Abstract
The synthesis of N-arylated pyrroles and indoles is documented, as well as their functionalization by deprotonative metallation using the base in situ prepared from LiTMP and ZnCl2·TMEDA (1/3 equiv). With N-phenylpyrrole and -indole, the reactions were carried out in hexane containing TMEDA which regioselectively afforded the 2-iodo derivatives after subsequent iodolysis. With pyrroles and indoles bearing N-substituents such as 2-thienyl, 3-pyridyl, 4-methoxyphenyl and 4-bromophenyl, the reactions all took place on the substituent, at the position either adjacent to the heteroatom (S, N) or ortho to the heteroatom-containing substituent (OMe, Br). The CH acidities of the substrates were determined in THF solution using the DFT B3LYP method in order to rationalize the experimental results.

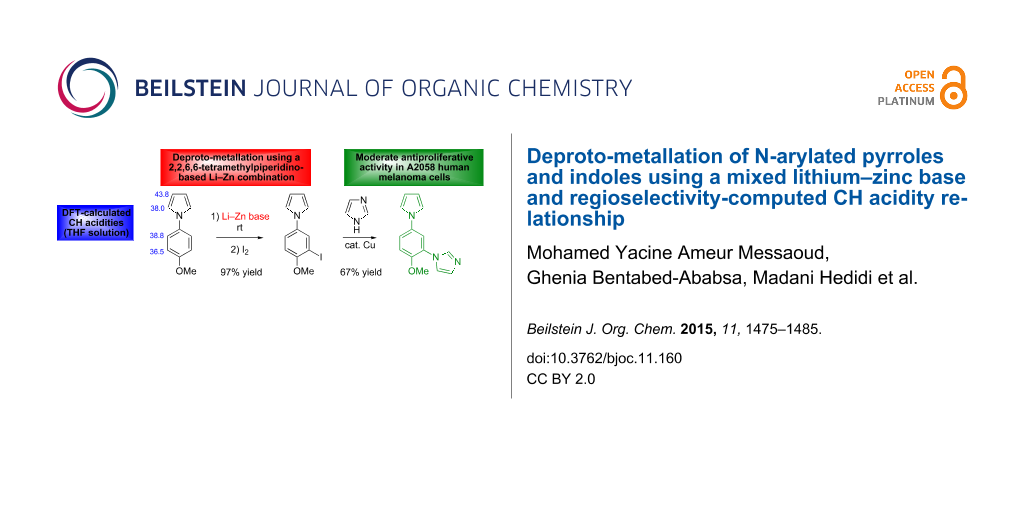
Graphical Abstract
Introduction
Pyrrole occurs in very important natural products such as tetrapyrrolic (linear) bilirubinoids, and (cyclic) porphyrins and corrins, as well as in pharmaceuticals (e.g., pyrrolnitrin, zomepirac) and polymers (e.g., photovoltaic cells). Indole is similarly present in numerous natural products (e.g., tryptophan, melanin, bufotenin, psilocin, indican) including bioactive products (e.g., strychnine, brucine, yohimbine, reserpine, vincamine, ergotamine, lysergic acid), as well as in pharmaceuticals (e.g., indomethacin, iprindole), agrochemicals (e.g., auxins, pyroquilon), and dyes and pigments (e.g., indigo, indocyanines) [1,2].
The deprotonative metallation [3-7] is a valuable tool for the regioselective functionalization of aromatic heterocycles such as pyrroles [8] and indoles [9]. A few examples concern the reaction of N-arylpyrroles and -indoles.
Studies show that, depending on the reaction conditions, two protons of N-phenylpyrrole (at the 2 and 2’ position) can be abstracted by a base. Kinetic conditions employing butyllithium activated by N,N,N’,N’-tetramethylethylenediamine (TMEDA) in diethyl ether lead to the 2,2’-dilithiated product [10,11]. In contrast, monolithiation at the 2 position is noted by using (i) the same base in diethyl ether at room temperature and long reaction times or in refluxing hexane (thermodynamic conditions) [11,12] or (ii) with butyllithium activated by potassium tert-butoxide (LICKOR) in tetrahydrofuran (THF) at −75 °C [11,13]. N-Arylpyrroles substituted on their six-membered ring by methoxy [14], halogen [15,16], alkyl [17,18], or trifluoromethyl [19,20] groups have been the topic of more recent studies. The reactions are in general performed at low temperatures (between −75 and 0 °C) and do not tolerate the presence of reactive functional groups.
Mono- and dilithiation of N-phenylindole takes place by using TMEDA-activated butyllithium, respectively in toluene at 100 °C (1 equiv of base) [21] and in diethyl ether at −70 °C (2 equiv) [22].
In the course of the last fifteen years, combinations of lithium reagents and softer metal compounds have established themselves as appropriate tools to deproto-metallate sensitive aromatic compounds [23-30]. In the search of bases usable at room temperature, we developed pairs of metal amides able to behave synergically in such reactions. In particular, the lithium–zinc basic mixture obtained in situ by mixing ZnCl2·TMEDA and LiTMP (TMP = 2,2,6,6-tetramethylpiperidino) in a 1 to 3 ratio [31], and for which the 1:1 LiTMP·2LiCl(±TMEDA)–Zn(TMP)2 composition was given [32], proved to be a ‘superbase’. Indeed, its reactivity is higher than that of the separate LiTMP and Zn(TMP)2) when used to functionalize sensitive aromatic compounds such as heterocycles [31,33-41].
Herein, we report our efforts to functionalize N-arylated pyrroles and indoles through deproto-metallation using this mixed lithium–zinc base (Figure 1). We showed earlier, for related substrates, the impact of the different hydrogen acidities on the regioselectivity of the reaction [37,38,40,42,43]. Based on these results, we here use CH acidities of the aromatic substrates in THF (calculated using the homodesmic reaction approach within the density functional theory (DFT) framework) to attempt a rationalization of the practical results.
![[1860-5397-11-160-1]](/bjoc/content/figures/1860-5397-11-160-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Substrates involved in deproto-metallation reaction.
Figure 1: Substrates involved in deproto-metallation reaction.
Results
Synthetic aspects
In order to prepare the different pyrrole and indole substrates 1 and 2, the unsubstituted azoles were reacted with aryl and heteroaryl iodides under copper catalysis. As far as the derivatives 1a,b, 1d,e and 2a–e are concerned, the reactions were carried out by using 1.5 equiv of the azole, 0.2 equiv of copper, 2 equiv of caesium carbonate as the base, in acetonitrile at reflux [44] (Table 1, Figure 2, Supporting Information File 2). Varying yields were obtained, with aryl iodides substituted by electron-withdrawing groups in general favouring the reaction. In contrast, much lower yields were noted when using aryl bromides. For the synthesis of derivatives 1f and 2f, we rather employed 1.2 equiv of the aryl halide, 0.05 equiv of copper(I) iodide, 0.10 equiv of N,N’-dimethylethylenediamine (DMEDA), 2 equiv of tripotassium phosphate in dimethylformamide as the solvent at 120 °C [45]. Under these conditions, the N-(4-(trifluoromethyl)phenyl)-substituted azoles were isolated in high yields (Scheme 1).
Table 1: Synthesis of the azole substrates 1a,b, 1d,e and 2a–e.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-160-i9.svg?max-width=637&scale=1.0)
|
||||
| Entry | Ar | Azole | Time | Product, yield (%)a |
|---|---|---|---|---|
| 1 | 2-thienyl | pyrrole | 16 h | 1a, 75 (20)b |
| 2 | 3-pyridyl | pyrrole | 32 h | 1b, 65 (26)c |
| 3 | 4-MeOC6H4 | pyrrole | 72 h | 1d, 46 |
| 4 | 4-BrC6H4 | pyrrole | 72 h | 1e, 81 |
| 5 | 2-thienyl | indole | 48 h | 2a, 50 |
| 6 | 3-pyridyl | indole | 48 h | 2b, 70 |
| 7 | Ph | indole | 24 h | 2c, 30 |
| 8 | 4-MeOC6H4 | indole | 72 h | 2d, 15 |
| 9 | 4-BrC6H4 | indole | 56 h | 2e, 60 |
aAfter purification. bUsing 2-bromothiophene. cUsing 3-bromopyridine.
