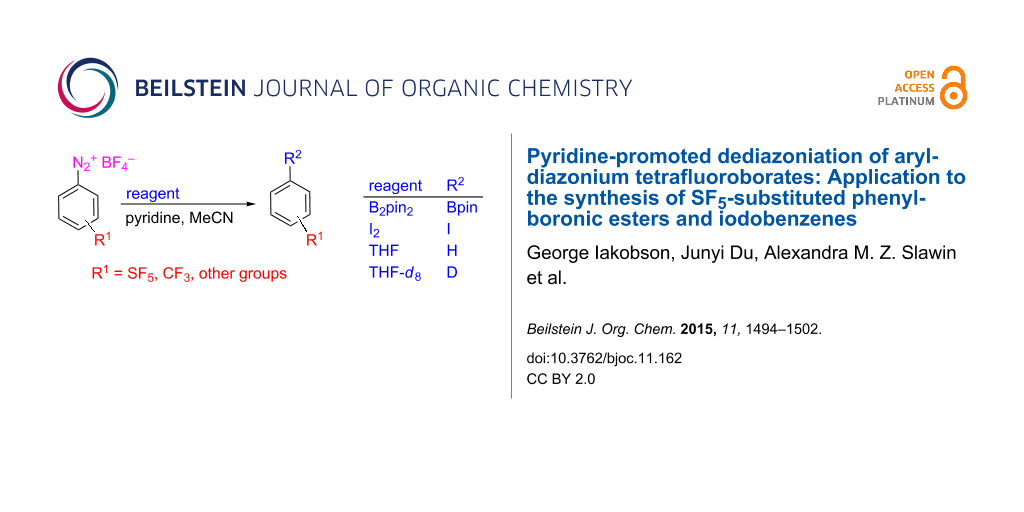
Abstract
Pyridine promotes dediazoniation of aryldiazonium tetrafluoroborates. The formed aryl radicals were trapped with B2pin2, iodine, or tetrahydrofuran to afford boronic esters, iodobenzenes and benzenes, respectively. The application to the synthesis of (pentafluorosulfanyl)phenylboronic esters, iodo(pentafluorosulfanyl)benzenes and (pentafluorosulfanyl)benzene is shown.

Graphical Abstract
Introduction
Pentafluorosulfanyl-containing compounds have been known for more than half a century [1-4]; however, for a long time they remained a relatively underdeveloped class of compounds [5,6]. The main reason for the slow development of the chemistry of SF5-containing compounds was the lack of availability of key building blocks. However, in recent years, the scientific community has been witnessing a renewed interest in this functional group. Synthetic methods towards aliphatic SF5-containing compounds are based on free radical addition of SF5Cl or SF5Br to unsaturated compounds [7-9], whereas aromatic derivatives are available either by the Umemoto’s two-step synthesis from diaryl disulfides or benzenethiols [10-12], or by the reaction of nitrophenyl disulfides with elemental fluorine [13-16]. Aromatic and heteroaromatic SF5 compounds are mostly prepared by the derivatization of commercial nitro-(pentafluorosulfanyl)benzenes [14,17-27] and approaches from SF5-aliphatics have also been studied [28-30]. The unique combination of properties the SF5 group imparts includes high chemical, thermal, and metabolic stability, strong electron-acceptor property, and high lipophilicity. Furthermore, applications of SF5 compounds in catalysis [31,32], life-science [6,18,33-38], and material sciences [5,19,38,39] are emerging.
Arylboronic acids and arylboronates represent versatile building blocks in organic synthesis [40]. They have found wide applications in transition metal-catalyzed cross-coupling reactions [41,42]. These boron compounds are accessed mainly by the reactions of arylmagnesium or aryllithium species with trialkylboronates [43,44], Pd- or Cu-catalyzed borylations of aryl halides using B2pin2, H-Bpin [45-50] or R2N-BH2 [51], direct borylations via aromatic C–H bond activations [52-58], Lewis acid catalyzed electrophilic borylations of electron-rich arenes [59-62], and Sandmeyer-type borylation of arylamines or diazonium salts with B2pin2 [63-67], B2(OH)4 [68] or R2N-BH2 [69]. Several attempts were made to synthesize the SF5-phenylboronates. Patent literature describes the synthesis of 3- or 4-(pentafluorosulfanyl)phenylboronates or boronic acids from SF5-bromobenzenes via lithiation or magnesiation. These approaches suffer from low yields and other drawbacks [70,71]. For lithiation of the aryl bromide, t-BuLi had to be used and the formation of Grignard reagents is inefficient. On the other hand, Shibata and co-workers have recently reported the synthesis of 3,5-bis(pentafluorosulfanyl)phenylboronic acid from the corresponding aryl bromide, trimethyl borate and iPrMgBr [32]. Finally, Joliton and Carreira have recently shown efficient Ir-catalyzed C–H borylation of several 1-substituted-3-(pentafluorosulfanyl)benzenes and applied the products of borylation to the Pd-catalyzed Suzuki–Miyaura reaction with aryl bromides or iodides. However, the reaction is limited to borylations in position five of 1-substituted-3-(pentafluorosulfanyl)benzenes [72].
Straightforward access to SF5-phenylboronic acids or boronates would be highly desirable since it would allow easy installation of the SF5-phenyl group by the subsequent Suzuki–Miyaura reaction. Nitro-(pentafluorosulfanyl)benzenes are the primary industrial SF5-aromatics, therefore the easiest access to SF5-phenylboronates appears to be starting from readily available SF5-substituted anilines or diazonium salts rather than SF5-containing halobenzenes. Herein, we report a new protocol for efficient borylation, iodination and hydrodediazoniation of SF5-phenyldiazonium tetrafluoroborates in the presence of pyridine. The generality of the borylation and iodination reactions was demonstrated on several examples.
