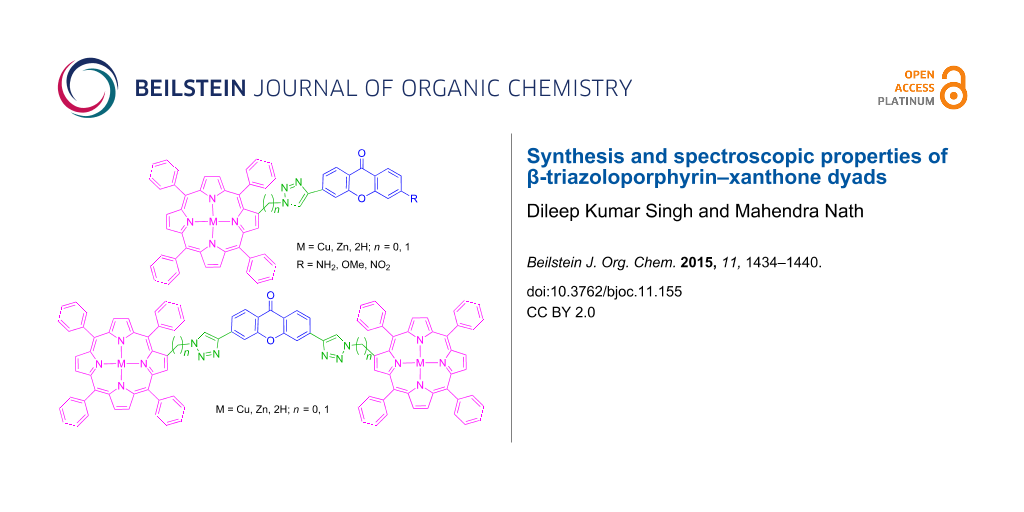
Abstract
A novel series of β-triazoloporphyrin–xanthone conjugates and xanthone-bridged β-triazoloporphyrin dyads has been synthesized in moderate to good yields through Cu(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction of copper(II) 2-azido-5,10,15,20-tetraphenylporphyrin or zinc(II) 2-azidomethyl-5,10,15,20-tetraphenylporphyrin with various alkyne derivatives of xanthones in DMF containing CuSO4 and ascorbic acid at 80 °C. Furthermore, these metalloporphyrins underwent demetalation under acidic conditions to afford the corresponding free-base porphyrins in good to excellent yields. After successful spectroscopic characterization, these porphyrins have been evaluated for their photophysical properties. The preliminary results revealed a bathochromic shift in the UV–vis and fluorescence spectra of these porphyrin–xanthone dyads.

Graphical Abstract
Introduction
In the past few decades, porphyrin macrocycles have emerged as a unique class of heterocyclic compounds and as most attractive building blocks for supramolecular arrays. These molecules provide various desirable properties such as a rigid planar geometry, highly conjugated structures, intense electronic absorption and emission properties and small HOMO–LUMO energy gaps [1,2]. Additionally, the electronic properties of porphyrins can be modulated by introducing diverse functionalities on their periphery or changing the metal ions in the porphyrin core [3,4]. Due to the high thermal stability and extended π-electron system, porphyrins are very useful for the construction of molecular switches [5,6] and other organic photoelectric materials [7,8]. In addition, porphyrins are potentially used as photosensitizers in photodynamic therapy to treat various types of tumors [9,10]. In recent years, many hybrid molecules including porphyrin–C60 [11], porphyrin–quinones [12] and porphyrin–cyclodextrin [13] conjugates were synthesized and evaluated for their photophysical properties. These compounds have successfully demonstrated the occurrence of an excited electron transfer from the porphyrin subunit to the attached acceptor moieties. On the other hand, some photoinduced electron transfer systems such as porphyrin–tetrathiafulvalenes [14], porphyrin–cyanines [15], porphyrin–carotenes [16], porphyrin–arene diimide [17] and porphyrin–fluorocene or rhodamine [18] have also been synthesized in which the porphyrin unit acts as an electron acceptor.
Recently, the 1,2,3-triazole scaffold has been successfully employed to connect porphyrins with diverse functionalities such as quinolone [19], ferrocene [20], carbohydrate [21] and fullerene [22] through a copper(I)-catalyzed Huisgen–Sharpless–Meldal 1,3-dipolar cycloaddition reaction [23,24]. Some of these triazolo-bridged porphyrin dyads have shown an efficient intramolecular energy transfer between the porphyrin part and the attached subunit. Moreover, the 1,4-disubstituted triazoles are found to be very useful for various applications including modification of cell surfaces [25], synthesis of new glycoproteins [26], specific labeling of virus particles [27] and synthesis of diporphyrin analogues [20,28-30].
Xanthene-9H-ones are an important class of oxygen-containing heterocycles and are mainly found as secondary metabolites in higher plants and microorganisms. The naturally occurring and synthetic xanthones possess diverse pharmacological activities [31-34] including anti-oxidative [35], antihypertensive [36], anti-inflammatory [37] and antiplatelet agents [38]. They have also been used as fluorophores and exhibited good fluorescence properties when attached to a triazole ring [39,40]. Owing to the biological significance of porphyrins, 1,2,3-triazoles and xanthones, it was contemplated to incorporate these heterocyclic scaffolds in a single molecular framework to construct novel β-triazolo–porphyrin–xanthone conjugates and their diporphyrin analogues which may prove useful as photosensitizers for photodynamic therapy applications.