![[1860-5397-11-160-2]](/bjoc/content/figures/1860-5397-11-160-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram (30% probability) of 2e.
Figure 2: ORTEP diagram (30% probability) of 2e.
![[1860-5397-11-160-i1]](/bjoc/content/inline/1860-5397-11-160-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the azole substrates 1f and 2f.
Scheme 1: Synthesis of the azole substrates 1f and 2f.
In a previous study [33], we have shown that hexane containing N,N,N’,N’-tetramethylethylenediamine (TMEDA) is a suitable solvent to perform the C2 deproto-metallation with the TMP-based lithium–zinc as the base of commercially available N-phenylpyrrole (1c). The result has been evidenced by subsequent iodolysis (Scheme 2). Nevertheless, for numerous substrates, THF is an alternative solvent that allows the reactions to be finished after 2 h contact at room temperature [33,34,36].
![[1860-5397-11-160-i2]](/bjoc/content/inline/1860-5397-11-160-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Deproto-metallation of 1c followed by iodolysis [33].
Scheme 2: Deproto-metallation of 1c followed by iodolysis [33].
Among the different methods of trapping that can be employed after deproto-zincation (interception with aldehydes, phenyl disulfide and allyl bromide, or palladium-catalyzed cross-coupling with aryl halides) [35], we chose the iodolysis, which is the most efficient quench. In addition, it offers the possibility of a subsequent functionalization of the formed aryl iodides by recourse to transition metal-catalyzed coupling reactions [37].
When bis-heterocyclic compound 1a was reacted in THF for 2 h at room temperature with the lithium–zinc base, in situ prepared from ZnCl2·TMEDA (0.5 equiv) and LiTMP (1.5 equiv), a selective deprotonation at the thiophene ring took place. After subsequent interception with iodine, N-(5-iodo-2-thienyl)pyrrole (3a) was isolated in nearly quantitative yield. Under the same reaction conditions, the corresponding indole substrate 2a was similarly converted into N-(5-iodo-2-thienyl)indole (4a) (Scheme 3).
![[1860-5397-11-160-i3]](/bjoc/content/inline/1860-5397-11-160-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deproto-metallation of 1a and 2a followed by iodolysis.
Scheme 3: Deproto-metallation of 1a and 2a followed by iodolysis.
Starting from the bis-heterocycles 1b and 2b, better results were obtained by doubling the amount of base (using 1 equiv of ZnCl2·TMEDA and 3 equiv of LiTMP). Under the reaction conditions employed above, the deprotonation occurred on the pyridine ring, at the 2 position adjacent to the azole substituent. The corresponding monoiodides 3b and 4b were isolated in 59 and 71% yield, respectively along with unreacted starting material in both cases (Scheme 4).
![[1860-5397-11-160-i4]](/bjoc/content/inline/1860-5397-11-160-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Deproto-metallation of 1b and 2b followed by iodolysis.
Scheme 4: Deproto-metallation of 1b and 2b followed by iodolysis.
As previously observed for N-phenylpyrrole (1c) [33], the best solvent for the deprotonation of N-phenylindole (2c) is hexane containing TMEDA (5 equiv). Optimization of the base amount led to employ ZnCl2·TMEDA (0.75 equiv) and LiTMP (2.25 equiv). After 2 h contact at room temperature and subsequent iodolysis, the N-phenylated 2-iodopyrrole 3c and 2-iodoindole 4c were produced in 86 and 92% yield, respectively (Scheme 5). The structure of 4c was unambiguously identified by X-ray diffraction (Figure 3, left, see Supporting Information File 2).
![[1860-5397-11-160-i5]](/bjoc/content/inline/1860-5397-11-160-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Deproto-metallation of 1c and 2c followed by iodolysis.
Scheme 5: Deproto-metallation of 1c and 2c followed by iodolysis.
![[1860-5397-11-160-3]](/bjoc/content/figures/1860-5397-11-160-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP diagrams (30% probability) of 4c, 3d and 3e.
Figure 3: ORTEP diagrams (30% probability) of 4c, 3d and 3e.
It is known that anisole can be ortho-deprotonated using the lithium–zinc base in THF [35]. It was thus of interest to involve N-(4-methoxyphenyl)-substituted azole substrates in the deprotonation–iodination sequence. Under similar conditions, deprotonation of 1d and 2d takes place next to the methoxy group, affording the monoiodides 3d and 4d in satisfying yields after iodolysis. Increasing the amount of base to 1 equiv of zinc in the case of 2d (in order to reduce the amount of recovered starting material at the end of the reaction) led to the isolation of the diiodide 4d’, resulting from a two-fold deprotonation at both the 2 and 3’ position (the rest being the monoiodide 4d) (Scheme 6). The iodide 3d was identified by X-ray diffraction from suitable crystals (Figure 3, middle, see Supporting Information File 2).
![[1860-5397-11-160-i6]](/bjoc/content/inline/1860-5397-11-160-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Deproto-metallation of 1d and 2d followed by iodolysis.
Scheme 6: Deproto-metallation of 1d and 2d followed by iodolysis.
Whereas various attempts to deprotonate bromobenzene using the lithium–zinc base failed, a result probably due to degradation of the deproto-metallated compound through benzyne formation [46], it proved possible to accumulate at room temperature the arylmetal compounds formed from the N-(4-bromophenyl)azoles 1e and 2e, as demonstrated by trapping with iodine. The best results (minimum of degradation) were observed by using ZnCl2·TMEDA (1 equiv) and LiTMP (3 equiv). Under these conditions the iodides 3e and 4e were obtained in 58 and 57% yield, respectively (Scheme 7). The structure of product 3e was unequivocally identified by X-ray diffraction (Figure 3, right, see Supporting Information File 2).
![[1860-5397-11-160-i7]](/bjoc/content/inline/1860-5397-11-160-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Deproto-metallation of 1e and 2e followed by iodolysis.
Scheme 7: Deproto-metallation of 1e and 2e followed by iodolysis.
The reactions starting from 1a–e and 2a–e led to predominant products. In contrast, in the case of 1f and 2f, complex mixtures containing two monoiodides and one diiodide (unidentified regioselectivity) were obtained together with recovered starting material.
In order to test the functionalization of the above synthesized aryl iodides through a subsequent transition-metal-catalyzed coupling reaction we subjected three of them to a copper-catalyzed N-arylation reaction. Thus, the iodides 3b, 3d and 4d were reacted with 2 equiv of imidazole in the presence of 0.1 equiv of copper(I) oxide, 2 equiv of caesium carbonate as the base in dimethylsulfoxide (DMSO) at 110 °C [47]. Under these conditions, the imidazole-substituted compounds 5b, 5d and 6d were generated in 85, 67 and 40% yield, respectively (Scheme 8, Figure 4, see Supporting Information File 2).
![[1860-5397-11-160-i8]](/bjoc/content/inline/1860-5397-11-160-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: N-arylation of the iodides 3b, 3d and 4d.
Scheme 8: N-arylation of the iodides 3b, 3d and 4d.
![[1860-5397-11-160-4]](/bjoc/content/figures/1860-5397-11-160-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP diagram (30% probability) of 5d.
Figure 4: ORTEP diagram (30% probability) of 5d.
Computational aspects
There is a lack of data on the acidity of N-substituted pyrroles and indoles in the literature. NH acidity of unsubstituted pyrrole and indole in DMSO was measured by Bordwell [48] and computed by means of semi-empirical AM1 [49] and ab initio methods [50]. The pKa values for N-methylindole, N-methylpyrrole and N-(dimethylamino)pyrrole as CH acids in THF were experimentally determined by Fraser et al [51]. The latter are more related to our goals and were found to be in good agreement with those computed within the DFT framework recently [42,52]. The results of quantum chemical calculations on CH acidity of the different N-arylated pyrroles 1 and indoles 2, obtained both for THF solution (Figure 5) and gas phase (see Supporting Information File 1), are presented in the current paper.
![[1860-5397-11-160-5]](/bjoc/content/figures/1860-5397-11-160-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Calculated values of pKa(THF) of the compounds 1 and 2, and bromobenzene.
Figure 5: Calculated values of pKa(THF) of the compounds 1 and 2, and bromobenzene.