Results and Discussion
At the onset of our investigation, Sandmeyer-type borylation of 3- and 4-(pentafluorosulfanyl)anilines (1a and 1b, respectively) to pinacolboronates 2a and 2b according to Wang and co-corkers was studied [64,65] (Table 1). The borylation of 1a took place in a reasonable yield in the presence of catalytic amounts of benzoyl peroxide (BPO, Table 1, entry 1), while for 1b, heating without any additives was preferable; however, the yield of 2b was only moderate (Table 1, entry 3).
Table 1: Synthesis of boronates 2 from aniline derivatives 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-162-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | 1 | Additive | Temp. (°C) | 2, Yield (%)b |
|---|---|---|---|---|
| 1 | 1a, 3-SF5 | BPO | rt | 2a, 70 |
| 2 | 1b, 4-SF5 | BPO | rt | 2b, traces |
| 3 | 1b, 4-SF5 | — | 80 | 2b, 55c |
aReaction conditions: 1 (1 mmol), t-BuONO (1.5 mmol), B2pin2 (1.1 mmol), additive (2 mol %), MeCN (4 mL). bIsolated yield. c82% purity.
For a detailed investigation of the borylation reaction, the diazonium tetrafluoroborates 3a and 3b were prepared and isolated according to Doyle conditions [73]. High yields (above 90%) of both isomers of the diazonium tetrafluoroborates 3a and 3b were obtained on 0.3–6 g scale (Table 2). Crystal structures of 3a (CCDC 1009848) and 3b (CCDC 1009849) were determined confirming the nature of the products. During the course of our studies, Okazaki and co-workers reported the synthesis of 3b in 84% yield under similar conditions and have shown its reactivity in various cross-coupling reactions with varied degree of success. The most efficient cross-coupling reactions were the Heck reactions with alkenes, a biaryl homocoupling reaction, an azo coupling to electron-rich arenes, and a dediazoniation with TMSN3 in an ionic liquid medium [74,75].
Table 2: Synthesis of diazonium tetrafluoroborates 3a and boronates 2b.
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-162-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 3 (mmol) | Reagents (equiv), solvent | Temp. (°C) | Time (h) | 2, Yield (%)c |
|---|---|---|---|---|---|
| 1 | 3a (1) | Pd(OAc)2 (0.01), L (0.02), THF | rt | 18 | 2a, 43 |
| 2 | 3b (1) | Pd(OAc)2 (0.01), L (0.02), THF | rt | 18 | 2b, 57 |
| 3 | 3a (1) | NaOAc (2), MeCN | −50 to rt | 3 | 2a, 73 |
| 4 | 3b (1) | NaOAc (2), MeCN | −50 to rt | 3 | 2b, 49 |
| 5 | 3b (1) | CuBr (0.05), MeCN/H2O (3:1) | rt | 330 | 2b, 23 |
| 6 | 3a (1) | —, MeOH | rt | 24 | 2a, 32 |
| 7 | 3b (1) | —, pyridined | −30 to rt | 2 | 2b, 55 |
| 8 | 3b (1) | pyridine (4), MeCNd | −30 to rt | 2 | 2b, 77 |
| 9 | 3b (3) | pyridine (4), MeCNd | −30 to rt | 2 | 2b, 77 |
| 10 | 3a (2) | pyridine (4), MeCNd | −30 to rt | 2 | 2a, 80 |
aReaction conditions: 1 (1–28 mmol), BF3·OEt2 (2.1 equiv), t-BuONO (1 equiv), CH2Cl2 or Et2O (3 mL/1 mmol of 1), 30 min. bReaction conditions: 3 (1–3 mmol), B2pin2 (1 equiv), reagents, solvent (2 mL/1 mmol of 3) under N2. cIsolated yield. dThe reaction was conducted under air.
An efficient borylation of aryldiazonium tetrafluoroborates with NHC-Pd catalysts was reported recently [63]. When applied to 3a and 3b using Pd(OAc)2 and NHC ligand precursors L, the borylated products 2a and 2b were isolated in only moderate yields (Table 2, entries 1 and 2). However, it was found that the Pd catalyst was not required for an efficient reaction. Alkali metal acetates are known to facilitate decomposition of aryldiazonium salts by the formation of diazoacetates and diazo anhydrides, which decompose to aryl radicals [76]. These additives were used in Meerwein arylation of isopropenyl acetate [77]. In our case, two-fold excess of sodium acetate in acetonitrile afforded 2a and 2b in good and moderate yields, respectively (Table 2 ,entries 3 and 4). The conditions reported by Yu and co-workers [66] (Table 2, entry 5) provided a mixture with starting 3b as the major component. While the diazonium salts 3a and 3b were found to be stable in acetonitrile, we observed slow decomposition in methanol and in the presence of B2pin2 under strictly Pd-free conditions (new Teflon stirring bar and glassware), the borylation took place with low conversion (Table 2, entry 6) [78]. In pyridine, however, the decomposition of 3b was very fast and a vigorous evolution of nitrogen was observed affording the borylated product in a moderate yield together with a mixture of SF5-pyridines in ca. 10% GC–MS yield (Table 2, entry 7). Finally, the use of a 4-fold excess of pyridine in acetonitrile was found to be optimal. Conducting the reaction on a gram scale proceeded without a notable loss of efficiency and the reaction can be performed in air (Table 2, entries 8–10).