Results and Discussion
In continuation of our interest to develop new synthetic methods for porphyrin analogues [41-46], we focus our attention on the construction of various novel β-substituted triazoloporphyrin–xanthone conjugates. For the preparation of these molecules, the alkynes, 3-amino-6-ethynylxanthen-9-one (3), 3-ethynyl-6-nitroxanthen-9-one (4), and 3-ethynyl-6-methoxyxanthen-9-one (5) were synthesized by using the literature procedures [39,40,47-49]. In addition, copper(II) 2-azido-5,10,15,20-tetraphenylporphyrin (1) [50] was synthesized in good yield after the treatment of copper(II) 2-amino-5,10,15,20-tetraphenylporphyrin with NaNO2 in THF in the presence of H2SO4 followed by the reaction with NaN3 [28]. The corresponding zinc(II) 2-azidomethyl-5,10,15,20-tetraphenylporphyrin (2) was prepared in 85% yield from zinc(II) 2-hydroxymethyl-5,10,15,20-tetraphenylporphyrin after the reaction with NaN3 and BF3·Et2O in 1,4-dioxane at 80 °C for 2 hours [51].
Initially, the copper(II) and zinc(II) derivatives of β-triazoloporphyrin–xanthone conjugates 6a,d,g and 7a,c were synthesized in 60–76% yields through a copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction between copper(II) 2-azido-5,10,15,20-tetraphenylporphyrin (1) or zinc(II) 2-azidomethyl-5,10,15,20-tetraphenylporphyrin (2) with ethynyl-substituted xanthones 3, 4 or 5, using copper sulfate and ascorbic acid in DMF at 80 °C (Scheme 1). Further, the demetalation of the copper(II) porphyrins 6a,d,g with conc. H2SO4 at 0 °C and zinc(II) derivatives 7a,c with conc. HCl at 25 °C and neutralization with 5% aq NaHCO3 afforded the corresponding free-base porphyrins 6b,e,h and 7b,d, respectively in 78–83% yields. In addition, the zinc porphyrins 6c,f,i and copper porphyrin 7e were prepared in 90–93% yields from free-base porphyrins 6b,e,h and 7d by their treatment with zinc acetate and copper acetate respectively, in a CHCl3–MeOH mixture at room temperature (Scheme 1).
![[1860-5397-11-155-i1]](/bjoc/content/inline/1860-5397-11-155-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of β-substituted triazoloporphyrin–xanthone conjugates 6a–i and 7a–e.
Scheme 1: Synthesis of β-substituted triazoloporphyrin–xanthone conjugates 6a–i and 7a–e.
Further, this methodology was extended to the preparation of symmetrical xanthone-bridged triazolodiporphyrins (12a,b and 13a–c). For the synthesis of these compounds, 3,6-diethynylxanthen-9-one (11) was synthesized in three steps from 3,6-dihydroxyxanthen-9-one (8) as a starting material. The first step involved the reaction of xanthone (8) with triflic anhydride in CH2Cl2 containing pyridine at 0 °C to afford 3,6-di-OTf-xanthone [52] (9). Subsequent Sonogashira coupling with trimethylsilylacetylene followed by the deprotection of the trimethylsilyl group in the presence of aqueous NaOH at room temperature produced the desired product 3,6-diethynylxanthen-9-one (11) in 81% yield (Scheme 2) [39].
![[1860-5397-11-155-i2]](/bjoc/content/inline/1860-5397-11-155-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 3,6-diethynyl-xanthen-9-one (11).
Scheme 2: Synthesis of 3,6-diethynyl-xanthen-9-one (11).
Finally, the copper and zinc derivatives of the xanthone-bridged triazolodiporphyrins (12a and 13a) were synthesized in good yields by the “click reaction” of 3,6-diethynylxanthen-9-one (11) with copper and zinc azidoporphyrins, respectively. Further, the free-base diporphyrins 12b and 13b were obtained in 81% and 84% yields, respectively after the treatment of 12a with conc. H2SO4 at 0 °C and 13a with conc. HCl at 25 °C in chloroform. In addition, the copper(II) bisporphyrin 13c was obtained in almost quantitative yield by reacting the free-base porphyrin 13b with Cu(OAc)2 in a CHCl3–MeOH mixture at room temperature (Scheme 3).
![[1860-5397-11-155-i3]](/bjoc/content/inline/1860-5397-11-155-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of xanthone-bridged triazolo-bisporphyrins 12a,b and 13a–c.
Scheme 3: Synthesis of xanthone-bridged triazolo-bisporphyrins 12a,b and 13a–c.
All the newly synthesized products were well purified by column chromatography and characterized by NMR, IR, UV–vis and mass spectral data in addition to elemental analyses. The 1H NMR spectrum of xanthone 10 showed a characteristic singlet of 18 protons at δ 0.28 ppm due to trimethylsilyl group which revealed the formation of compound 10. A characteristic singlet of alkyne protons in xanthones 4 and 11 were found at δ 4.69 ppm and δ 4.66 ppm, respectively. In addition, the IR spectra of all the three xanthones 4, 10 and 11 showed a C≡C-bond stretching at around 2100–2157 cm−1 which confirms the attachment of the alkyne moiety to the xanthone core.
