not stable in the presence of the copper(II) chloride catalyst, and triethylsilylacetylene did not convert cleanly to product. Triisopropylsilyl (TIPS) acetylene was found to be superior to tert-butyldimethylsilylacetylene as a source of silylated tetrasubstituted propargylic amines.
![[1860-5397-11-154-i1]](/bjoc/content/inline/1860-5397-11-154-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: KA2 coupling followed by tandem silyl deprotection and triazole formation.
Scheme 1: KA2 coupling followed by tandem silyl deprotection and triazole formation.
Although TMS-protected alkynes have been converted to triazoles via a one-pot silyl deprotection CuAAC reaction [30-33], TIPS-protected alkynes have not. As the triisopropylsilyl protecting group is more difficult to remove than the less hindered trimethylsilyl, conditions for TIPS deprotection include 1.5 equiv of AgF or Cu(OAc)2 combined with syringe pump addition of TBAF [34,35]. An additional difficulty is that Ellman’s alpha-tetrasubstituted triazoles are synthesized by CuAAC reaction with desilylated, purified tetrasubstituted propargylic amines [6-8]. Therefore, the goal was to develop the second portion of the sequence in Scheme 1: a tandem deprotection–cycloaddition of tetrasubstituted TIPS-protected propargylamines 4 that would allow them to react in situ with various azides 5 to give hindered triazoles 6.
Table 1: Optimization of silyl deprotection/cycloaddition.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-154-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solventa | Catalystb | GC yield (%), 1 h | GC yield (%), 18 h |
|---|---|---|---|---|
| 1 | t-BuOH/H2O (1:1 v/v) | CuCl | 21 | 2 |
| 2 | DMSO/H2O (2:1 v/v) | CuCl | 0 | 0 |
| 3 | DMF/H2O (1:2 v/v) | CuCl | 0 | 0 |
| 4 | THF/MeOH (1:1 v/v) | CuCl | 4 | 34 |
| 5 | t-BuOH | CuCl | 6 | 62 |
| 6 | MeOH | CuCl | 13 | 65 |
| 7 | MeOH | CuBr | 62 | 63 |
| 8 | MeOH | Cu Powder | 24 | 24 |
| 9 | MeOH | CuCl2 | 72 | 90 |
| 10 | MeOH | CuBr2 | 65 | 86 |
| 11 | MeOH | CuF2·2H2O | 69 | 88 |
| 12 | MeOH | CuSO4·5H2O | 49 | 79 |
| 13 | MeOH | Cu(OAc)2·H2O | 46 | 82 |
| 14 | MeOH | Cu(OTf)2 | 39 | 99 |
aEntries 7–14 were carried out under an atmosphere of argon. bEntries 9–14 with copper(II) sources include 5 mol % of sodium ascorbate reductant.
As a copper(I) catalyst is required for azide–alkyne cycloaddition, the development of a method for the one-pot deprotection/CuAAC began with Cu(I) chloride and a survey of solvents reported [2] for triazole formation (Table 1). TIPS-protected propargylamine 4a and benzyl azide (5a) are heated in the presence of 1.5 equivalents of TBAF (tetrabutylammonium fluoride), 5 mol % CuCl, and the solvent(s) indicated (Table 1, entries 1–6). Aqueous solvent mixtures produce only trace amounts of product at 1 h and 18 h, but methanol and tert-butanol provide two-thirds conversion to triazole 6a after 18 h.
To increase the rate of reaction and to induce complete conversion to 6a, copper(I) as well as copper(II) sources with an equal amount of sodium ascorbate as the reducing agent were tested in MeOH (Table 1, entries 6–14). All combinations of copper(II) salts with reductant provide higher GC yields (79–99%, Table 1, entries 9–14) than CuCl alone (65%, Table 1, entry 6) at 18 h – corroborating reports that CuAACs are often more efficient with copper(I) catalysts formed from the in situ reduction of copper(II) [2]. After 18 h, copper(II) chloride provides 90% GC yield (Table 1, entry 9), and copper(II) triflate unexpectedly results in quantitative GC yield of 6a (Table 1, entry 14). Rare in CuAAC reactions, copper(II) triflate is known to form protic acid in solution, which could cause proto-desilylation [36], but this is not observed.