A potential acidity of the methoxy groups in substrates 1d and 2d was ignored here since there was no sign of their deprotonation under the experimental conditions and as a consequence their acidity was expected to be substantially lower. The data in Figure 5 refer to the most stable rotamer of the compounds. Thus, potential energy surface (PES) scans allowed us to state that both 2a and 2b are likely to adopt a form in which the heteroatoms are far away from the benzo-fragment.
The calculated values of gas-phase acidity of the investigated compounds (see Supporting Information File 1) lie within the range of 364.4–394.3 kcal mol−1 which is typical for weak CH acidic compounds while the pKa values in THF solution (Figure 5) covered a 28.8–45.0 span. These data allow the assignment of potential deprotonation sites in the investigated substrates. Also the correlation between gas-phase ΔGacid and the pKa(THF) values can be tracked. In all cases, the CH acidity increases when changing from electron-donating groups to more electron-withdrawing ones. One can easily see that for both pyrroles and indoles the most acidic hydrogen is at the 2 position of the azole part, while for an aryl substituent it depends on its nature.
Discussion
The results observed in the course of these deproto-metallation reactions complete previously reported studies using alkali metal bases with similar substrates [36-41]. The calculations of the CH acidities in THF (Figure 5) allow us to comment on the regioselectivities observed in the different studies.
To our knowledge, the N-(2-thienyl) azoles 1a and 2a (Scheme 3 and Figure 5) have never been subjected to deproto-metallation before. If we only consider the reaction of 1a, it is conceivable that the regioselectivity only results from an attack of the lithium–zinc base at the most acidic site. In contrast, the regioselectivity observed in case of substrate 2a cannot be explained by the pKa values as the only reason. Otherwise, deprotonation would have been also observed at the 2 position of the indole ring. Whereas the azole nitrogen in 2a cannot coordinate a metal through its electron lone pair (it is delocalized within the aromatic π-electron ring system), a coordination by the sulfur atom of the thiophene substituent could take place with a possible decrease of neighbouring pKa values.
As for 1a and 2a, deproto-metallation has not yet been reported for 1b and 2b (Scheme 4 and Figure 5). In the case of these substrates, it is clear that deprotonation takes place after coordination of the pyridine nitrogen to a metal; otherwise, the reactions would have been observed at the 2 position of the azole ring. Among both positions adjacent to the pyridine nitrogen, the most acidic is attacked.
N-Phenylpyrrole (1c, Scheme 5 and Figure 5) does not have any atom capable of coordinating a metal. Thus, under thermodynamic conditions (using TMEDA-activated butyllithium either in diethyl ether at room temperature after long reaction times or in refluxing hexane [11,12]; using LICKOR in THF at −75 °C [11,13]), the most acidic site next to the azole nitrogen is affected. Using TMEDA-activated butyllithium in diethyl ether under kinetic conditions leads to 2,2’-dilithiation [10,11]. In this case, the intermediate 2-lithiated compound can be involved in aggregates favouring deprotonation at a neighbouring site to afford the 2,2’-dilithiated derivative. The 2,2’-dimetallated derivative of 1c has also been noted using the lithium–zinc base, for example when the reaction is performed in THF (favoured at reflux temperature) [33]. In this case, the dimetallated compound does not correspond to a kinetic but to a thermodynamic compound. This could be due to the formation of a stable zinc metallacycle, as suggested in an earlier paper [37]. As previously reported [33], dideprotonation can be suppressed by using hexane containing TMEDA instead of THF and both compounds 1c and 2c are functionalized at their most acidic position (Scheme 5 and Figure 5).
The deprotolithiation of N-(4-methoxyphenyl)pyrrole (1d, Scheme 6 and Figure 5) by using chelates of butyllithium in THF at −75 °C for 1 h was documented by Faigl and co-workers in 1997 [14]. Whereas N,N,N’,N”,N”-pentamethyldiethylenetriamine (PMDETA) employed as ligand leads to reaction at the (maybe less hindered) 2 position (46% yield after conversion to the corresponding carboxylic acid), the most acidic position 3’ is attacked in the presence of TMEDA (18% yield after similar conversion) [14]. Using the lithium–zinc base also gives the 3’-metallated derivative, but more efficiently. N-(4-Methoxyphenyl)indole (2d, Scheme 6 and Figure 5) has two similarly acidified sites at C2 and C3’. The regioselective reaction at C3’ could result from a coordination of the methoxy group to a metal prior to deprotonation. Nevertheless, when the amount of base is increased, competitive dideprotonation takes place to furnish diiodides as previously noted with other azoles [37,38].
Compared with a methoxy group, the bromo substituent exhibits a stronger acidifying effect. By itself, this effect is not sufficient to enable the accumulation of a 2-metallated bromobenzene. However, when this ortho-bromine effect is combined with the meta-effect of a pyrrolyl group, the corresponding CH acidities are increased (see Figure 5), and things become different. Thus, whereas the direct deproto-lithiation of bromobenzene leads to highly unstable 2-bromophenyllithium, that of N-(4-bromophenyl)pyrrole (1e, Scheme 7 and Figure 5) is possible. By using LiTMP in THF at −75 °C for 1 h, Faigl and co-workers evidenced deprotonation next to the bromo group (21% yield after conversion to the corresponding carboxylic acid), i.e. at the most acidic position [16]. With the lithium–zinc base, the reactions from the N-(4-bromophenyl) azoles 1e and 2e (Scheme 7 and Figure 5) occurred with the same regioselectivity, and proved possible with acceptable yields at room temperature.
The deprotolithiation of N-(4-(trifluoromethyl)phenyl)pyrrole (1f, Figure 5) was reported by Faigl and co-workers in 1999 [19]. By employing TMEDA-activated butyllithium in THF at −75 °C, the authors mainly noted proton abstraction next to the trifluoromethyl group. A competitive minor reaction at the 2 position of the pyrrole group can be rationalized by similar pKa values at C2 and C3’ [19]. Indeed, when compared with a bromo substituent, the trifluoromethyl group similarly (and maybe more strongly) exhibits a long range acidifying effect, but less acidifies the ortho positions [53,54]. The lack of regioselectivity observed in the course of the reaction between the lithium–zinc base and the N-(4-(trifluoromethyl)phenyl)azole 1f or 2f (Figure 5) could be in relation with a reaction temperature (too high) not suitable to discriminate between similarly acidified positions.
Antiproliferative activity in A2058 melanoma cells
The N-arylated pyrroles and indoles exerted low to moderate antiproliferative activity in A2058 melanoma cells (Figure 6). The best result was obtained with 1f at 10−5 M, which induced 31.9 ± 0.1% growth inhibition in cells treated for 72 h. The linear N-arylated pyrroles 1b and 1e were poorly active. Arylation by a thiophene moiety (1a and 2a) moderately increased the antiproliferative activity, with no significant difference between the pyrrole and indole derivatives.
![[1860-5397-11-160-6]](/bjoc/content/figures/1860-5397-11-160-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Antiproliferative activity (growth inhibition) of the tested compounds 1a,b,e,f, 2a,b and 5d at concentration 10−5 M and 72 h in A2058 human melanoma cells.
Figure 6: Antiproliferative activity (growth inhibition) of the tested compounds 1a,b,e,f, 2a,b and 5d at con...
Conclusion
Unlike other azoles such as pyrazole and triazoles [37,38,40], pyrrole and indole do not possess any atom capable of coordinating metals. As a consequence, the corresponding CH acidities in THF solution, which were calculated using a continuum solvation model, better help in rationalizing the outcome of the deproto-metallation reactions.
In addition, whereas N-(4-substituted phenyl)pyrazoles and -triazoles (e.g., with methoxy as substituent, Figure 7) can be deprotonated at C2’ for the same reason [37,38,40], the corresponding N-(4-substituted phenyl)pyrroles and -indoles are rather functionalized at C3’, next to the substituent.
![[1860-5397-11-160-7]](/bjoc/content/figures/1860-5397-11-160-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Iodides previously formed as major products from the corresponding N-(4-methoxyphenyl)azoles using the lithium–zinc base.
Figure 7: Iodides previously formed as major products from the corresponding N-(4-methoxyphenyl)azoles using ...