In the 1H NMR spectra, 7 β-pyrrolic protons of porphyrin–xanthone conjugates (6b,c,e,f,h,i and 7a–d) and 14 β-pyrrolic protons of bisporphyrins (12b and 13a,b) were found in the down-field region between δ 8.44–9.07 ppm as either singlet, doublet or multiplet. The 20 meso-phenyl protons of compounds 6b,c,e,f,h,i and 7a–d and the 40 meso-phenyl protons of bisporphyrins 12b and 13a,b were assigned to doublets and multiplets in the region between δ 6.99–8.26 ppm. In addition, 6 xanthone protons in the case of all porphyrin–xanthone dyads were found between δ 6.47–8.41 ppm as doublet, double doublet or multiplet. A characteristic singlet of two protons at around δ −2.7 ppm was assigned for internal NH protons of the porphyrin core in the case of all the free-base porphyrins. In porphyrins 7a–d and 13a,b, a singlet for the methylene protons was observed between δ 5.76–5.82 ppm. Similarly, a singlet of three protons due to the presence of methoxy group was observed in the upfield region at δ ~3.9 ppm in porphyrins 6h,i and 7c,d. The two protons for the amino group in porphyrins 6b and 6c appeared as broad singlet at δ 4.33 and 4.89 ppm, respectively, whereas these protons are down-field shifted to δ ~6.52 ppm in the case of porphyrins 7a and 7b. The IR spectra of the compounds containing amino groups showed a NH2 bond stretching band between 3348–3433 cm−1, whereas all the free-base porphyrins showed absorptions between 3322–3327 cm−1 due to the internal NH groups of the porphyrin core. In addition, a strong band was observed at 1602–1654 cm−1 due to the presence of the carbonyl groups in the porphyrin–xanthone dyads. The mass spectra of all compounds further supported the assigned structures by showing a molecular ion peak either as [M + H]+ or [M + Na]+ or [M + K]+.
The electronic absorption spectra of all the newly prepared porphyrin–xanthone conjugates were taken in CHCl3 (1.5 × 10−6 M) at room temperature. The copper(II) β-triazoloporphyrin–xanthone conjugates 6a,d,g and 12a exhibited a typical Soret band at around 420–421 nm and two Q-bands at ~544 and 578 nm, which were found to be red-shifted by about 3–5 nm as compared to the starting porphyrin Cu-TPP (Soret band at 416 nm and Q-bands at 541 and 574 nm). Further, the free-base β-triazoloporphyrin–xanthone conjugates 6b,e,h and 12b display their Soret bands at around 424–425 nm and four Q-bands around ~521, 556, 596 and 652 nm which were also red-shifted by about 5–6 nm when compared to the TPP (Soret band at 419 nm and Q-bands at 516, 551, 590 and 646 nm). In addition, zinc(II) β-triazoloporphyrin–xanthone dyads 6c,f,i exhibited their Soret bands between 430–431 nm and two Q-bands at ~561 and 602 nm which were found to be red-shifted by about 5–6 nm as compared to the Zn-TPP (Soret band at 425 nm and Q-bands at 555 and 597 nm). In contrast, zinc(II) β-triazolomethylporphyrin–xanthone dyads (7a,c,13a), free-base β-triazolomethylporphyrin–xanthone dyads (7b,d,13b) and copper(II) β-triazolomethylporphyrin–xanthone dyads (7e,13c) exhibited only a 2 nm red shift in their Soret and Q-bands when compared with the Zn-TPP, TPP and Cu-TPP, respectively. Thus, the electronic absorption spectra of β-triazoloporphyrin–xanthone conjugates exhibited 2–4 nm red shifts in their Soret and Q-bands as compared to the β-triazolomethylporphyrin–xanthone conjugates. In addition, the absorption intensity of symmetrical xanthone-bridged β-triazolodiporphyrins (12a,b and 13a–c) was quite larger than their corresponding monoporphyrin–xanthone conjugates. The electronic absorption spectra of copper porphyrins (Cu-TPP, 6a, 7e, 12a and 13c), free-base porphyrins (TPP, 6b, 7b, 12b and 13b), and zinc porphyrins (Zn-TPP, 6c, 6f, 7a and 13a) are shown in Figure 1a–c.
![[1860-5397-11-155-1]](/bjoc/content/figures/1860-5397-11-155-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Electronic absorption spectra of Cu-TPP, 6a, 7e, 12a and 13c. (b) Electronic absorption spectra of TPP, 6b, 7b, 12b and 13b. (c) Electronic absorption spectra of Zn-TPP, 6c, 6f, 7a and 13a in CHCl3 (1.5 × 10−6 mol L−1) at 298 K and inset shows the expanded UV–vis spectra with Q-bands. (d) Fluorescence spectra of porphyrins TPP, Zn-TPP, 6b,c, 7a,b, and 13a,b in CHCl3 (1.5 × 10−6 mol L−1) at 298 K, λex = 420 nm.
Figure 1: (a) Electronic absorption spectra of Cu-TPP, 6a, 7e, 12a and 13c. (b) Electronic absorption spectra...
Besides the Soret and Q-bands, an additional absorption band was also observed at ~340 nm in the UV–vis spectra of these porphyrin–xanthone conjugates due to the presence of xanthone moiety (Figure 1a–c) which suggests that there is no significant interaction between the xanthone and porphyrin moieties in the ground state [53-55].