Pyridine is known to induce decomposition of aryldiazonium salts. Zollinger and Abramovitch studied the interaction of pyridine with aryldiazonium tetrafluoroborates and suggested the formation of diazopyridinium salts which homolytically decompose to aryl radicals, nitrogen and a pyridinium tetrafluoroborate radical [79,80]. Tanaka and co-workers have used the combination of PhN2+BF4− and pyridine for arylations of silylenol ethers [81]. They have observed the formation of large amounts of phenylpyridines. In our case, we detected SF5-phenylpyridines in trace amounts only during borylation using pyridine as a solvent. The major products in the borylation reaction apart from 2a and 2b were found to be F-Bpin and pyridine·BF3 complexes. Both compounds are easily hydrolyzable but they were observed by NMR of the crude reaction mixture and compared to the synthetized authentic samples. On the other hand, we were not able to observe a pyridine·B2pin2 complex by 11B NMR in CD3CN or CDCl3. The borylation was extended to several other aryldiazonium tetrafluoroborates showing that both electron-donor and electron-acceptor substituted phenyldiazonium tetrafluoroborates undergo efficient borylation with an equimolar amount of B2pin2 (Scheme 1); however, ortho-substituted phenyldiazonium salts were found to be either not efficient substrates (3f) or completely unreactive (3g), presumably due to a large steric demand of B2pin2.
![[1860-5397-11-162-i1]](/bjoc/content/inline/1860-5397-11-162-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Borylation of aryldiazonium tetrafluoroborates 3. Reaction conditions: 3 (1 mmol), B2pin2 (1 mmol), pyridine (4 mmol), MeCN (2 mL), 2 h.
Scheme 1: Borylation of aryldiazonium tetrafluoroborates 3. Reaction conditions: 3 (1 mmol), B2pin2 (1 mmol),...
The mechanism of this borylation reaction remains to be elucidated. Based on experimental results and literature precedent, we propose the following free-radical mechanism (Scheme 2). Aryldiazonium salt 3 reacts with pyridine to form aryldiazopyridinium 4 which decomposes to an aryl radical, a pyridinium tetrafluoroborate radical and nitrogen. The aryl radical reacts with B2pin2 to form the borylated product 2 and the Bpin radical (likely to be stabilized by pyridine) [82]. The byproducts pyridine·BF3 and F-Bpin are formed by the reaction of the pyridinium tetrafluoroborate radical and the Bpin radical.
![[1860-5397-11-162-i2]](/bjoc/content/inline/1860-5397-11-162-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Subjecting the diazonium salt 3i to the borylation conditions gave further indirect evidence for the formation of aryl radicals (Scheme 3). Full conversion of 3i was observed affording a mixture of products 2i, 2i’, 5i (two isomers of unknown configuration) and 6i in 16:31:28:25 GC–MS ratio. The presence of these products can be explained only by the formation of the substituted SF5-phenyl radical which undergoes borylation to 2i, hydrogen atom transfer followed by borylation to 2i’, intramolecular cyclization to 5i or hydrogen abstraction to 6i.
![[1860-5397-11-162-i3]](/bjoc/content/inline/1860-5397-11-162-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of diazonium salt 3i under borylation conditions.
Scheme 3: Reaction of diazonium salt 3i under borylation conditions.
Starting from aniline derivative 1b, a one pot diazotization–borylation sequence using different acids afforded the corresponding borylated product 2b in good yields (Table 3).
Table 3: One pot diazotization-borylation of 1ba.
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-162-i9.svg?max-width=637&scale=1.0)
|
|||
| Entry | Acid (equiv) | Time (h) | 2b, Yield (%)b |
|---|---|---|---|
| 1c | p-TsOH·H2O (1) | 15 | 51 |
| 2 | aq HBF4 (1.7) | 1 | 81 |
| 3 | aq HCl (1.7) | 1 | 78 |
aReaction conditions: 1b (1 mmol), acid, t-BuONO (1 mmol), MeCN (3 mL), B2pin2 (1.0–1.1 mmol), pyridine (4 mmol). bIsolated yield. cReaction temperature for steps 1 and 2 was rt.
Suzuki–Miyaura cross-coupling reactions of boronates 2a and 2b with aryl iodides using a simple system without any optimization proceeded in satisfactory yields considering the electron-deficient character of the boronates and consequently less efficient transmetallation step (Scheme 4).
![[1860-5397-11-162-i4]](/bjoc/content/inline/1860-5397-11-162-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Suzuki–Miyaura reaction of boronates 2a and 2b with aryl iodides. Reaction conditions: 2 (1 mmol), ArI (1.1 mmol), Pd(PPh3)4 (5 mol %), LiOH·H2O (4 mmol), 1,4-dioxane (2 mL), water (1 mL), 3.5 h.
Scheme 4: Suzuki–Miyaura reaction of boronates 2a and 2b with aryl iodides. Reaction conditions: 2 (1 mmol), ...
Transformation to SF5-phenylboronic acid 8b and potassium trifluoroborates 9 was straightforward under standard conditions (Scheme 5). Similar potassium SF5-phenyltrifluoroborates were found to be highly reactive with a variety of aryl bromides and iodides in the presence of catalytic amounts of PdCl2(dppf)·CH2Cl2 or Pd(OAc)2 [72]. The recently published synthesis of arylboronic acids from anilines or aryldiazonium tetrafluoroborates using B2(OH)4 [68] applied to 3b provided 8b in only 25% 19F NMR yield.