Experimental
General methods. All the reactions were performed under argon atmosphere. THF was distilled over sodium/benzophenone. ZnCl2·TMEDA was prepared as described previously [35]. Column chromatography was performed on silica gel (40–63 μm). Melting points were measured on a Kofler apparatus. IR spectra were taken on a Perkin-Elmer Spectrum 100 spectrometer. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III spectrometer at 300 and 75 MHz, respectively. 1H chemical shifts (δ) are given in ppm relative to the solvent residual peak, 13C chemical shifts are relative to the central peak of the solvent signal [55]. Mass spectra measurements were performed using a HP 6890 instrument.
Crystallography. The single crystals were studied with graphite monochromatized MoKα radiation (λ = 0.71073 Å). X-ray diffraction data were collected at T = 150(2) K using an APEXII Bruker-AXS diffractometer. The structure was solved by direct methods using the SIR97 program [56], and then refined with full-matrix least-square methods based on F2 (SHELX-97) [57] with the aid of the WINGX program [58]. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Hydrogen atoms were finally included in their calculated positions. Molecular diagrams were generated by ORTEP-3 (version 2.02) [59].
Procedure 1 for the synthesis of the N-arylated pyrroles and indoles 1a,b,d,e and 2a–e [44]. To azole (6.0 mmol) and aryl halide (4.0 mmol) in acetonitrile (20 mL) were successively added Cu (50 mg, 0.80 mmol), Cs2CO3 (2.6 g, 8.0 mmol) and, in the case of aryl bromides, KI (99 mg, 6.0 mmol). The mixture was stirred under argon at acetonitrile reflux temperature (the reaction time is given in the product description) before dilution with AcOEt (40 mL) and filtration. Concentration under reduced pressure and purification by chromatography on silica gel (the eluent is given in the product description) led to the expected compounds.
Procedure 2 for the synthesis of the N-arylated pyrroles and indoles 1f and 2f [45]. To azole (10 mmol) and aryl halide (12 mmol) in DMF (5 mL) were successively added CuI (95 mg, 0.50 mmol), K3PO4 (4.2 g, 20 mmol) and DMEDA (0.11 mL, 1.0 mmol). The mixture was stirred under argon at 120 °C (the reaction time is given in the product description) before dilution with AcOEt (40 mL) and filtration. Concentration under reduced pressure and purification by chromatography on silica gel (the eluent is given in the product description) led to the compounds described below.
General procedure 3 for the deprotonative metallation followed by iodination (analogous to that described in [60]). To a stirred, cooled (0 °C) solution of 2,2,6,6-tetramethylpiperidine (0.25 mL, 1.5 mmol) in THF (2–3 mL) were successively added BuLi (about 1.6 M hexanes solution, 1.5 mmol) and, 5 min later, ZnCl2·TMEDA (0.13 g, 0.50 mmol). The mixture was stirred for 15 min at 0 °C before introduction of the substrate (1.0 mmol) at 0–10 °C. After 2 h at room temperature, a solution of I2 (0.38 g, 1.5 mmol) in THF (4 mL) was added. The mixture was stirred overnight before addition of an aqueous saturated solution of Na2S2O3 (4 mL) and extraction with AcOEt (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification by chromatography on silica gel (the eluent is given in the product description) led to the compounds 3a and 4a.
General procedure 4 for the deprotonative metallation followed by iodination (analogous to that described in [60]). To a stirred, cooled (0 °C) solution of 2,2,6,6-tetramethylpiperidine (0.25 mL, 1.5 mmol) in THF (2-3 mL) were successively added BuLi (about 1.6 M hexanes solution, 1.5 mmol) and, 5 min later, ZnCl2·TMEDA (0.13 g, 0.50 mmol). The mixture was stirred for 15 min at 0 °C before introduction of the substrate (0.5 mmol) at 0–10 °C. After 2 h at room temperature, a solution of I2 (0.38 g, 1.5 mmol) in THF (4 mL) was added. The mixture was stirred overnight before addition of an aqueous saturated solution of Na2S2O3 (4 mL) and extraction with AcOEt (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification by chromatography on silica gel (the eluent is given in the product description) led to the compounds 3b,e and 4b,d',e.
General procedure 5 for the deprotonative metallation followed by iodination (analogous to that described in [60]). To a stirred, cooled (0 °C) solution of 2,2,6,6-tetramethylpiperidine (0.25 mL, 1.5 mmol) in hexane (2–3 mL) containing TMEDA (0.50 mL, 3.3 mmol) were successively added BuLi (about 1.6 M hexanes solution, 1.5 mmol) and, 5 min later, ZnCl2·TMEDA (0.13 g, 0.50 mmol). The mixture was stirred for 15 min at 0 °C before introduction of the substrate (0.67 mmol) at 0–10 °C. After 2 h at room temperature, a solution of I2 (0.38 g, 1.5 mmol) in THF (4 mL) was added. The mixture was stirred overnight before addition of an aqueous saturated solution of Na2S2O3 (4 mL) and extraction with AcOEt (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification by chromatography on silica gel (the eluent is given in the product description) led to the compounds 3c and 4c.
General procedure 6 for the deprotonative metallation followed by iodination (analogous to that described in [60]). To a stirred, cooled (0 °C) solution of 2,2,6,6-tetramethylpiperidine (0.25 mL, 1.5 mmol) in THF (2–3 mL) were successively added BuLi (about 1.6 M hexanes solution, 1.5 mmol) and, 5 min later, ZnCl2·TMEDA (0.13 g, 0.50 mmol). The mixture was stirred for 15 min at 0 °C before introduction of the substrate (0.67 mmol) at 0–10 °C. After 2 h at room temperature, a solution of I2 (0.38 g, 1.5 mmol) in THF (4 mL) was added. The mixture was stirred overnight before addition of an aqueous saturated solution of Na2S2O3 (4 mL) and extraction with AcOEt (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. Purification by chromatography on silica gel (the eluent is given in the product description) led to the compounds 3d and 4d.
Procedure 7 for the N-arylation of imidazole [47]. A mixture of the prepared iodide (1.0 mmol), Cu2O (0.10 g, 0.10 mmol), Cs2CO3 (0.65 g, 2.0 mmol), imidazole (0.14 g, 2.0 mmol) and DMSO (0.5 mL) was stirred for 24 h at 110 °C under argon. After cooling to room temperature, the mixture was diluted with AcOEt (10 mL) and filtered over celite®. Washing with AcOEt, removal of the solvent and purification by chromatography on silica gel (the eluent is given in the product description) led to the compounds 5b,d and 6d.
Computational procedure. The DFT calculations were performed using GAUSSIAN 03W package [61]. The B3LYP formalism was employed. All optimized geometries were obtained using the 6-31G(d) basis set without any symmetry constraints implied. Vibrational frequencies were calculated at the same level of theory in order to characterize stationary points and to calculate zero-point vibrational energies (ZPVE) and thermal corrections. The total energy of species was found using the 6-311+G(d,p) basis set and tight convergence criteria. Further, the gas-phase Gibbs energies (G0298) were calculated using the following equation:
G0298 = E + ZPVE + H00→298 – TS0298
The gas-phase acidity ΔGacid was defined as the Gibbs energy of deprotonation of the particular substrate R–H (R–H(g) → R−(g) + H+(g)):
ΔGacid = G0298(R−) + G0298(H+) − G0298(R–H).
The solvent influence was simulated within the polarized continuum model (PCM) with the default parameters for THF. The cavity was built up using atomic radii from the UFF force field. The PCM energies EPCM were calculated at the B3LYP/6-311+G(d,p) level using geometries optimized for isolated structures. The Gibbs energy in solution Gsol was calculated for each species by the formula:
Gsol = G0298 + EPCM − E.