A broader range of green alcohol solvents [37] were assessed in the presence of the most active catalyst, in situ reduced Cu(OTf)2. Isopropanol was found to provide faster conversion than methanol, ethanol, and tert-butanol at 1 h. A control reaction in isopropanol with in situ reduced copper(II) sources confirms that the triflate is more active than the chloride at 6 h: 67% versus 26% GC yield. Spiking the copper(II) chloride reaction with TfOH provides a similar 30% GC yield. With isopropanol as the solvent, the effect of ambient atmosphere was compared to inert atmosphere. Argon is not necessary, and an atmosphere of nitrogen provides 24% higher GC yield than air. Testing reaction concentration found no significant difference between 0.5 M to 1.0 M isopropanol but increasing the molarity to 2.0 M nearly doubles the GC yield at 18 h from 49% to 87%. Although a lower temperature of 40 °C results in incomplete conversion at 18 h, evidence of product decomposition appears at a higher temperature of 80 °C. A temperature of 60 °C provides two-thirds conversion at 1 h and 85% GC yield of triazole 6a at 18 h. Finally, when the copper loading is varied from 2.5 mol % to 25 mol % copper(II) triflate with an equivalent loading of sodium ascorbate (NaAsc), GC yields range from 53–63%. However, isolated yields are highest with 10 mol % Cu(OTf)2 plus 10 mol % NaAsc as the in situ reductant.
Substrate scope
With optimized conditions in hand, Table 2 shows the range of triisopropylsilyl cyclohexyl propargylamines that are cleanly converted to the corresponding triazole. Test substrate 6a is isolated in 66% yield under standard conditions: 10 mol % Cu(OTf)2, 10 mol % NaAsc, and 1.5 equiv TBAF in isopropanol (2.0 M) at 65 °C for 6 hours. The N,N-diallyl variant 6b recrystallized in 67% yield is readily deprotectable [38]. This would allow for further elaboration in a similar fashion to the synthesis of cathepsin S inhibitor in Figure 1; N-deprotection followed by acylation forms the triazole amide product [6-8]. N-[4-(Trifluoromethyl)benzyl]triazole 6c and morpholinyltriazole 6d are isolated in 76% and 72% yield, respectively. Cyclopentylamine-derived 6e and N-methylpiperazine-derived 6f provide additional choices for amine substituents.
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-154-i5.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-154-i6.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-154-i7.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-154-i8.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-154-i9.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-154-i10.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-154-i11.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-154-i12.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-154-i13.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-154-i14.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-154-i15.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-154-i16.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-154-i17.svg?max-width=637&scale=1.0)
Alternate organic azides were synthesized according to known methods [39-41]. 4-Methylbenzyl azide forms triazole 6g in a comparable yield to 6a, but electron-poor 4-(trifluoromethyl)benzyl azide forms triazole 6h in lower yield. Aryl and alkyl azides display parallel reactivity in the formation of triazoles 6i and 6j, and the HCl salt of lipophilic 6j is isolated cleanly in 56% yield. As TIPS-protected propargylamine 4a is utilized throughout Table 3, the lower yields observed in some cases are attributed to the intrinsic efficiency of the azide participant.
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-154-i18.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-154-i19.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-154-i20.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-154-i21.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-154-i22.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-154-i23.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-154-i24.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-154-i25.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-154-i26.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-154-i27.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-154-i28.svg?max-width=637&scale=1.0)
Control reactions were carried out to assess whether conditions developed for in situ silyl deprotection would otherwise affect the azide when reacting with a simpler terminal alkyne (Scheme 2). As the copper-catalyzed cycloaddition of tert-butylacetylene and benzyl azide proceeds in 92% yield, decomposition of the benzyl azide or non-silyl alkyne appears unlikely. Furthermore, this reaction (Scheme 2) proceeds in 92% yield without TBAF. This indicates that the sensitivity of the silyl propargylamine component causes a lower yield of the propargylamine-derived product when reacting with the same benzyl azide under the identical conditions.