The fluorescence spectra of compounds 6b,c, 7a,b, and 13a,b depicted in Figure 1d showed two emission bands of free-base porphyrin–xanthone conjugates between 656–725 nm which are slightly quenched, but red-shifted about 5–10 nm as compared to the TPP (emission bands at 650 and 715 nm). Similarly, the two emission bands of zinc porphyrins were also observed between 607–662 nm and they were also quenched and red-shifted by about 5–10 nm as compared to the Zn-TPP (emission bands at 602 and 651 nm). In contrast, the copper porphyrins did not show any significant emission due to the paramagnetic nature of the Cu(II) ions [56].
Conclusion
In summary, we have successfully synthesized and characterized two new alkyne-substituted xanthones, 3-ethynyl-6-nitroxanthen-9-one and 3,6-diethynylxanthen-9-one. In addition, a new series of various β-triazole-linked porphyrin–xanthone conjugates and xanthone-bridged triazoloporphyrin dyads were synthesized through click chemistry in moderate to good yields. The preliminary photophysical evaluation of these π-conjugated molecules revealed a bathochromic shift in their electronic absorption and fluorescence spectra as compared to the meso-tetraarylporphyrins. These results are significantly encouraging and henceforth may be useful for the development of new porphyrin materials for various applications including photosensitizers for photodynamic applications.
Acknowledgements
This work has been supported by University of Delhi, India under the scheme to strengthen R&D Doctoral Research Program. We are thankful to CIF, University of Delhi and SAIF, CDRI, Lucknow, India for providing NMR and mass data. Dileep Kumar Singh is grateful to CSIR, New Delhi, India for providing SRF.
References
-
Perepichka, D. F.; Bryce, M. R. Angew. Chem., Int. Ed. 2005, 44, 5370–5373. doi:10.1002/anie.200500413
Return to citation in text: [1] -
Ferreira, J. A. B.; Serra, V. V.; Sánchez-Coronilla, A.; Pires, S. M. G.; Faustino, M. A. F.; Silva, A. M. S.; Neves, M. G. P. M. S.; Cavaleiro, J. A. S.; Costa, S. M. B. Chem. Commun. 2013, 49, 8809–8811. doi:10.1039/c3cc44925d
Return to citation in text: [1] -
Liao, M.-S.; Scheiner, S. J. Chem. Phys. 2002, 117, 205–219. doi:10.1063/1.1480872
Return to citation in text: [1] -
Yang, L.; Fang, W.; Zhang, Y. Chem. Commun. 2012, 48, 3842–3844. doi:10.1039/c2cc31016c
Return to citation in text: [1] -
Straight, S. D.; Andréasson, J.; Kodis, G.; Moore, A. L.; Moore, T. A.; Gust, D. J. Am. Chem. Soc. 2005, 127, 2717–2724. doi:10.1021/ja044128i
Return to citation in text: [1] -
Wallin, S.; Monnereau, C.; Blart, E.; Gankou, J.-R.; Odobel, F.; Hammarström, L. J. Phys. Chem. A 2010, 114, 1709–1721. doi:10.1021/jp907824d
Return to citation in text: [1] -
Li, L.-L.; Diau, E. W.-G. Chem. Soc. Rev. 2013, 42, 291–304. doi:10.1039/C2CS35257E
Return to citation in text: [1] -
Li, X.; Liu, L.; Kang, S.-Z.; Mu, J.; Li, G. Appl. Surf. Sci. 2011, 257, 5950–5956. doi:10.1016/j.apsusc.2011.01.058
Return to citation in text: [1] -
Vicente, M. G. H. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 175–194. doi:10.2174/1568011013354769
Return to citation in text: [1] -
Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R. K. Chem. Soc. Rev. 2011, 40, 340–362. doi:10.1039/B915149B
Return to citation in text: [1] -
Fungo, F.; Milanesio, M. E.; Durantini, E. N.; Otero, L.; Dittrich, T. J. Mater. Chem. 2007, 17, 2107–2112. doi:10.1039/b617944d
Return to citation in text: [1] -
Banala, S.; Rühl, T.; Wurst, K.; Kräutler, B. Angew. Chem., Int. Ed. 2009, 48, 599–603. doi:10.1002/anie.200804143
Return to citation in text: [1] -
Králová, J.; Kejík, Z.; Bříza, T.; Poučková, P.; Král, A.; Martásek, P.; Král, V. J. Med. Chem. 2010, 53, 128–138. doi:10.1021/jm9007278
Return to citation in text: [1] -
Sadaike, S.-i.; Takimiya, K.; Aso, Y.; Otsubo, T. Tetrahedron Lett. 2003, 44, 161–165. doi:10.1016/S0040-4039(02)02474-7
Return to citation in text: [1] -
Ogawa, K.; Nagatsuka, Y.; Kobuke, Y. J. Porphyrins Phthalocyanines 2011, 15, 678–685. doi:10.1142/S1088424611003616
Return to citation in text: [1] -
Fungo, F.; Otero, L.; Durantini, E.; Thompson, W. J.; Silber, J. J.; Moore, T. A.; Moore, A. L.; Gust, D.; Sereno, L. Phys. Chem. Chem. Phys. 2003, 5, 469–475. doi:10.1039/b209694c