The following homodesmic reaction was composed for the pKa values calculation:
R–H(s) + Het−(s) → R−(s) + Het–H(s),
where Het–H is N-methylindole. The latter was chosen as reference compound due to its structural similarity and since its pKa(THF) = 38.1 found by Fraser et al [51] was expected to be close to those for our substrates. Consequently, the Gibbs energy of the homodesmic reaction (ΔGr,sol) and the pKa value are related by the following equation:
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-160-i10.svg?max-width=637&scale=1.18182)
Biological evaluation. The antiproliferative activity of N-arylated pyrrole and indole derivatives was studied in the A2058 (ATCC® CRL-11147) cell line as described previously [60]. A2058 cells are highly invasive human epithelial adherent melanoma cells, derived from lymph nodes metastatic cells obtained from a 43 year old male patient. They are tumorigenic at 100% frequency in nude mice, and considered as very resistant to anticancer drugs. All cell culture experiments were performed at 37 °C. Cells were grown to confluence in 75 cm² flasks in DMEM supplemented with 10% fetal calf serum (FCS) and 1% penicillin–streptomycin (Dominique Dutscher, France), in a 5% CO2 humidified atmosphere. Molecules were solubilized in DMSO at 10−3 M and diluted in the cell culture medium to obtain 2·10−5 M solutions. Confluent cells were trypsinized and centrifuged in FCS at 1500g for 5 min. The supernatant containing trypsin was discarded and the cell pellet was resuspended in cell culture medium to obtain a 4·104 cell·mL−1 suspension. At t0, 50 µL of the 2·10−5 M test solutions were deposited in a 96-wells flat bottom microplate, and 50 µL of the cell suspension were added. The 2000 cells were then grown for 72 h in the cell culture medium containing 10−5 M molecules. At t72, 20 µL of a 5 g·L−1 MTT solution were added to each well of the microplate, allowing living cells containing a functional mitochondrial succinate dehydrogenase to metabolize MTT to the corresponding blue formazan salt for 4 h. The cell culture medium was removed using an Eppendorf epMotion 5070 pipeting robot (Eppendorf, France) and formazan crystals were dissolved in 200 µL DMSO. Microplates were placed at 37 °C for 5 min to solubilize formazan crystals and absorbance was read at 550 nm using a VERSAmax microplate reader (Molecular devices, France). The percentage of growth inhibition was calculated as GI (%) = 100 − ((A550nm sample − A550nm BG)/(A550nm control − A550nm BG))·100, with:
- A550 nm sample as median absorbance of 8 wells containing cells treated with 10−5 M compound solution
- A550 nm BG as median background absorbance of 8 wells containing control cell culture medium + 1% DMSO
- A550 nm control as median absorbance of 8 wells containing cells grown in control cell culture medium + 1% DMSO.
The data are expressed as GI (%) + sem (%) from 3 independent assays.
Supporting Information
| Supporting Information File 1: Experimental description of the synthesized compounds, 1H and 13C NMR spectra, calculated values of the Gibbs energies ΔGacid [kcal·mo1−1] for deprotonation, selected Cartesian coordinates of molecular geometry for the most stable rotamer forms optimized at B3LYP/6-31G(d) level of theory. | ||
| Format: PDF | Size: 3.1 MB | Download |
| Supporting Information File 2: CIF files of 2e (CCDC1402111), 4c (1402112), 3d (1402113), 3e (1402114), and 5d (1402115). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. | ||
| Format: CIF | Size: 619.2 KB | Download |
Acknowledgements
We are grateful to the Institut Universitaire de France, Rennes Métropole and the French Cancer League (Comité 17) for financial support. We thank Thermo Fischer for generous gift of 2,2,6,6-tetramethylpiperidine. We also thank the “Cancéropôle Grand Ouest, axe Valorisation des produits de la mer en cancérologie” for scientific support.
References
-
Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: New York, NY, U.S.A., 2003.
Return to citation in text: [1] -
Gribble, G. W. Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Springer: Berlin, Germany, 2010.
Return to citation in text: [1] -
Gschwend, H. W.; Rodriguez, H. R. Org. React. 2005, 26, 1–360. doi:10.1002/0471264180.or026.01
Return to citation in text: [1] -
Beak, P.; Snieckus, V. Acc. Chem. Res. 1982, 15, 306–312. doi:10.1021/ar00082a002
Return to citation in text: [1] -
Snieckus, V. Chem. Rev. 1990, 90, 879–933. doi:10.1021/cr00104a001
Return to citation in text: [1] -
Gant, T. G.; Meyers, A. I. Tetrahedron 1994, 50, 2297–2360. doi:10.1016/S0040-4020(01)86953-2
Return to citation in text: [1] -
Schlosser, M. Organometallics in Synthesis, 2nd ed.; Wiley-VCH: New York, NY, U.S.A., 2002.
Chapter I.
Return to citation in text: [1] -
Kishbaugh, T. L. S. Top. Heterocycl. Chem. 2012, 29, 1–45. doi:10.1007/7081_2012_76
Return to citation in text: [1] -
Pelkey, E. T. Top. Heterocycl. Chem. 2010, 26, 141–191. doi:10.1007/7081_2010_56
Return to citation in text: [1] -
Cheeseman, G. W. H.; Greenberg, S. G. J. Organomet. Chem. 1979, 166, 139–152. doi:10.1016/s0022-328x(00)91628-0
Return to citation in text: [1] [2] -
Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Shirley, D. A.; Gross, B. H.; Roussel, P. A. J. Org. Chem. 1955, 20, 225–231. doi:10.1021/jo01120a012
Return to citation in text: [1] [2] -
Thurner, A.; Faigl, F.; Ágai, B.; Töke, L. Synth. Commun. 1998, 28, 443–449. doi:10.1080/00397919808005098
Return to citation in text: [1] [2] -
Faigl, F.; Fogassy, K.; Thurner, A.; Töke, L. Tetrahedron 1997, 53, 4883–4888. doi:10.1016/S0040-4020(97)00183-X
Return to citation in text: [1] [2] [3] -
Faigl, F.; Fogassy, K.; Szántó, Z.; Lopata, A.; Töke, L. Tetrahedron 1998, 54, 4367–4374. doi:10.1016/S0040-4020(98)00150-1
Return to citation in text: [1] -
Fogassy, K.; Kovács, K.; Keserű, G. M.; Tőke, L.; Faigl, F. J. Chem. Soc., Perkin Trans. 1 2001, 1039–1043. doi:10.1039/b100008j
Return to citation in text: [1] [2] -
Faigl, F.; Feldhoffer, B. V.; Thurner, A. Synth. Commun. 2006, 36, 2841–2849. doi:10.1080/00397910600770672
Return to citation in text: [1] -
Faigl, F.; Vas-Feldhoffer, B.; Kubinyi, M.; Pál, K.; Tárkányi, G.; Czugler, M. Tetrahedron: Asymmetry 2009, 20, 98–103. doi:10.1016/j.tetasy.2009.01.010
Return to citation in text: [1] -