![[1860-5397-11-154-i2]](/bjoc/content/inline/1860-5397-11-154-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Silyl deprotection/click conditions applied to tert-butylacetylene. An identical yield is observed without TBAF.
Scheme 2: Silyl deprotection/click conditions applied to tert-butylacetylene. An identical yield is observed ...
This two-step sequence converts commercially available starting materials into alpha-tetrasubstituted amines via three reactions. It is important to note that triazole 6a is formed in 65% overall yield (Scheme 3). The first step simply involves heating 5 mol % CuCl2 with equimolar amounts of three starting materials, cyclohexanone (1), 4-methylpiperidine (2a), and TIPS-acetylene (3). As there are no added ligands, solvents, promoters, or excess starting materials, the sole byproduct in the formation of tetrasubstituted propargylamine 4a is one equivalent of water [25]. The second step links silyl-deprotection with azide–alkyne cycloaddition, producing hindered triazoles in 6 hours. This tandem reaction was optimal with a copper(II) triflate and sodium ascorbate (NaAsc) as a mild reductant. The outcome that copper(II) triflate provides the highest yields for the one-pot deprotection/click reaction was unexpected as it is not known as a catalyst for azide–alkyne cycloaddition reactions [1,2].
![[1860-5397-11-154-i3]](/bjoc/content/inline/1860-5397-11-154-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: High overall yield of 1,2,3-triazole fully-substituted at the 4-position.
Scheme 3: High overall yield of 1,2,3-triazole fully-substituted at the 4-position.
Conclusion
A tandem copper-catalyzed silyl deprotection/azide cycloaddition was developed for TIPS-protected tetrasubstituted propargylamines. These substrates are synthesized by a copper-catalyzed ketone–amine–alkyne (KA2) coupling that proceeds efficiently with no additives such that an equivalent of water is the sole byproduct. This two-step sequence allows for the incorporation of tetrasubstituted carbons bearing amines at the 4-position of the 1,2,3-triazole core. The overall yield of this three-reaction sequence is high and provides rapid access to hindered triazoles in two steps from commercially available starting materials. Both steps are copper-catalyzed and convert inexpensive starting materials into high-value adducts. As the catalyst combination of copper(II) triflate and sodium ascorbate allows for the inclusion of sensitive and hindered substrates, this unprecedented method should be applicable to the robust and less hindered substrates more common in triazole literature. The power of copper catalysis will accelerate further exploration of the therapeutic potential of alpha-tetrasubstituted triazoles.
Supporting Information
The Supporting Information features experimental details, compound characterization, and copies of 1H and 13C NMR spectra of triazoles 6.