![[1860-5397-11-162-i5]](/bjoc/content/inline/1860-5397-11-162-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Syntesis of boronic acid 8b and trifluoroborates 9. Reaction conditions for the synthesis of 8b: 2 (2 mmol), NaIO4 (8 mmol), THF (8 mL), H2O (2 mL), rt, 1 h. Reaction conditions for the synthesis of 9: 2 (2 mmol), KHF2 (10 mmol), MeOH (8 mL), H2O (3.6 mL), rt, 20 min.
Scheme 5: Syntesis of boronic acid 8b and trifluoroborates 9. Reaction conditions for the synthesis of 8b: 2 ...
To extend the synthetic utility of the pyridine-mediated derivatization of aryldiazonium tetrafluoroborates we investigated the reaction with iodobenzene and iodine as efficient scavengers of aryl radicals [83,84]. A competitive experiment starting from 3a and equimolar amounts of B2pin2 and iodobenzene in the presence of pyridine (4 equiv) in MeCN afforded a mixture of 2a (72% yield) and 1-iodo-3-(pentafluorosulfanyl)benzene (10a, 23% yield). Additionally, a reaction of 3b with PhI (4 equiv) in the absence of B2pin2 gave 1-iodo-4-(pentafluorosulfanyl)benzene (10b) in 35% yield. Both experiments point to the formation of aryl radicals during the reaction and suggest a possibility to conduct practical aromatic iodination. Indeed, the iodination reaction with I2 proved to be more efficient than with PhI (Scheme 6). In contrast to borylation, iodination with I2 shows a higher sensitivity to electronic properties of substituents on the aromatic ring. Electron-acceptor substituted aryldiazonium compounds are excellent substrates while those with electron-donor groups react much less efficiently. The substitution of pyridine with collidine (2,4,6-trimethylpyridine) gave similar yields. Unlike borylations, the iodination reactions were not sensitive to ortho substitutions. In the case of 3i, compound 10i was the sole product; no product of hydrogen atom transfer or cyclization was observed, demonstrating that the reaction with I2 is much faster than with B2pin2. Importantly, the yields of SF5-phenyl iodides 10a and 10b using our two-step diazotization–iodination method significantly exceed those obtained by classical Sandmeyer reaction (ca. 80% yield over two steps compared to 63% for 10a and 50% for 10b by one-pot Sandmeyer procedure requiring 10 fold excess of KI) [14]. The side-product in the iodination of 3 was bis(pyridine)iodonium tetrafluoroborate, which can be easily isolated from the reaction mixture by precipitation upon addition of diethyl ether. This iodonium salt was first synthetized by Barluenga [85] and later used for mild iodination of alkenes, alkynes and aromatics [85-87]. Its formation can be explained by the reaction of the pyridinium tetrafluoroborate radical (Scheme 2), pyridine and I2 or iodine radical. Bromination of 3b with Br2 was attempted under conditions similar to iodination but the reaction was slow and inefficient resulting in a mixture of products with expected 1-bromo-4-(pentafluorosulfanyl)benzene as a minor product. With equimolar NBS instead of bromine, the reaction is much cleaner but slow; after overnight at ambient temperature the bromo product was isolated in 35% yield.
![[1860-5397-11-162-i6]](/bjoc/content/inline/1860-5397-11-162-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Iodination of aryldiazonium tetrafluoroborates 3. Reaction conditions: 3 (1 mmol), I2 (1.1 mmol), pyridine (4 mmol), MeCN (3 mL), 2 h. aYields in the presence of collidine instead of pyridine.
Scheme 6: Iodination of aryldiazonium tetrafluoroborates 3. Reaction conditions: 3 (1 mmol), I2 (1.1 mmol), p...
Finally, hydrodediazoniation using tributyltin hydride or THF was tested and THF proved to be a more efficient hydrogen atom donor. The addition of excess pyridine to MeCN/THF solution of diazonium tetrafluoroborates 3a or 3b led to an efficient hydrodediazoniation and the formation of (pentafluorosulfanyl)benzene (6). Deuteration experiments established that the hydrogen atom in the product comes exclusively from THF and not from pyridine or MeCN (Table 4). The observed deuterium enrichment using THF-d8 was around 80%. Thermal decomposition of aryldiazonium salts prepared from immobilized triazene precursors and the formation of deuterated aromatics using THF-d8 was reported [88]. We explain the reduced yield of 6-D (48% yield) and the formation of tar products by hydrogen atom abstraction from 3 or 6 and subsequent polymerization. No significant amounts of double deuterated products were detected. The kinetic isotope effect was determined from intermolecular competition experiment using 3b and a 1:1 mixture of THF and THF-d8 giving KIE = 5.5 (6:6-D ratio determined by GC–MS) and combined yield of 62%. This means that the hydrogen abstraction is much faster than the deuterium abstraction and suggests the C-H(D) bond formation as the rate-limiting step. For unambiguous identification of the rate-limiting step, individual rate constants kH and kD in two parallel reactions would have to be determined [89]. Dihydrofuran (11) and pyridinium tetrafluoroborate were identified as byproducts of the dediazoniation reactions. Similarly to the previous processes, we presume the formation of aryldiazopyridinium 4 and its decomposition to an aryl radical and the pyridinium tetrafluoroborate radical. The aryl radical abstracts a hydrogen atom from THF forming 6 and a THF radical. The THF radical then transfers the hydrogen atom to the pyridinium tetrafluoroborate radical giving dihydrofuran (11) and pyridinium salt.
Table 4: Hydrodediazoniation of 3a and 3b with THFa.