Faigl, F.; Fogassy, K.; Szűcs, E.; Kovács, K.; Keserű, G. M.; Harmat, V.; Böcskei, Z.; Tőke, L. Tetrahedron 1999, 55, 7881–7892. doi:10.1016/s0040-4020(99)00398-1
Return to citation in text: [1] [2] [3] -
Faigl, F.; Mátravölgyi, B.; Deák, S.; Holczbauer, T.; Czugler, M.; Balázs, L.; Hermecz, I. Tetrahedron 2012, 68, 4259–4266. doi:10.1016/j.tet.2012.03.070
Return to citation in text: [1] -
Rataboul, F.; Zapf, A.; Jackstell, R.; Harkal, S.; Riermeier, T.; Monsees, A.; Dingerdissen, U.; Beller, M. Chem. – Eur. J. 2004, 10, 2983–2990. doi:10.1002/chem.200306026
Return to citation in text: [1] -
Artemova, N. V.; Chevykalova, M. N.; Luzikov, Y. N.; Nifant'ev, I. E.; Nifant'ev, E. E. Tetrahedron 2004, 60, 10365–10370. doi:10.1016/j.tet.2004.07.082
Return to citation in text: [1] -
Mulvey, R. E. Organometallics 2006, 25, 1060–1075. doi:10.1021/om0510223
Return to citation in text: [1] -
Mulvey, R. E.; Mongin, F.; Uchiyama, M.; Kondo, Y. Angew. Chem., Int. Ed. 2007, 46, 3802–3824. doi:10.1002/anie.200604369
Return to citation in text: [1] -
Mulvey, R. E. Acc. Chem. Res. 2009, 42, 743–755. doi:10.1021/ar800254y
Return to citation in text: [1] -
Haag, B.; Mosrin, M.; Ila, H.; Malakhov, V.; Knochel, P. Angew. Chem., Int. Ed. 2011, 50, 9794–9824. doi:10.1002/anie.201101960
Return to citation in text: [1] -
Mongin, F.; Uchiyama, M. Curr. Org. Chem. 2011, 15, 2340–2361. doi:10.2174/138527211796150651
Return to citation in text: [1] -
Mongin, F.; Harrison-Marchand, A. Chem. Rev. 2013, 113, 7563–7727. doi:10.1021/cr3002966
Return to citation in text: [1] -
Mulvey, R. E. Dalton Trans. 2013, 42, 6676–6693. doi:10.1039/c3dt00053b
Return to citation in text: [1] -
Harford, P. J.; Peel, A. J.; Chevallier, F.; Takita, R.; Mongin, F.; Uchiyama, M.; Wheatley, A. E. H. Dalton Trans. 2014, 43, 14181–14203. doi:10.1039/c4dt01130a
Return to citation in text: [1] -
L'Helgoual'ch, J. M.; Seggio, A.; Chevallier, F.; Yonehara, M.; Jeanneau, E.; Uchiyama, M.; Mongin, F. J. Org. Chem. 2008, 73, 177–183. doi:10.1021/jo7020345
Return to citation in text: [1] [2] -
García-Álvarez, P.; Mulvey, R. E.; Parkinson, J. A. Angew. Chem., Int. Ed. 2011, 50, 9668–9671. doi:10.1002/anie.201104297
Return to citation in text: [1] -
Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Seggio, A.; Chevallier, F.; Vaultier, M.; Mongin, F. J. Org. Chem. 2007, 72, 6602–6605. doi:10.1021/jo0708341
Return to citation in text: [1] [2] -
Snégaroff, K.; Komagawa, S.; Chevallier, F.; Gros, P. C.; Golhen, S.; Roisnel, T.; Uchiyama, M.; Mongin, F. Chem. – Eur. J. 2010, 16, 8191–8201. doi:10.1002/chem.201000543
Return to citation in text: [1] [2] [3] [4] -
Snégaroff, K.; Nguyen, T. T.; Marquise, N.; Halauko, Y. S.; Harford, P. J.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Chevallier, F.; Wheatley, A. E. H.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2011, 17, 13284–13297. doi:10.1002/chem.201101993
Return to citation in text: [1] [2] [3] -
Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Kadiyala, R. R.; Tilly, D.; Nagaradja, E.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Halauko, Y. S.; Chevallier, F.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2013, 19, 7944–7960. doi:10.1002/chem.201300552
Return to citation in text: [1] [2] -
Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Mongin, F. Org. Biomol. Chem. 2014, 12, 1475–1487. doi:10.1039/c3ob42380h
Return to citation in text: [1] [2] [3] [4] [5] -
Marquise, N.; Bretel, G.; Lassagne, F.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Gros, P. C.; Mongin, F. RSC Adv. 2014, 4, 19602–19612. doi:10.1039/c4ra02583k
Return to citation in text: [1] [2] -
Matulis, V. E.; Halauko, Y. S.; Ivashkevich, O. A.; Gaponik, P. N. J. Mol. Struct.: THEOCHEM 2009, 909, 19–24. doi:10.1016/j.theochem.2009.05.024
Return to citation in text: [1] [2] -
Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Grigoriev, Y. V.; Gaponik, P. N. Tetrahedron 2010, 66, 3415–3420. doi:10.1016/j.tet.2010.03.053
Return to citation in text: [1] -
Zhu, R.; Xing, L.; Wang, X.; Cheng, C.; Su, D.; Hu, Y. Adv. Synth. Catal. 2008, 350, 1253–1257. doi:10.1002/adsc.200700535
Return to citation in text: [1] [2] -
Antilla, J. C.; Baskin, J. M.; Barder, T. E.; Buchwald, S. L. J. Org. Chem. 2004, 69, 5578–5587. doi:10.1021/jo049658b
Return to citation in text: [1] [2] -
Huisgen, R.; Mack, W.; Herbig, K.; Ott, N.; Anneser, E. Chem. Ber. 1960, 93, 412–414. doi:10.1002/cber.19600930222
Return to citation in text: [1] -
Teo, Y.-C.; Yong, F.-F.; Sim, S. Tetrahedron 2013, 69, 7279–7284. doi:10.1016/j.tet.2013.06.095
Return to citation in text: [1] [2] -
Bordwell, F. G. Pure Appl. Chem. 1977, 49, 963–968. doi:10.1351/pac197749070963
Return to citation in text: [1] -
Del Valle, J. C.; De Paz, J. L. G. J. Mol. Struct.: THEOCHEM 1992, 254, 481–491. doi:10.1016/0166-1280(92)80091-Y
Return to citation in text: [1] -
Vianello, R.; Maksić, Z. B. Mol. Phys. 2005, 103, 209–219. doi:10.1080/00268970512331316184
Return to citation in text: [1] -
Fraser, R. R.; Mansour, T. S.; Savard, S. Can. J. Chem. 1985, 63, 3505–3509. doi:10.1139/v85-574
Return to citation in text: [1] [2] -
Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. Tetrahedron 2007, 63, 1568–1576. doi:10.1016/j.tet.2006.12.032
Return to citation in text: [1] -
Mongin, F.; Schlosser, M. Tetrahedron Lett. 1997, 38, 1559–1562. doi:10.1016/S0040-4039(97)00131-7
Return to citation in text: [1] -
Schlosser, M. Angew. Chem., Int. Ed. 1998, 37, 1496–1513. doi:10.1002/(SICI)1521-3773(19980619)37:11<1496::AID-ANIE1496>3.0.CO;2-U
Return to citation in text: [1] -
Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v
Return to citation in text: [1] -
Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G. L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A. G. G.; Polidori, G.; Spagna, R. J. Appl. Crystallogr. 1999, 32, 115–119. doi:10.1107/S0021889898007717
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, A64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/S0021889899006020
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117
Return to citation in text: [1] -
Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Bioorg. Med. Chem. 2014, 22, 3498–3507. doi:10.1016/j.bmc.2014.04.028
Return to citation in text: [1] [2] [3] [4] [5] -
Gaussian 03, Revision C.01; Gaussian, Inc.: Pittsburgh, PA, U.S.A., 2003.