Return to citation in text: [1] -
Redmore, N. P.; Rubtsov, I. V.; Therien, M. J. J. Am. Chem. Soc. 2003, 125, 8769–8778. doi:10.1021/ja021278p
Return to citation in text: [1] -
Sun, X.; Li, D.; Chen, G.; Zhang, J. Dyes Pigm. 2006, 71, 118–122. doi:10.1016/j.dyepig.2005.06.008
Return to citation in text: [1] -
da C. Santos, F.; Cunha, A. C.; de Souza, M. C. B. V.; Tomé, A. C.; Neves, M. G. P. M. S.; Ferreira, V. F.; Cavaleiro, J. A. S. Tetrahedron Lett. 2008, 49, 7268–7270. doi:10.1016/j.tetlet.2008.10.024
Return to citation in text: [1] -
Shetti, V. S.; Ravikanth, M. Eur. J. Org. Chem. 2010, 494–508. doi:10.1002/ejoc.200901070
Return to citation in text: [1] [2] -
Locos, O. B.; Heindl, C. C.; Corral, A.; Senge, M. O.; Scanlan, E. M. Eur. J. Org. Chem. 2010, 1026–1028. doi:10.1002/ejoc.200901292
Return to citation in text: [1] -
de Miguel, G.; Wielopolski, M.; Schuster, D. I.; Fazio, M. A.; Lee, O. P.; Haley, C. K.; Ortiz, A. L.; Echegoyen, L.; Clark, T.; Guldi, D. M. J. Am. Chem. Soc. 2011, 133, 13036–13054. doi:10.1021/ja202485s
Return to citation in text: [1] -
Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5
Return to citation in text: [1] -
Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128–1137. doi:10.1016/S1359-6446(03)02933-7
Return to citation in text: [1] -
Deiters, A.; Cropp, T. A.; Mukherji, M.; Chin, J. W.; Anderson, J. C.; Schultz, P. G. J. Am. Chem. Soc. 2003, 125, 11782–11783. doi:10.1021/ja0370037
Return to citation in text: [1] -
Chan, T. R.; Hilgraf, R.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2004, 6, 2853–2855. doi:10.1021/ol0493094
Return to citation in text: [1] -
Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e
Return to citation in text: [1] -
Shen, D.-M.; Liu, C.; Chen, Q.-Y. Eur. J. Org. Chem. 2007, 1419–1422. doi:10.1002/ejoc.200601046
Return to citation in text: [1] [2] -
Séverac, M.; Le Pleux, L.; Scarpaci, A.; Blart, E.; Odobel, F. Tetrahedron Lett. 2007, 48, 6518–6522. doi:10.1016/j.tetlet.2007.07.049
Return to citation in text: [1] -
Punidha, S.; Sinha, J.; Kumar, A.; Ravikanth, M. J. Org. Chem. 2008, 73, 323–326. doi:10.1021/jo702018s
Return to citation in text: [1] -
Vieira, L. M. M.; Kijjoa, A. Curr. Med. Chem. 2005, 12, 2413–2446. doi:10.2174/092986705774370682
Return to citation in text: [1] -
Sousa, M. E.; Pinto, M. M. M. Curr. Med. Chem. 2005, 12, 2447–2479. doi:10.2174/092986705774370736
Return to citation in text: [1] -
Pinto, M. M. M.; Sousa, M. E.; Nascimento, M. S. J. Curr. Med. Chem. 2005, 12, 2517–2538. doi:10.2174/092986705774370691
Return to citation in text: [1] -
Fotie, J.; Bohle, D. S. Anti-Infect. Agents Med. Chem. 2006, 5, 15–31. doi:10.2174/187152106774755563
Return to citation in text: [1] -
Blanco-Ayala, T.; Lugo-Huitrón, R.; Serrano-López, E. M.; Reyes-Chilpa, R.; Rangel-López, E.; Pineda, B.; Medina-Campos, O. N.; Sánchez-Chapul, L.; Pinzón, E.; Cristina, T.-S.; Silva-Adaya, D.; Pedraza-Chaverrí, J.; Ríos, C.; de la Cruz, V. P.; Torres-Ramos, M. BMC Complementary Altern. Med. 2013, 13, 262–277. doi:10.1186/1472-6882-13-262
Return to citation in text: [1] -
Wang, L.-W.; Kang, J.-J.; Chen, I.-J.; Teng, C.-M.; Lin, C.-N. Bioorg. Med. Chem. 2002, 10, 567–572. doi:10.1016/S0968-0896(01)00315-7
Return to citation in text: [1] -
Librowski, T.; Czarnecki, R.; Czekaj, T.; Marona, H. Medicina (Kaunas) 2005, 41, 54–58.
Return to citation in text: [1] -
Correia-da-Silva, M.; Sousa, E.; Duarte, B.; Marques, F.; Carvalho, F.; Cunha-Ribeiro, L. M.; Pinto, M. M. M. J. Med. Chem. 2011, 54, 5373–5384. doi:10.1021/jm2006589
Return to citation in text: [1] -
Li, J.; Hu, M.; Yao, S. Q. Org. Lett. 2009, 11, 3008–3011. doi:10.1021/ol9010344
Return to citation in text: [1] [2] [3] -
Li, J.; Yao, S. Q. Org. Lett. 2009, 11, 405–408. doi:10.1021/ol802700w
Return to citation in text: [1] [2] -
Singh, D. K.; Nath, M. Org. Biomol. Chem. 2015, 13, 1836–1845. doi:10.1039/C4OB02370F
Return to citation in text: [1] -
Singh, D. K.; Nath, M. Beilstein J. Org. Chem. 2014, 10, 808–813. doi:10.3762/bjoc.10.76
Return to citation in text: [1] -
Sharma, S.; Nath, M. Beilstein J. Org. Chem. 2013, 9, 496–502. doi:10.3762/bjoc.9.53
Return to citation in text: [1] -
Bhatt, R. K.; Singh, D. K.; Nath, M. J. Indian Chem. Soc. 2013, 90, 1493–1496.