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479
Return to citation in text: [1] [2] [3] -
Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j
Return to citation in text: [1] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1] -
Brik, A.; Muldoon, J.; Lin, Y.-C.; Elder, J. H.; Goodsell, D. S.; Olson, A. J.; Fokin, V. V.; Sharpless, K. B.; Wong, C.-H. ChemBioChem 2003, 4, 1246–1248. doi:10.1002/cbic.200300724
Return to citation in text: [1] -
Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m
Return to citation in text: [1] [2] [3] [4] -
Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s
Return to citation in text: [1] [2] [3] [4] [5] -
Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230
Return to citation in text: [1] [2] [3] [4] -
Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s
Return to citation in text: [1] -
Pagliai, F.; Pirali, T.; Del Grosso, E.; Di Brisco, R.; Tron, G. C.; Sorba, G.; Genazzani, A. A. J. Med. Chem. 2006, 49, 467–470. doi:10.1021/jm051118z
Return to citation in text: [1] [2] -
Srinivasan, R.; Uttamchandani, M.; Yao, S. Q. Org. Lett. 2006, 8, 713–716. doi:10.1021/ol052895w
Return to citation in text: [1] -
Tron, G. C.; Pirali, T.; Billington, R. A.; Canonico, P. L.; Sorba, G.; Genazzani, A. A. Med. Res. Rev. 2008, 28, 278–308. doi:10.1002/med.20107
Return to citation in text: [1] -
Huisgen, R. Pure Appl. Chem. 1989, 61, 613–628. doi:10.1351/pac198961040613
Return to citation in text: [1] -
Gupte, A.; Boshoff, H. I.; Wilson, D. J.; Neres, J.; Labello, N. P.; Somu, R. V.; Xing, C.; Barry, C. E., III; Aldrich, C. C. J. Med. Chem. 2008, 51, 7495–7507. doi:10.1021/jm8008037
Return to citation in text: [1] -
Ferreira, S. B.; Sodero, A. C. R.; Cardoso, M. F. C.; Lima, E. S.; Kaiser, C. R.; Silva, F. P., Jr.; Ferreira, V. F. J. Med. Chem. 2010, 53, 2364–2375. doi:10.1021/jm901265h
Return to citation in text: [1] -
Van Poecke, S.; Negri, A.; Gago, F.; Van Daele, I.; Solaroli, N.; Karlsson, A.; Balzarini, J.; Van Calenbergh, S. J. Med. Chem. 2010, 53, 2902–2912. doi:10.1021/jm901532h
Return to citation in text: [1] -
Li, Y.; Shen, M.; Zhang, Z.; Luo, J.; Pan, X.; Lu, X.; Long, H.; Wen, D.; Zhang, F.; Leng, F.; Li, Y.; Tu, Z.; Ren, X.; Ding, K. J. Med. Chem. 2012, 55, 10033–10046. doi:10.1021/jm301188x
Return to citation in text: [1] -
Testa, C.; Scrima, M.; Grimaldi, M.; D’Ursi, A. M.; Dirain, M. L.; Lubin-Germain, N.; Singh, A.; Haskell-Luevano, C.; Chorev, M.; Rovero, P.; Papini, A. M. J. Med. Chem. 2014, 57, 9424–9434. doi:10.1021/jm501027w
Return to citation in text: [1] -
Brik, A.; Muldoon, J.; Lin, Y.-C.; Elder, J. H.; Goodsell, D. S.; Olson, A. J.; Fokin, V. V.; Sharpless, K. B.; Wong, C.-H. ChemBioChem 2003, 4, 1246–1248. doi:10.1002/cbic.200300724
Return to citation in text: [1] -
Blay, G.; Monleon, A.; Pedro, J. R. Curr. Org. Chem. 2009, 13, 1498–1539. doi:10.2174/138527209789177734
Return to citation in text: [1] -
Yoo, W. J.; Zhao, L.; Li, C.-J. Aldrichimica Acta 2011, 44, 43–51.