Return to citation in text: [1]
| 46. | Huisgen, R.; Mack, W.; Herbig, K.; Ott, N.; Anneser, E. Chem. Ber. 1960, 93, 412–414. doi:10.1002/cber.19600930222 |
| 47. | Teo, Y.-C.; Yong, F.-F.; Sim, S. Tetrahedron 2013, 69, 7279–7284. doi:10.1016/j.tet.2013.06.095 |
| 48. | Bordwell, F. G. Pure Appl. Chem. 1977, 49, 963–968. doi:10.1351/pac197749070963 |
| 11. | Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1 |
| 13. | Thurner, A.; Faigl, F.; Ágai, B.; Töke, L. Synth. Commun. 1998, 28, 443–449. doi:10.1080/00397919808005098 |
| 10. | Cheeseman, G. W. H.; Greenberg, S. G. J. Organomet. Chem. 1979, 166, 139–152. doi:10.1016/s0022-328x(00)91628-0 |
| 11. | Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1 |
| 36. | Snégaroff, K.; Nguyen, T. T.; Marquise, N.; Halauko, Y. S.; Harford, P. J.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Chevallier, F.; Wheatley, A. E. H.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2011, 17, 13284–13297. doi:10.1002/chem.201101993 |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 38. | Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e |
| 39. | Kadiyala, R. R.; Tilly, D.; Nagaradja, E.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Halauko, Y. S.; Chevallier, F.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2013, 19, 7944–7960. doi:10.1002/chem.201300552 |
| 40. | Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Mongin, F. Org. Biomol. Chem. 2014, 12, 1475–1487. doi:10.1039/c3ob42380h |
| 41. | Marquise, N.; Bretel, G.; Lassagne, F.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Gros, P. C.; Mongin, F. RSC Adv. 2014, 4, 19602–19612. doi:10.1039/c4ra02583k |
| 11. | Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1 |
| 12. | Shirley, D. A.; Gross, B. H.; Roussel, P. A. J. Org. Chem. 1955, 20, 225–231. doi:10.1021/jo01120a012 |
| 51. | Fraser, R. R.; Mansour, T. S.; Savard, S. Can. J. Chem. 1985, 63, 3505–3509. doi:10.1139/v85-574 |
| 42. | Matulis, V. E.; Halauko, Y. S.; Ivashkevich, O. A.; Gaponik, P. N. J. Mol. Struct.: THEOCHEM 2009, 909, 19–24. doi:10.1016/j.theochem.2009.05.024 |
| 52. | Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. Tetrahedron 2007, 63, 1568–1576. doi:10.1016/j.tet.2006.12.032 |
| 49. | Del Valle, J. C.; De Paz, J. L. G. J. Mol. Struct.: THEOCHEM 1992, 254, 481–491. doi:10.1016/0166-1280(92)80091-Y |
| 50. | Vianello, R.; Maksić, Z. B. Mol. Phys. 2005, 103, 209–219. doi:10.1080/00268970512331316184 |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 53. | Mongin, F.; Schlosser, M. Tetrahedron Lett. 1997, 38, 1559–1562. doi:10.1016/S0040-4039(97)00131-7 |
| 54. | Schlosser, M. Angew. Chem., Int. Ed. 1998, 37, 1496–1513. doi:10.1002/(SICI)1521-3773(19980619)37:11<1496::AID-ANIE1496>3.0.CO;2-U |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 38. | Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e |
| 40. | Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Mongin, F. Org. Biomol. Chem. 2014, 12, 1475–1487. doi:10.1039/c3ob42380h |
| 19. | Faigl, F.; Fogassy, K.; Szűcs, E.; Kovács, K.; Keserű, G. M.; Harmat, V.; Böcskei, Z.; Tőke, L. Tetrahedron 1999, 55, 7881–7892. doi:10.1016/s0040-4020(99)00398-1 |
| 19. | Faigl, F.; Fogassy, K.; Szűcs, E.; Kovács, K.; Keserű, G. M.; Harmat, V.; Böcskei, Z.; Tőke, L. Tetrahedron 1999, 55, 7881–7892. doi:10.1016/s0040-4020(99)00398-1 |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 38. | Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e |
| 16. | Fogassy, K.; Kovács, K.; Keserű, G. M.; Tőke, L.; Faigl, F. J. Chem. Soc., Perkin Trans. 1 2001, 1039–1043. doi:10.1039/b100008j |
| 14. | Faigl, F.; Fogassy, K.; Thurner, A.; Töke, L. Tetrahedron 1997, 53, 4883–4888. doi:10.1016/S0040-4020(97)00183-X |
| 14. | Faigl, F.; Fogassy, K.; Thurner, A.; Töke, L. Tetrahedron 1997, 53, 4883–4888. doi:10.1016/S0040-4020(97)00183-X |
| 35. | Snégaroff, K.; Komagawa, S.; Chevallier, F.; Gros, P. C.; Golhen, S.; Roisnel, T.; Uchiyama, M.; Mongin, F. Chem. – Eur. J. 2010, 16, 8191–8201. doi:10.1002/chem.201000543 |
| 55. | Gottlieb, H. E.; Kotlyar, V.; Nudelman, A. J. Org. Chem. 1997, 62, 7512–7515. doi:10.1021/jo971176v |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 38. | Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e |
| 40. | Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Mongin, F. Org. Biomol. Chem. 2014, 12, 1475–1487. doi:10.1039/c3ob42380h |
| 1. | Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles, 2nd ed.; Wiley-VCH: New York, NY, U.S.A., 2003. |
| 2. | Gribble, G. W. Heterocyclic Scaffolds II: Reactions and Applications of Indoles; Springer: Berlin, Germany, 2010. |
| 10. | Cheeseman, G. W. H.; Greenberg, S. G. J. Organomet. Chem. 1979, 166, 139–152. doi:10.1016/s0022-328x(00)91628-0 |
| 11. | Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1 |
| 31. | L'Helgoual'ch, J. M.; Seggio, A.; Chevallier, F.; Yonehara, M.; Jeanneau, E.; Uchiyama, M.; Mongin, F. J. Org. Chem. 2008, 73, 177–183. doi:10.1021/jo7020345 |
| 60. | Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Bioorg. Med. Chem. 2014, 22, 3498–3507. doi:10.1016/j.bmc.2014.04.028 |
| 9. | Pelkey, E. T. Top. Heterocycl. Chem. 2010, 26, 141–191. doi:10.1007/7081_2010_56 |
| 32. | García-Álvarez, P.; Mulvey, R. E.; Parkinson, J. A. Angew. Chem., Int. Ed. 2011, 50, 9668–9671. doi:10.1002/anie.201104297 |
| 8. | Kishbaugh, T. L. S. Top. Heterocycl. Chem. 2012, 29, 1–45. doi:10.1007/7081_2012_76 |
| 22. | Artemova, N. V.; Chevykalova, M. N.; Luzikov, Y. N.; Nifant'ev, I. E.; Nifant'ev, E. E. Tetrahedron 2004, 60, 10365–10370. doi:10.1016/j.tet.2004.07.082 |
| 44. | Zhu, R.; Xing, L.; Wang, X.; Cheng, C.; Su, D.; Hu, Y. Adv. Synth. Catal. 2008, 350, 1253–1257. doi:10.1002/adsc.200700535 |
| 3. | Gschwend, H. W.; Rodriguez, H. R. Org. React. 2005, 26, 1–360. doi:10.1002/0471264180.or026.01 |
| 4. | Beak, P.; Snieckus, V. Acc. Chem. Res. 1982, 15, 306–312. doi:10.1021/ar00082a002 |
| 5. | Snieckus, V. Chem. Rev. 1990, 90, 879–933. doi:10.1021/cr00104a001 |
| 6. | Gant, T. G.; Meyers, A. I. Tetrahedron 1994, 50, 2297–2360. doi:10.1016/S0040-4020(01)86953-2 |
| 7. |
Schlosser, M. Organometallics in Synthesis, 2nd ed.; Wiley-VCH: New York, NY, U.S.A., 2002.