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-162-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry | 3 (mmol) | D source | D enrichmentb | 6, Yield (%)c |
|---|---|---|---|---|
| 1 | 3a (0.5) | — | n/a | 6, 75 |
| 2 | 3b (0.5) | — | n/a | 6, 70 |
| 3 | 3b (0.5) |
THF-d8
(0.5 mL) |
77–82 | 6-D, 48 |
| 4 | 3b (0.25) |
C5D5N
(4 mmol) |
0 | 6, 70 |
| 5 | 3b (0.25) |
CD3CN
(0.75 mL) |
0 | 6, 67 |
aReaction conditions: 3 (0.25–0.5 mmol), pyridine (4 equiv), THF (1 mL/1 mmol of 3), MeCN (3–4 mL/1 mmol of 3), 2 h. bBased on GC–MS. cBased on 19F NMR using 1-nitro-4-(pentafluorosulfanyl)benzene as an internal standard.
Conclusion
In conclusion, a novel dediazoniation–borylation methodology was developed based on the reaction of aryldiazonium tetrafluoroborates with pyridine and B2pin2 to give arylpinacolborates. Particular emphasis was on the synthesis of SF5-phenylboronates where our methodology represents a considerable improvement in reaction efficiency compared to previously published syntheses. Furthermore, no transition metals are needed and mild reaction conditions are used. The borylation is applicable to a variety of aryldiazonium tetrafluoroborates with electron-donor or acceptor groups while ortho-substituted substrates are less reactive. A mechanism involving aryl radicals is suggested. The Suzuki–Miyaura reaction of SF5-phenylboronates with aryl iodides provided the cross-coupling biaryl products. In analogy to the borylation reaction, iodination of aryldiazonium tetrafluoroborates with pyridine and iodine resulted in aryl iodides. An efficient reaction was observed with electron-acceptor substituted aromatic compounds even with ortho-substituted derivatives. In the case of SF5-substituted iodobenzenes, the method is much more efficient than the classical Sandmeyer reaction starting from SF5-containing aniline derivatives. Finally, hydrodediazoniation of SF5-phenyldiazonium tetrafluoroborates by hydrogen atom abstraction from THF in the presence of pyridine provided (pentafluorosulfanyl)benzene.
Supporting Information
Synthesis and characterization of all products, copies of 1H, 13C, and 19F NMR spectra of newly synthesized products, and X-ray crystallographic files of the compounds 3a and 3b.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.9 MB | Download |
| Supporting Information File 2: Crystal structure of compound 3a. | ||
| Format: CIF | Size: 11.6 KB | Download |
| Supporting Information File 3: Crystal structure of compound 3b. | ||
| Format: CIF | Size: 20.3 KB | Download |
References
-
Clifford, A. F.; El-Shamy, H. K.; Emeleus, H. J.; Haszeldine, R. N. J. Chem. Soc. 1953, 2372–2375. doi:10.1039/jr9530002372
Return to citation in text: [1] -
Hoffmann, F. W.; Simmons, T. C.; Beck, R. B.; Holler, H. V.; Katz, T.; Koshar, R. J.; Larsen, E. R.; Mulvaney, J. E.; Rogers, F. E.; Singleton, B.; Sparks, R. S. J. Am. Chem. Soc. 1957, 79, 3424–3429. doi:10.1021/ja01570a029
Return to citation in text: [1] -
Dresdner, R. D.; Mao, T. J.; Young, J. A. J. Am. Chem. Soc. 1959, 81, 574–577. doi:10.1021/ja01545a027
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1960, 82, 4751–4752. doi:10.1021/ja01502a083
Return to citation in text: [1] -
Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u
Return to citation in text: [1] [2] -
Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030
Return to citation in text: [1] [2] -
Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003
Return to citation in text: [1] -
Vida, N.; Pastýříková, T.; Klepetářová, B.; Beier, P. J. Org. Chem. 2014, 79, 8906–8911. doi:10.1021/jo501562z
Return to citation in text: [1] -
Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53
Return to citation in text: [1] -
Lummer, K.; Ponomarenko, M. V.; Röschenthaler, G.-V.; Bremer, M.; Beier, P. J. Fluorine Chem. 2014, 157, 79–83. doi:10.1016/j.jfluchem.2013.10.009
Return to citation in text: [1] -
Kanishchev, O. S.; Dolbier, W. R., Jr. Angew. Chem., Int. Ed. 2015, 127, 282–286. doi:10.1002/ange.201409990
Return to citation in text: [1] -
Bowden, R. D.; Greenhall, M. P.; Moilliet, J. S.; Thomson, J. Chem. Abstr. 1997, 126, 199340.
The preparation of fluorinated organic compounds, WO 9705106, 13 Feb 1997.
Return to citation in text: [1] -
Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8
Return to citation in text: [1] [2] [3] -
Chambers, R. D.; Spink, R. C. H. Chem. Commun. 1999, 883–884. doi:10.1039/A901473J
Return to citation in text: [1] -
Sipyagin, A. M.; Bateman, C. P.; Matsev, A. V.; Waterfeld, A.; Jilek, R. E.; Key, C. D.; Szulczewski, G. J.; Thrasher, J. S. J. Fluorine Chem. 2014, 167, 203–210. doi:10.1016/j.jfluchem.2014.07.031
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006
Return to citation in text: [1] -
Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142.