Return to citation in text: [1] -
Pereshivko, O. P.; Peshkov, V. A.; Van der Eycken, E. V. Org. Lett. 2010, 12, 2638–2641. doi:10.1021/ol1008312
Return to citation in text: [1] -
Cheng, M.; Zhang, Q.; Hu, X.-Y.; Li, B.-G.; Ji, J.-X.; Chan, A. S. C. Adv. Synth. Catal. 2011, 353, 1274–1278. doi:10.1002/adsc.201000914
Return to citation in text: [1] -
Pierce, C. J.; Larsen, C. H. Green Chem. 2012, 14, 2672–2676. doi:10.1039/c2gc35713e
Return to citation in text: [1] [2] [3] -
Palchak, Z. L.; Lussier, D. J.; Pierce, C. J.; Larsen, C. H. Green Chem. 2015, 17, 1802–1810. doi:10.1039/c4gc02318h
Return to citation in text: [1] [2] [3] -
Pierce, C. J.; Nguyen, M.; Larsen, C. H. Angew. Chem., Int. Ed. 2012, 51, 12289–12292. doi:10.1002/anie.201206674
Return to citation in text: [1] -
Ma, X.; Gang, D. R. Nat. Prod. Rep. 2004, 21, 752–772. doi:10.1039/B409720N
Return to citation in text: [1] -
Clayden, J.; Donnard, M.; Lefranc, J.; Tetlow, D. J. Chem. Commun. 2011, 47, 4624–4639. doi:10.1039/C1CC00049G
Return to citation in text: [1] -
Vardanyan, R. S.; Hruby, V. J. Future Med. Chem. 2014, 6, 385–412. doi:10.4155/fmc.13.215
Return to citation in text: [1] -
Aucagne, V.; Leigh, D. A. Org. Lett. 2006, 8, 4505–4507. doi:10.1021/ol061657d
Return to citation in text: [1] -
Fletcher, J. T.; Bumgarner, B. J.; Engels, N. D.; Skoglund, D. A. Organometallics 2008, 27, 5430–5433. doi:10.1021/om800768k
Return to citation in text: [1] -
Fletcher, J. T.; Walz, S. E.; Keeney, M. E. Tetrahedron Lett. 2008, 49, 7030–7032. doi:10.1016/j.tetlet.2008.09.136
Return to citation in text: [1] -
Beltrán, E.; Serrano, J. L.; Sierra, T.; Giménez, R. Org. Lett. 2010, 12, 1404–1407. doi:10.1021/ol902900y
Return to citation in text: [1] -
Escamilla, I. V.; Ramos, L. F. R.; Escalera, J. S.; Hernandez, A. A. J. Mex. Chem. Soc. 2011, 55, 133–136.
Return to citation in text: [1] -
Heuft, M. A.; Collins, S. K.; Yap, G. P. A.; Fallis, A. G. Org. Lett. 2001, 3, 2883–2886. doi:10.1021/ol016414u
Return to citation in text: [1] -
Meyet, C. E.; Pierce, C. J.; Larsen, C. H. Org. Lett. 2012, 14, 964–967. doi:10.1021/ol2029492
Return to citation in text: [1] -
Jessop, P. G. Green Chem. 2011, 13, 1391–1398. doi:10.1039/c0gc00797h
Return to citation in text: [1] -
Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M
Return to citation in text: [1] -
Shinde, S. S.; Chi, H. M.; Lee, B. S.; Chi, D. Y. Tetrahedron Lett. 2009, 50, 6654–6657. doi:10.1016/j.tetlet.2009.09.064
Return to citation in text: [1] -
Asano, K.; Matsubara, S. Org. Lett. 2010, 12, 4988–4991. doi:10.1021/ol101990d
Return to citation in text: [1] -
Rossy, C.; Majimel, J.; Delapierre, M. T.; Fouquet, E.; Felpin, F.-X. J. Organomet. Chem. 2014, 755, 78–85. doi:10.1016/j.jorganchem.2014.01.006
Return to citation in text: [1]
| 1. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 2. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 3. | Tornøe, C. W.; Christensen, C.; Meldal, M. J. Org. Chem. 2002, 67, 3057–3064. doi:10.1021/jo011148j |
| 4. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 5. | Brik, A.; Muldoon, J.; Lin, Y.-C.; Elder, J. H.; Goodsell, D. S.; Olson, A. J.; Fokin, V. V.; Sharpless, K. B.; Wong, C.-H. ChemBioChem 2003, 4, 1246–1248. doi:10.1002/cbic.200300724 |