Chapter I. |
| 23. | Mulvey, R. E. Organometallics 2006, 25, 1060–1075. doi:10.1021/om0510223 |
| 24. | Mulvey, R. E.; Mongin, F.; Uchiyama, M.; Kondo, Y. Angew. Chem., Int. Ed. 2007, 46, 3802–3824. doi:10.1002/anie.200604369 |
| 25. | Mulvey, R. E. Acc. Chem. Res. 2009, 42, 743–755. doi:10.1021/ar800254y |
| 26. | Haag, B.; Mosrin, M.; Ila, H.; Malakhov, V.; Knochel, P. Angew. Chem., Int. Ed. 2011, 50, 9794–9824. doi:10.1002/anie.201101960 |
| 27. | Mongin, F.; Uchiyama, M. Curr. Org. Chem. 2011, 15, 2340–2361. doi:10.2174/138527211796150651 |
| 28. | Mongin, F.; Harrison-Marchand, A. Chem. Rev. 2013, 113, 7563–7727. doi:10.1021/cr3002966 |
| 29. | Mulvey, R. E. Dalton Trans. 2013, 42, 6676–6693. doi:10.1039/c3dt00053b |
| 30. | Harford, P. J.; Peel, A. J.; Chevallier, F.; Takita, R.; Mongin, F.; Uchiyama, M.; Wheatley, A. E. H. Dalton Trans. 2014, 43, 14181–14203. doi:10.1039/c4dt01130a |
| 45. | Antilla, J. C.; Baskin, J. M.; Barder, T. E.; Buchwald, S. L. J. Org. Chem. 2004, 69, 5578–5587. doi:10.1021/jo049658b |
| 15. | Faigl, F.; Fogassy, K.; Szántó, Z.; Lopata, A.; Töke, L. Tetrahedron 1998, 54, 4367–4374. doi:10.1016/S0040-4020(98)00150-1 |
| 16. | Fogassy, K.; Kovács, K.; Keserű, G. M.; Tőke, L.; Faigl, F. J. Chem. Soc., Perkin Trans. 1 2001, 1039–1043. doi:10.1039/b100008j |
| 19. | Faigl, F.; Fogassy, K.; Szűcs, E.; Kovács, K.; Keserű, G. M.; Harmat, V.; Böcskei, Z.; Tőke, L. Tetrahedron 1999, 55, 7881–7892. doi:10.1016/s0040-4020(99)00398-1 |
| 20. | Faigl, F.; Mátravölgyi, B.; Deák, S.; Holczbauer, T.; Czugler, M.; Balázs, L.; Hermecz, I. Tetrahedron 2012, 68, 4259–4266. doi:10.1016/j.tet.2012.03.070 |
| 58. | Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/S0021889899006020 |
| 14. | Faigl, F.; Fogassy, K.; Thurner, A.; Töke, L. Tetrahedron 1997, 53, 4883–4888. doi:10.1016/S0040-4020(97)00183-X |
| 21. | Rataboul, F.; Zapf, A.; Jackstell, R.; Harkal, S.; Riermeier, T.; Monsees, A.; Dingerdissen, U.; Beller, M. Chem. – Eur. J. 2004, 10, 2983–2990. doi:10.1002/chem.200306026 |
| 59. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117 |
| 11. | Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1 |
| 13. | Thurner, A.; Faigl, F.; Ágai, B.; Töke, L. Synth. Commun. 1998, 28, 443–449. doi:10.1080/00397919808005098 |
| 56. | Altomare, A.; Burla, M. C.; Camalli, M.; Cascarano, G. L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A. G. G.; Polidori, G.; Spagna, R. J. Appl. Crystallogr. 1999, 32, 115–119. doi:10.1107/S0021889898007717 |
| 11. | Faigl, F.; Schlosser, M. Tetrahedron 1993, 49, 10271–10278. doi:10.1016/S0040-4020(01)80556-1 |
| 12. | Shirley, D. A.; Gross, B. H.; Roussel, P. A. J. Org. Chem. 1955, 20, 225–231. doi:10.1021/jo01120a012 |
| 17. | Faigl, F.; Feldhoffer, B. V.; Thurner, A. Synth. Commun. 2006, 36, 2841–2849. doi:10.1080/00397910600770672 |
| 18. | Faigl, F.; Vas-Feldhoffer, B.; Kubinyi, M.; Pál, K.; Tárkányi, G.; Czugler, M. Tetrahedron: Asymmetry 2009, 20, 98–103. doi:10.1016/j.tetasy.2009.01.010 |
| 57. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, A64, 112–122. doi:10.1107/S0108767307043930 |
| 44. | Zhu, R.; Xing, L.; Wang, X.; Cheng, C.; Su, D.; Hu, Y. Adv. Synth. Catal. 2008, 350, 1253–1257. doi:10.1002/adsc.200700535 |
| 31. | L'Helgoual'ch, J. M.; Seggio, A.; Chevallier, F.; Yonehara, M.; Jeanneau, E.; Uchiyama, M.; Mongin, F. J. Org. Chem. 2008, 73, 177–183. doi:10.1021/jo7020345 |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 34. | Seggio, A.; Chevallier, F.; Vaultier, M.; Mongin, F. J. Org. Chem. 2007, 72, 6602–6605. doi:10.1021/jo0708341 |
| 35. | Snégaroff, K.; Komagawa, S.; Chevallier, F.; Gros, P. C.; Golhen, S.; Roisnel, T.; Uchiyama, M.; Mongin, F. Chem. – Eur. J. 2010, 16, 8191–8201. doi:10.1002/chem.201000543 |
| 36. | Snégaroff, K.; Nguyen, T. T.; Marquise, N.; Halauko, Y. S.; Harford, P. J.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Chevallier, F.; Wheatley, A. E. H.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2011, 17, 13284–13297. doi:10.1002/chem.201101993 |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 38. | Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e |
| 39. | Kadiyala, R. R.; Tilly, D.; Nagaradja, E.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Halauko, Y. S.; Chevallier, F.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2013, 19, 7944–7960. doi:10.1002/chem.201300552 |
| 40. | Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Mongin, F. Org. Biomol. Chem. 2014, 12, 1475–1487. doi:10.1039/c3ob42380h |
| 41. | Marquise, N.; Bretel, G.; Lassagne, F.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Gros, P. C.; Mongin, F. RSC Adv. 2014, 4, 19602–19612. doi:10.1039/c4ra02583k |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 38. | Chevallier, F.; Blin, T.; Nagaradja, E.; Lassagne, F.; Roisnel, T.; Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2012, 10, 4878–4885. doi:10.1039/c2ob25554e |
| 40. | Nagaradja, E.; Chevallier, F.; Roisnel, T.; Dorcet, V.; Halauko, Y. S.; Ivashkevich, O. A.; Matulis, V. E.; Mongin, F. Org. Biomol. Chem. 2014, 12, 1475–1487. doi:10.1039/c3ob42380h |
| 42. | Matulis, V. E.; Halauko, Y. S.; Ivashkevich, O. A.; Gaponik, P. N. J. Mol. Struct.: THEOCHEM 2009, 909, 19–24. doi:10.1016/j.theochem.2009.05.024 |
| 43. | Halauko, Y. S.; Matulis, V. E.; Ivashkevich, O. A.; Grigoriev, Y. V.; Gaponik, P. N. Tetrahedron 2010, 66, 3415–3420. doi:10.1016/j.tet.2010.03.053 |
| 60. | Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Bioorg. Med. Chem. 2014, 22, 3498–3507. doi:10.1016/j.bmc.2014.04.028 |
| 47. | Teo, Y.-C.; Yong, F.-F.; Sim, S. Tetrahedron 2013, 69, 7279–7284. doi:10.1016/j.tet.2013.06.095 |
| 60. | Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Bioorg. Med. Chem. 2014, 22, 3498–3507. doi:10.1016/j.bmc.2014.04.028 |
| 60. | Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Bioorg. Med. Chem. 2014, 22, 3498–3507. doi:10.1016/j.bmc.2014.04.028 |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 35. | Snégaroff, K.; Komagawa, S.; Chevallier, F.; Gros, P. C.; Golhen, S.; Roisnel, T.; Uchiyama, M.; Mongin, F. Chem. – Eur. J. 2010, 16, 8191–8201. doi:10.1002/chem.201000543 |
| 35. | Snégaroff, K.; Komagawa, S.; Chevallier, F.; Gros, P. C.; Golhen, S.; Roisnel, T.; Uchiyama, M.; Mongin, F. Chem. – Eur. J. 2010, 16, 8191–8201. doi:10.1002/chem.201000543 |
| 37. | Chevallier, F.; Halauko, Y. S.; Pecceu, C.; Nassar, I. F.; Dam, T. U.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Mongin, F. Org. Biomol. Chem. 2011, 9, 4671–4684. doi:10.1039/c1ob05267e |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 34. | Seggio, A.; Chevallier, F.; Vaultier, M.; Mongin, F. J. Org. Chem. 2007, 72, 6602–6605. doi:10.1021/jo0708341 |
| 36. | Snégaroff, K.; Nguyen, T. T.; Marquise, N.; Halauko, Y. S.; Harford, P. J.; Roisnel, T.; Matulis, V. E.; Ivashkevich, O. A.; Chevallier, F.; Wheatley, A. E. H.; Gros, P. C.; Mongin, F. Chem. – Eur. J. 2011, 17, 13284–13297. doi:10.1002/chem.201101993 |
| 60. | Hedidi, M.; Bentabed-Ababsa, G.; Derdour, A.; Roisnel, T.; Dorcet, V.; Chevallier, F.; Picot, L.; Thiéry, V.; Mongin, F. Bioorg. Med. Chem. 2014, 22, 3498–3507. doi:10.1016/j.bmc.2014.04.028 |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 45. | Antilla, J. C.; Baskin, J. M.; Barder, T. E.; Buchwald, S. L. J. Org. Chem. 2004, 69, 5578–5587. doi:10.1021/jo049658b |
| 33. | Seggio, A.; Lannou, M. I.; Chevallier, F.; Nobuto, D.; Uchiyama, M.; Golhen, S.; Roisnel, T.; Mongin, F. Chem. – Eur. J. 2007, 13, 9982–9989. doi:10.1002/chem.200700608 |
| 51. | Fraser, R. R.; Mansour, T. S.; Savard, S. Can. J. Chem. 1985, 63, 3505–3509. doi:10.1139/v85-574 |
© 2015 Messaoud et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)