Return to citation in text: [1] -
Sharma, S.; Nath, M. New J. Chem. 2011, 35, 1630–1639. doi:10.1039/c1nj20248k
Return to citation in text: [1] -
Sharma, S.; Nath, M. Dyes Pigm. 2012, 92, 1241–1249. doi:10.1016/j.dyepig.2011.07.022
Return to citation in text: [1] -
Chang, Y.-T.; Ahn, Y.-H. Combinatorial fluorescent rosamine library and uses thereof. U.S. Patent. Appl. US 2008/0124751 A1, May 29, 2008.
Return to citation in text: [1] -
Šebej, P.; Wintner, J.; Müller, P.; Slanina, T.; Al Anshori, J.; Antony, L. A. P.; Klán, P.; Wirz, J. J. Org. Chem. 2013, 78, 1833–1843. doi:10.1021/jo301455n
Return to citation in text: [1] -
Piazzi, L.; Belluti, F.; Bisi, A.; Gobbi, S.; Rizzo, S.; Bartolini, M.; Andrisano, V.; Recanatini, M.; Rampa, A. Bioorg. Med. Chem. 2007, 15, 575–585. doi:10.1016/j.bmc.2006.09.026
Return to citation in text: [1] -
Fenwick, D. Azide substituted porphyrins with crosslinking applications for use in photodynamic therapy. Ph.D. Thesis, The University of British Columbia, Canada, 2002.
Return to citation in text: [1] -
Garg, A. Synthesis and spectroscopic properties of novel β-functionalized 5,10,15,20-tetraarylporphyrins and diporphyrin analogues. Ph.D. Thesis, University of Delhi, Delhi, India, 2012.
Return to citation in text: [1] -
Wu, L.; Burgess, K. Org. Lett. 2008, 10, 1779–1782. doi:10.1021/ol800526s
Return to citation in text: [1] -
Tao, M.; Liu, L.; Liu, D.; Zhou, X. Dyes Pigm. 2010, 85, 21–26. doi:10.1016/j.dyepig.2009.09.012
Return to citation in text: [1] -
Lazarides, T.; Charalambidis, G.; Vuillamy, A.; Réglier, M.; Klontzas, E.; Froudakis, G.; Kuhri, S.; Guldi, D. M.; Coutsolelos, A. G. Inorg. Chem. 2011, 50, 8926–8936. doi:10.1021/ic201052k
Return to citation in text: [1] -
Zhou, X.; Liu, D.; Wang, T.; Hu, X.; Guo, J.; Weerasinghe, K. C.; Wang, L.; Li, W. J. Photochem. Photobiol., A: Chem. 2014, 274, 57–63. doi:10.1016/j.jphotochem.2013.09.018
Return to citation in text: [1] -
Szintay, G.; Horváth, A.; Grampp, G. J. Photochem. Photobiol., A 1999, 126, 83–89. doi:10.1016/S1010-6030(99)00130-6
Return to citation in text: [1]
| 39. | Li, J.; Hu, M.; Yao, S. Q. Org. Lett. 2009, 11, 3008–3011. doi:10.1021/ol9010344 |
| 40. | Li, J.; Yao, S. Q. Org. Lett. 2009, 11, 405–408. doi:10.1021/ol802700w |
| 41. | Singh, D. K.; Nath, M. Org. Biomol. Chem. 2015, 13, 1836–1845. doi:10.1039/C4OB02370F |
| 42. | Singh, D. K.; Nath, M. Beilstein J. Org. Chem. 2014, 10, 808–813. doi:10.3762/bjoc.10.76 |
| 43. | Sharma, S.; Nath, M. Beilstein J. Org. Chem. 2013, 9, 496–502. doi:10.3762/bjoc.9.53 |
| 44. | Bhatt, R. K.; Singh, D. K.; Nath, M. J. Indian Chem. Soc. 2013, 90, 1493–1496. |
| 45. | Sharma, S.; Nath, M. New J. Chem. 2011, 35, 1630–1639. doi:10.1039/c1nj20248k |
| 46. | Sharma, S.; Nath, M. Dyes Pigm. 2012, 92, 1241–1249. doi:10.1016/j.dyepig.2011.07.022 |
| 39. | Li, J.; Hu, M.; Yao, S. Q. Org. Lett. 2009, 11, 3008–3011. doi:10.1021/ol9010344 |
| 40. | Li, J.; Yao, S. Q. Org. Lett. 2009, 11, 405–408. doi:10.1021/ol802700w |
| 47. | Chang, Y.-T.; Ahn, Y.-H. Combinatorial fluorescent rosamine library and uses thereof. U.S. Patent. Appl. US 2008/0124751 A1, May 29, 2008. |
| 48. | Šebej, P.; Wintner, J.; Müller, P.; Slanina, T.; Al Anshori, J.; Antony, L. A. P.; Klán, P.; Wirz, J. J. Org. Chem. 2013, 78, 1833–1843. doi:10.1021/jo301455n |