| 6. | Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m |
| 7. | Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s |
| 8. | Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230 |
| 9. | Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998–15999. doi:10.1021/ja054114s |
| 10. | Pagliai, F.; Pirali, T.; Del Grosso, E.; Di Brisco, R.; Tron, G. C.; Sorba, G.; Genazzani, A. A. J. Med. Chem. 2006, 49, 467–470. doi:10.1021/jm051118z |
| 11. | Srinivasan, R.; Uttamchandani, M.; Yao, S. Q. Org. Lett. 2006, 8, 713–716. doi:10.1021/ol052895w |
| 12. | Tron, G. C.; Pirali, T.; Billington, R. A.; Canonico, P. L.; Sorba, G.; Genazzani, A. A. Med. Res. Rev. 2008, 28, 278–308. doi:10.1002/med.20107 |
| 2. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 24. | Pierce, C. J.; Larsen, C. H. Green Chem. 2012, 14, 2672–2676. doi:10.1039/c2gc35713e |
| 25. | Palchak, Z. L.; Lussier, D. J.; Pierce, C. J.; Larsen, C. H. Green Chem. 2015, 17, 1802–1810. doi:10.1039/c4gc02318h |
| 10. | Pagliai, F.; Pirali, T.; Del Grosso, E.; Di Brisco, R.; Tron, G. C.; Sorba, G.; Genazzani, A. A. J. Med. Chem. 2006, 49, 467–470. doi:10.1021/jm051118z |
| 14. | Gupte, A.; Boshoff, H. I.; Wilson, D. J.; Neres, J.; Labello, N. P.; Somu, R. V.; Xing, C.; Barry, C. E., III; Aldrich, C. C. J. Med. Chem. 2008, 51, 7495–7507. doi:10.1021/jm8008037 |
| 15. | Ferreira, S. B.; Sodero, A. C. R.; Cardoso, M. F. C.; Lima, E. S.; Kaiser, C. R.; Silva, F. P., Jr.; Ferreira, V. F. J. Med. Chem. 2010, 53, 2364–2375. doi:10.1021/jm901265h |
| 16. | Van Poecke, S.; Negri, A.; Gago, F.; Van Daele, I.; Solaroli, N.; Karlsson, A.; Balzarini, J.; Van Calenbergh, S. J. Med. Chem. 2010, 53, 2902–2912. doi:10.1021/jm901532h |
| 17. | Li, Y.; Shen, M.; Zhang, Z.; Luo, J.; Pan, X.; Lu, X.; Long, H.; Wen, D.; Zhang, F.; Leng, F.; Li, Y.; Tu, Z.; Ren, X.; Ding, K. J. Med. Chem. 2012, 55, 10033–10046. doi:10.1021/jm301188x |
| 18. | Testa, C.; Scrima, M.; Grimaldi, M.; D’Ursi, A. M.; Dirain, M. L.; Lubin-Germain, N.; Singh, A.; Haskell-Luevano, C.; Chorev, M.; Rovero, P.; Papini, A. M. J. Med. Chem. 2014, 57, 9424–9434. doi:10.1021/jm501027w |
| 19. | Brik, A.; Muldoon, J.; Lin, Y.-C.; Elder, J. H.; Goodsell, D. S.; Olson, A. J.; Fokin, V. V.; Sharpless, K. B.; Wong, C.-H. ChemBioChem 2003, 4, 1246–1248. doi:10.1002/cbic.200300724 |
| 30. | Aucagne, V.; Leigh, D. A. Org. Lett. 2006, 8, 4505–4507. doi:10.1021/ol061657d |
| 31. | Fletcher, J. T.; Bumgarner, B. J.; Engels, N. D.; Skoglund, D. A. Organometallics 2008, 27, 5430–5433. doi:10.1021/om800768k |
| 32. | Fletcher, J. T.; Walz, S. E.; Keeney, M. E. Tetrahedron Lett. 2008, 49, 7030–7032. doi:10.1016/j.tetlet.2008.09.136 |
| 33. | Beltrán, E.; Serrano, J. L.; Sierra, T.; Giménez, R. Org. Lett. 2010, 12, 1404–1407. doi:10.1021/ol902900y |
| 28. | Clayden, J.; Donnard, M.; Lefranc, J.; Tetlow, D. J. Chem. Commun. 2011, 47, 4624–4639. doi:10.1039/C1CC00049G |
| 1. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 2. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 29. | Vardanyan, R. S.; Hruby, V. J. Future Med. Chem. 2014, 6, 385–412. doi:10.4155/fmc.13.215 |