Return to citation in text: [1] [2] -
Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<1989::AID-ANIE1989>3.0.CO;2-K
Return to citation in text: [1] [2] -
Frischmuth, A.; Unsinn, A.; Groll, K.; Stadtmüller, H.; Knochel, P. Chem. – Eur. J. 2012, 18, 10234–10238. doi:10.1002/chem.201201485
Return to citation in text: [1] -
Beier, P.; Pastýříková, T.; Vida, N.; Iakobson, G. Org. Lett. 2011, 13, 1466–1469. doi:10.1021/ol2001478
Return to citation in text: [1] -
Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p
Return to citation in text: [1] -
Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011
Return to citation in text: [1] -
Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127
Return to citation in text: [1] -
Vida, N.; Beier, P. J. Fluorine Chem. 2012, 143, 130–134. doi:10.1016/j.jfluchem.2012.04.001
Return to citation in text: [1] -
Iakobson, G.; Pošta, M.; Beier, P. Synlett 2013, 24, 855–859. doi:10.1055/s-0032-1318452
Return to citation in text: [1] -
Wang, C.; Yu, Y.-B.; Fan, S.; Zhang, X. Org. Lett. 2013, 15, 5004–5007. doi:10.1021/ol4023326
Return to citation in text: [1] -
Sergeeva, T. A.; Dolbier, W. R., Jr. Org. Lett. 2004, 6, 2417–2419. doi:10.1021/ol0491991
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Zheng, Z. J. Fluorine Chem. 2011, 132, 389–393. doi:10.1016/j.jfluchem.2011.03.017
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Zheng, Z. J. Org. Chem. 2009, 74, 5626–5628. doi:10.1021/jo9007699
Return to citation in text: [1] -
Lee, J.-W.; List, B. J. Am. Chem. Soc. 2012, 134, 18245–18248. doi:10.1021/ja3096202
Return to citation in text: [1] -
Yang, Y.-D.; Lu, X.; Tokunaga, E.; Shibata, N. J. Fluorine Chem. 2012, 143, 204–209. doi:10.1016/j.jfluchem.2012.06.007
Return to citation in text: [1] [2] -
Chia, P. W.; Brennan, S. C.; Slawin, A. M. Z.; Riccardi, D.; O'Hagan, D. Org. Biomol. Chem. 2012, 10, 7922–7927. doi:10.1039/c2ob26402a
Return to citation in text: [1] -
Sun, L.; Li, J.; Bera, H.; Dolzhenko, A. V.; Chiu, G. N. C.; Chui, W. K. Eur. J. Med. Chem. 2013, 70, 400–410. doi:10.1016/j.ejmech.2013.10.022
Return to citation in text: [1] -
Stump, B.; Eberle, C.; Schweizer, W. B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R. L.; Lentz, D.; Diederich, F. ChemBioChem 2009, 10, 79–83. doi:10.1002/cbic.200800565
Return to citation in text: [1] -
Altomonte, S.; Baillie, G. L.; Ross, R. A.; Riley, J.; Zanda, M. RSC Adv. 2014, 4, 20164–20176. doi:10.1039/c4ra01212g
Return to citation in text: [1] -
Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059
Return to citation in text: [1] -
Nakayama, H.; Nishida, J.-i.; Takada, N.; Sato, H.; Yamashita, Y. Chem. Mater. 2012, 24, 671–676. doi:10.1021/cm202650u
Return to citation in text: [1] [2] -
Winter, R.; Nixon, P. G.; Gard, G. L.; Graham, D. J.; Castner, D. G.; Holcomb, N. R.; Grainger, D. W. Langmuir 2004, 20, 5776–5781. doi:10.1021/la040011w
Return to citation in text: [1] -
Hall, D. G., Ed. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley-VCH: Weinheim, Germany, 2005.
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Miyaura, N. Top. Curr. Chem. 2002, 219, 11–59. doi:10.1007/3-540-45313-X
Return to citation in text: [1] -
Brown, H. C.; Cole, T. E. Organometallics 1983, 2, 1316–1319. doi:10.1021/om50004a009
Return to citation in text: [1] -
Brown, H. C.; Srebnik, M.; Cole, T. E. Organometallics 1986, 5, 2300–2303. doi:10.1021/om00142a020
Return to citation in text: [1] -
Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508–7510. doi:10.1021/jo00128a024
Return to citation in text: [1] -
Murata, M.; Watanabe, S.; Masuda, Y. J. Org. Chem. 1997, 62, 6458–6459. doi:10.1021/jo970963p
Return to citation in text: [1] -