| 49. | Piazzi, L.; Belluti, F.; Bisi, A.; Gobbi, S.; Rizzo, S.; Bartolini, M.; Andrisano, V.; Recanatini, M.; Rampa, A. Bioorg. Med. Chem. 2007, 15, 575–585. doi:10.1016/j.bmc.2006.09.026 |
| 1. | Perepichka, D. F.; Bryce, M. R. Angew. Chem., Int. Ed. 2005, 44, 5370–5373. doi:10.1002/anie.200500413 |
| 2. | Ferreira, J. A. B.; Serra, V. V.; Sánchez-Coronilla, A.; Pires, S. M. G.; Faustino, M. A. F.; Silva, A. M. S.; Neves, M. G. P. M. S.; Cavaleiro, J. A. S.; Costa, S. M. B. Chem. Commun. 2013, 49, 8809–8811. doi:10.1039/c3cc44925d |
| 9. | Vicente, M. G. H. Curr. Med. Chem. - Anti-Cancer Agents 2001, 1, 175–194. doi:10.2174/1568011013354769 |
| 10. | Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R. K. Chem. Soc. Rev. 2011, 40, 340–362. doi:10.1039/B915149B |
| 20. | Shetti, V. S.; Ravikanth, M. Eur. J. Org. Chem. 2010, 494–508. doi:10.1002/ejoc.200901070 |
| 56. | Szintay, G.; Horváth, A.; Grampp, G. J. Photochem. Photobiol., A 1999, 126, 83–89. doi:10.1016/S1010-6030(99)00130-6 |
| 7. | Li, L.-L.; Diau, E. W.-G. Chem. Soc. Rev. 2013, 42, 291–304. doi:10.1039/C2CS35257E |
| 8. | Li, X.; Liu, L.; Kang, S.-Z.; Mu, J.; Li, G. Appl. Surf. Sci. 2011, 257, 5950–5956. doi:10.1016/j.apsusc.2011.01.058 |
| 21. | Locos, O. B.; Heindl, C. C.; Corral, A.; Senge, M. O.; Scanlan, E. M. Eur. J. Org. Chem. 2010, 1026–1028. doi:10.1002/ejoc.200901292 |
| 5. | Straight, S. D.; Andréasson, J.; Kodis, G.; Moore, A. L.; Moore, T. A.; Gust, D. J. Am. Chem. Soc. 2005, 127, 2717–2724. doi:10.1021/ja044128i |
| 6. | Wallin, S.; Monnereau, C.; Blart, E.; Gankou, J.-R.; Odobel, F.; Hammarström, L. J. Phys. Chem. A 2010, 114, 1709–1721. doi:10.1021/jp907824d |
| 18. | Sun, X.; Li, D.; Chen, G.; Zhang, J. Dyes Pigm. 2006, 71, 118–122. doi:10.1016/j.dyepig.2005.06.008 |
| 39. | Li, J.; Hu, M.; Yao, S. Q. Org. Lett. 2009, 11, 3008–3011. doi:10.1021/ol9010344 |
| 3. | Liao, M.-S.; Scheiner, S. J. Chem. Phys. 2002, 117, 205–219. doi:10.1063/1.1480872 |
| 4. | Yang, L.; Fang, W.; Zhang, Y. Chem. Commun. 2012, 48, 3842–3844. doi:10.1039/c2cc31016c |
| 19. | da C. Santos, F.; Cunha, A. C.; de Souza, M. C. B. V.; Tomé, A. C.; Neves, M. G. P. M. S.; Ferreira, V. F.; Cavaleiro, J. A. S. Tetrahedron Lett. 2008, 49, 7268–7270. doi:10.1016/j.tetlet.2008.10.024 |
| 53. | Tao, M.; Liu, L.; Liu, D.; Zhou, X. Dyes Pigm. 2010, 85, 21–26. doi:10.1016/j.dyepig.2009.09.012 |
| 54. | Lazarides, T.; Charalambidis, G.; Vuillamy, A.; Réglier, M.; Klontzas, E.; Froudakis, G.; Kuhri, S.; Guldi, D. M.; Coutsolelos, A. G. Inorg. Chem. 2011, 50, 8926–8936. doi:10.1021/ic201052k |
| 55. | Zhou, X.; Liu, D.; Wang, T.; Hu, X.; Guo, J.; Weerasinghe, K. C.; Wang, L.; Li, W. J. Photochem. Photobiol., A: Chem. 2014, 274, 57–63. doi:10.1016/j.jphotochem.2013.09.018 |
| 14. | Sadaike, S.-i.; Takimiya, K.; Aso, Y.; Otsubo, T. Tetrahedron Lett. 2003, 44, 161–165. doi:10.1016/S0040-4039(02)02474-7 |
| 16. | Fungo, F.; Otero, L.; Durantini, E.; Thompson, W. J.; Silber, J. J.; Moore, T. A.; Moore, A. L.; Gust, D.; Sereno, L. Phys. Chem. Chem. Phys. 2003, 5, 469–475. doi:10.1039/b209694c |
| 51. | Garg, A. Synthesis and spectroscopic properties of novel β-functionalized 5,10,15,20-tetraarylporphyrins and diporphyrin analogues. Ph.D. Thesis, University of Delhi, Delhi, India, 2012. |
| 13. | Králová, J.; Kejík, Z.; Bříza, T.; Poučková, P.; Král, A.; Martásek, P.; Král, V. J. Med. Chem. 2010, 53, 128–138. doi:10.1021/jm9007278 |