| 20. | Blay, G.; Monleon, A.; Pedro, J. R. Curr. Org. Chem. 2009, 13, 1498–1539. doi:10.2174/138527209789177734 |
| 21. | Yoo, W. J.; Zhao, L.; Li, C.-J. Aldrichimica Acta 2011, 44, 43–51. |
| 24. | Pierce, C. J.; Larsen, C. H. Green Chem. 2012, 14, 2672–2676. doi:10.1039/c2gc35713e |
| 7. | Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s |
| 7. | Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s |
| 8. | Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230 |
| 6. | Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m |
| 22. | Pereshivko, O. P.; Peshkov, V. A.; Van der Eycken, E. V. Org. Lett. 2010, 12, 2638–2641. doi:10.1021/ol1008312 |
| 23. | Cheng, M.; Zhang, Q.; Hu, X.-Y.; Li, B.-G.; Ji, J.-X.; Chan, A. S. C. Adv. Synth. Catal. 2011, 353, 1274–1278. doi:10.1002/adsc.201000914 |
| 24. | Pierce, C. J.; Larsen, C. H. Green Chem. 2012, 14, 2672–2676. doi:10.1039/c2gc35713e |
| 25. | Palchak, Z. L.; Lussier, D. J.; Pierce, C. J.; Larsen, C. H. Green Chem. 2015, 17, 1802–1810. doi:10.1039/c4gc02318h |
| 26. | Pierce, C. J.; Nguyen, M.; Larsen, C. H. Angew. Chem., Int. Ed. 2012, 51, 12289–12292. doi:10.1002/anie.201206674 |
| 2. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 34. | Escamilla, I. V.; Ramos, L. F. R.; Escalera, J. S.; Hernandez, A. A. J. Mex. Chem. Soc. 2011, 55, 133–136. |
| 35. | Heuft, M. A.; Collins, S. K.; Yap, G. P. A.; Fallis, A. G. Org. Lett. 2001, 3, 2883–2886. doi:10.1021/ol016414u |
| 6. | Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m |
| 7. | Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s |
| 8. | Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230 |
| 25. | Palchak, Z. L.; Lussier, D. J.; Pierce, C. J.; Larsen, C. H. Green Chem. 2015, 17, 1802–1810. doi:10.1039/c4gc02318h |
| 1. | Meldal, M.; Tornøe, C. W. Chem. Rev. 2008, 108, 2952–3015. doi:10.1021/cr0783479 |
| 2. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 6. | Brak, K.; Doyle, P. S.; McKerrow, J. H.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 6404–6410. doi:10.1021/ja710254m |
| 7. | Patterson, A. W.; Wood, W. J. L.; Hornsby, M.; Lesley, S.; Spraggon, G.; Ellman, J. A. J. Med. Chem. 2006, 49, 6298–6307. doi:10.1021/jm060701s |
| 8. | Wood, W. J. L.; Patterson, A. W.; Tsuruoka, H.; Jain, R. K.; Ellman, J. A. J. Am. Chem. Soc. 2005, 127, 15521–15527. doi:10.1021/ja0547230 |
| 39. | Shinde, S. S.; Chi, H. M.; Lee, B. S.; Chi, D. Y. Tetrahedron Lett. 2009, 50, 6654–6657. doi:10.1016/j.tetlet.2009.09.064 |
| 40. | Asano, K.; Matsubara, S. Org. Lett. 2010, 12, 4988–4991. doi:10.1021/ol101990d |
| 41. | Rossy, C.; Majimel, J.; Delapierre, M. T.; Fouquet, E.; Felpin, F.-X. J. Organomet. Chem. 2014, 755, 78–85. doi:10.1016/j.jorganchem.2014.01.006 |
| 38. | Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M |
| 2. | Hein, J. E.; Fokin, V. V. Chem. Soc. Rev. 2010, 39, 1302–1315. doi:10.1039/b904091a |
| 36. | Meyet, C. E.; Pierce, C. J.; Larsen, C. H. Org. Lett. 2012, 14, 964–967. doi:10.1021/ol2029492 |
© 2015 Palchak et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)