Murata, M.; Oyama, T.; Watanabe, S.; Masuda, Y. J. Org. Chem. 2000, 65, 164–168. doi:10.1021/jo991337q
Return to citation in text: [1] -
Fürstner, A.; Seidel, G. Org. Lett. 2002, 4, 541–543. doi:10.1021/ol0171463
Return to citation in text: [1] -
Zhu, W.; Ma, D. Org. Lett. 2006, 8, 261–263. doi:10.1021/ol052633u
Return to citation in text: [1] -
Kleeberg, C.; Dang, L.; Lin, Z.; Marder, T. B. Angew. Chem., Int. Ed. 2009, 48, 5350–5354. doi:10.1002/anie.200901879
Return to citation in text: [1] -
Euzenat, L.; Horhant, D.; Ribourdouille, Y.; Duriez, C.; Alcaraz, G.; Vaultier, M. Chem. Commun. 2003, 2280–2281. doi:10.1039/B306874A
Return to citation in text: [1] -
Chen, H.; Schlecht, S.; Semple, T. C.; Hartwig, J. F. Science 2000, 287, 1995–1997. doi:10.1126/science.287.5460.1995
Return to citation in text: [1] -
Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390–391. doi:10.1021/ja0173019
Return to citation in text: [1] -
Ishiyama, T.; Takagi, J.; Hartwig, J. F.; Miyaura, N. Angew. Chem., Int. Ed. 2002, 41, 3056–3058. doi:10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#
Return to citation in text: [1] -
Preshlock, S. M.; Plattner, D. L.; Maligres, P. E.; Krska, S. W.; Maleczka, R. E., Jr.; Smith, M. R., III. Angew. Chem., Int. Ed. 2013, 52, 12915–12919. doi:10.1002/anie.201306511
Return to citation in text: [1] -
Dai, H.-X.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 134–137. doi:10.1021/ja2097095
Return to citation in text: [1] -
Kawamorita, S.; Miyazaki, T.; Ohmiya, H.; Iwai, T.; Sawamura, M. J. Am. Chem. Soc. 2011, 133, 19310–19313. doi:10.1021/ja208364a
Return to citation in text: [1] -
Ros, A.; Estepa, B.; López-Rodríguez, R.; Álvarez, E.; Fernández, R.; Lassaletta, J. M. Angew. Chem., Int. Ed. 2011, 50, 11724–11728. doi:10.1002/anie.201104544
Return to citation in text: [1] -
Ingleson, M. J. Synlett 2012, 23, 1411–1415. doi:10.1055/s-0031-1291147
Return to citation in text: [1] -
De Vries, T. S.; Prokofjevs, A.; Vedejs, E. Chem. Rev. 2012, 112, 4246–4282. doi:10.1021/cr200133c
Return to citation in text: [1] -
Del Grosso, A.; Singleton, P. J.; Muryn, C. A.; Ingleson, M. J. Angew. Chem., Int. Ed. 2011, 50, 2102–2106. doi:10.1002/anie.201006196
Return to citation in text: [1] -
Del Grosso, A.; Helm, M. D.; Solomon, S. A.; Caras-Quintero, D.; Ingleson, M. J. Chem. Commun. 2011, 47, 12459–12461. doi:10.1039/c1cc14226g
Return to citation in text: [1] -
Ma, Y.; Song, C.; Jiang, W.; Xue, G.; Cannon, J. F.; Wang, X.; Andrus, M. B. Org. Lett. 2003, 5, 4635–4638. doi:10.1021/ol035857q
Return to citation in text: [1] [2] -
Mo, F.; Jiang, Y.; Qiu, D.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 1846–1849. doi:10.1002/anie.200905824
Return to citation in text: [1] [2] -
Qiu, D.; Jin, L.; Zheng, Z.; Meng, H.; Mo, F.; Wang, X.; Zhang, Y.; Wang, J. J. Org. Chem. 2013, 78, 1923–1933. doi:10.1021/jo3018878
Return to citation in text: [1] [2] -
Yu, H.; Zhang, J.; Wang, X.; Ye, J. Synlett 2012, 23, 1394–1396. doi:10.1055/s-0031-1290960
Return to citation in text: [1] [2] -
Yu, J.; Zhang, L.; Yan, G. Adv. Synth. Catal. 2012, 354, 2625–2628. doi:10.1002/adsc.201200416
Return to citation in text: [1] -
Erb, W.; Hellal, A.; Albini, M.; Rouden, J.; Blanchet, J. Chem. – Eur. J. 2014, 20, 6608–6612. doi:10.1002/chem.201402487
Return to citation in text: [1] [2] -
Marciasini, L. D.; Richy, N.; Vaultier, M.; Pucheault, M. Adv. Synth. Catal. 2013, 355, 1083–1088. doi:10.1002/adsc.201200942
Return to citation in text: [1] -
Sherrington, J. Organoboron compound comprising sulphur pentafluoride. WO 123749 A1, Dec 29, 2005.
Return to citation in text: [1] -
Stamford, A. W.; Cumming, J. N. Pentafluorosulfur imino heterocyclic compounds as BACE-1 inhibitors, composition, and their use. WO 044184 A1, April 14, 2011.