| 17. | Redmore, N. P.; Rubtsov, I. V.; Therien, M. J. J. Am. Chem. Soc. 2003, 125, 8769–8778. doi:10.1021/ja021278p |
| 12. | Banala, S.; Rühl, T.; Wurst, K.; Kräutler, B. Angew. Chem., Int. Ed. 2009, 48, 599–603. doi:10.1002/anie.200804143 |
| 50. | Fenwick, D. Azide substituted porphyrins with crosslinking applications for use in photodynamic therapy. Ph.D. Thesis, The University of British Columbia, Canada, 2002. |
| 11. | Fungo, F.; Milanesio, M. E.; Durantini, E. N.; Otero, L.; Dittrich, T. J. Mater. Chem. 2007, 17, 2107–2112. doi:10.1039/b617944d |
| 15. | Ogawa, K.; Nagatsuka, Y.; Kobuke, Y. J. Porphyrins Phthalocyanines 2011, 15, 678–685. doi:10.1142/S1088424611003616 |
| 28. | Shen, D.-M.; Liu, C.; Chen, Q.-Y. Eur. J. Org. Chem. 2007, 1419–1422. doi:10.1002/ejoc.200601046 |
| 25. | Deiters, A.; Cropp, T. A.; Mukherji, M.; Chin, J. W.; Anderson, J. C.; Schultz, P. G. J. Am. Chem. Soc. 2003, 125, 11782–11783. doi:10.1021/ja0370037 |
| 22. | de Miguel, G.; Wielopolski, M.; Schuster, D. I.; Fazio, M. A.; Lee, O. P.; Haley, C. K.; Ortiz, A. L.; Echegoyen, L.; Clark, T.; Guldi, D. M. J. Am. Chem. Soc. 2011, 133, 13036–13054. doi:10.1021/ja202485s |
| 23. | Kolb, H. C.; Finn, M. G.; Sharpless, K. B. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. doi:10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5 |
| 24. | Kolb, H. C.; Sharpless, K. B. Drug Discovery Today 2003, 8, 1128–1137. doi:10.1016/S1359-6446(03)02933-7 |
| 37. | Librowski, T.; Czarnecki, R.; Czekaj, T.; Marona, H. Medicina (Kaunas) 2005, 41, 54–58. |
| 38. | Correia-da-Silva, M.; Sousa, E.; Duarte, B.; Marques, F.; Carvalho, F.; Cunha-Ribeiro, L. M.; Pinto, M. M. M. J. Med. Chem. 2011, 54, 5373–5384. doi:10.1021/jm2006589 |
| 35. | Blanco-Ayala, T.; Lugo-Huitrón, R.; Serrano-López, E. M.; Reyes-Chilpa, R.; Rangel-López, E.; Pineda, B.; Medina-Campos, O. N.; Sánchez-Chapul, L.; Pinzón, E.; Cristina, T.-S.; Silva-Adaya, D.; Pedraza-Chaverrí, J.; Ríos, C.; de la Cruz, V. P.; Torres-Ramos, M. BMC Complementary Altern. Med. 2013, 13, 262–277. doi:10.1186/1472-6882-13-262 |
| 36. | Wang, L.-W.; Kang, J.-J.; Chen, I.-J.; Teng, C.-M.; Lin, C.-N. Bioorg. Med. Chem. 2002, 10, 567–572. doi:10.1016/S0968-0896(01)00315-7 |
| 20. | Shetti, V. S.; Ravikanth, M. Eur. J. Org. Chem. 2010, 494–508. doi:10.1002/ejoc.200901070 |
| 28. | Shen, D.-M.; Liu, C.; Chen, Q.-Y. Eur. J. Org. Chem. 2007, 1419–1422. doi:10.1002/ejoc.200601046 |
| 29. | Séverac, M.; Le Pleux, L.; Scarpaci, A.; Blart, E.; Odobel, F. Tetrahedron Lett. 2007, 48, 6518–6522. doi:10.1016/j.tetlet.2007.07.049 |
| 30. | Punidha, S.; Sinha, J.; Kumar, A.; Ravikanth, M. J. Org. Chem. 2008, 73, 323–326. doi:10.1021/jo702018s |
| 31. | Vieira, L. M. M.; Kijjoa, A. Curr. Med. Chem. 2005, 12, 2413–2446. doi:10.2174/092986705774370682 |
| 32. | Sousa, M. E.; Pinto, M. M. M. Curr. Med. Chem. 2005, 12, 2447–2479. doi:10.2174/092986705774370736 |
| 33. | Pinto, M. M. M.; Sousa, M. E.; Nascimento, M. S. J. Curr. Med. Chem. 2005, 12, 2517–2538. doi:10.2174/092986705774370691 |
| 34. | Fotie, J.; Bohle, D. S. Anti-Infect. Agents Med. Chem. 2006, 5, 15–31. doi:10.2174/187152106774755563 |
| 26. | Chan, T. R.; Hilgraf, R.; Sharpless, K. B.; Fokin, V. V. Org. Lett. 2004, 6, 2853–2855. doi:10.1021/ol0493094 |
| 27. | Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192–3193. doi:10.1021/ja021381e |
© 2015 Singh and Nath; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)