Return to citation in text: [1] -
Joliton, A.; Carreira, E. M. Org. Lett. 2013, 15, 5147–5149. doi:10.1021/ol4025666
Return to citation in text: [1] [2] -
Doyle, M. P.; Bryker, W. J. J. Org. Chem. 1979, 44, 1572–1574. doi:10.1021/jo01323a048
Return to citation in text: [1] -
Okazaki, T.; Laali, K. K.; Bunge, S. D.; Adas, S. K. Eur. J. Org. Chem. 2014, 1630–1644. doi:10.1002/ejoc.201301538
Return to citation in text: [1] -
Okazaki, T.; Laali, K. K.; Reddy, A. S. J. Fluorine Chem. 2014, 165, 91–95. doi:10.1016/j.jfluchem.2014.06.021
Return to citation in text: [1] -
Galli, C. Chem. Rev. 1988, 88, 765–792. doi:10.1021/cr00087a004
Return to citation in text: [1] -
Molinaro, C.; Mowat, J.; Gosselin, F.; O'Shea, P. D.; Marcoux, J.-F.; Angelaud, R.; Davies, I. W. J. Org. Chem. 2007, 72, 1856–1858. doi:10.1021/jo062483g
Return to citation in text: [1] -
Zhao, C.-J.; Xue, D.; Jia, Z.-H.; Wang, C.; Xiao, J. Synlett 2014, 25, 1577–1584. doi:10.1055/s-0033-1339118
Return to citation in text: [1] -
Abramovitch, R. A.; Saha, J. G. Tetrahedron 1965, 21, 3297–3303. doi:10.1016/S0040-4020(01)96951-0
Return to citation in text: [1] -
Loewenschuss, H.; Wahl, G. H., Jr.; Zollinger, H. Helv. Chim. Acta 1976, 59, 1438–1448. doi:10.1002/hlca.19760590505
Return to citation in text: [1] -
Sakakura, T.; Hara, M.; Tanaka, M. J. Chem. Soc., Perkin Trans. 1 1994, 283–288. doi:10.1039/P19940000283
Return to citation in text: [1] -
Köster, R.; Bellut, H.; Ziegler, E. Angew. Chem., Int. Ed. 1967, 6, 255. doi:10.1002/anie.196702551
Return to citation in text: [1] -
Friedman, L.; Chlebowski, J. J. Org. Chem. 1968, 33, 1636–1638. doi:10.1021/jo01268a070
Return to citation in text: [1] -
Wojnarovits, L.; LaVerne, J. A. Radiat. Phys. Chem. 1996, 47, 99–101. doi:10.1016/0969-806X(95)00089-G
Return to citation in text: [1] -
Barluenga, J.; González, J. M.; Campos, P. J.; Asensio, G. Angew. Chem., Int. Ed. 1985, 24, 319–320. doi:10.1002/anie.198503191
Return to citation in text: [1] [2] -
Barluenga, J.; Rodriguez, M. A.; Campos, P. J. J. Org. Chem. 1990, 55, 3104–3106. doi:10.1021/jo00297a027
Return to citation in text: [1] -
Barluenga, J.; Gonzalez, J. M.; Garcia-Martin, M. A.; Campos, P. J.; Asensio, G. J. Org. Chem. 1993, 58, 2058–2060. doi:10.1021/jo00060a020
Return to citation in text: [1] -
Wanderheiden, S.; Bulat, B.; Zevaco, T.; Jung, N.; Bräse, S. Chem. Commun. 2011, 47, 9063–9065. doi:10.1039/c1cc12950c
Return to citation in text: [1] -
Simmons, E. M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. doi:10.1002/anie.201107334
Return to citation in text: [1]
| 77. | Molinaro, C.; Mowat, J.; Gosselin, F.; O'Shea, P. D.; Marcoux, J.-F.; Angelaud, R.; Davies, I. W. J. Org. Chem. 2007, 72, 1856–1858. doi:10.1021/jo062483g |
| 66. | Yu, H.; Zhang, J.; Wang, X.; Ye, J. Synlett 2012, 23, 1394–1396. doi:10.1055/s-0031-1290960 |
| 1. | Clifford, A. F.; El-Shamy, H. K.; Emeleus, H. J.; Haszeldine, R. N. J. Chem. Soc. 1953, 2372–2375. doi:10.1039/jr9530002372 |
| 2. | Hoffmann, F. W.; Simmons, T. C.; Beck, R. B.; Holler, H. V.; Katz, T.; Koshar, R. J.; Larsen, E. R.; Mulvaney, J. E.; Rogers, F. E.; Singleton, B.; Sparks, R. S. J. Am. Chem. Soc. 1957, 79, 3424–3429. doi:10.1021/ja01570a029 |
| 3. | Dresdner, R. D.; Mao, T. J.; Young, J. A. J. Am. Chem. Soc. 1959, 81, 574–577. doi:10.1021/ja01545a027 |
| 4. | Sheppard, W. A. J. Am. Chem. Soc. 1960, 82, 4751–4752. doi:10.1021/ja01502a083 |
| 13. |
Bowden, R. D.; Greenhall, M. P.; Moilliet, J. S.; Thomson, J. Chem. Abstr. 1997, 126, 199340.
The preparation of fluorinated organic compounds, WO 9705106, 13 Feb 1997. |
| 14. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8 |
| 15. | Chambers, R. D.; Spink, R. C. H. Chem. Commun. 1999, 883–884. doi:10.1039/A901473J |
| 16. | Sipyagin, A. M.; Bateman, C. P.; Matsev, A. V.; Waterfeld, A.; Jilek, R. E.; Key, C. D.; Szulczewski, G. J.; Thrasher, J. S. J. Fluorine Chem. 2014, 167, 203–210. doi:10.1016/j.jfluchem.2014.07.031 |
| 51. | Euzenat, L.; Horhant, D.; Ribourdouille, Y.; Duriez, C.; Alcaraz, G.; Vaultier, M. Chem. Commun. 2003, 2280–2281. doi:10.1039/B306874A |
| 83. | Friedman, L.; Chlebowski, J. J. Org. Chem. 1968, 33, 1636–1638. doi:10.1021/jo01268a070 |
| 84. | Wojnarovits, L.; LaVerne, J. A. Radiat. Phys. Chem. 1996, 47, 99–101. doi:10.1016/0969-806X(95)00089-G |
| 10. | Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53 |
| 11. | Lummer, K.; Ponomarenko, M. V.; Röschenthaler, G.-V.; Bremer, M.; Beier, P. J. Fluorine Chem. 2014, 157, 79–83. doi:10.1016/j.jfluchem.2013.10.009 |
| 12. | Kanishchev, O. S.; Dolbier, W. R., Jr. Angew. Chem., Int. Ed. 2015, 127, 282–286. doi:10.1002/ange.201409990 |
| 52. | Chen, H.; Schlecht, S.; Semple, T. C.; Hartwig, J. F. Science 2000, 287, 1995–1997. doi:10.1126/science.287.5460.1995 |
| 53. | Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N.; Anastasi, N. R.; Hartwig, J. F. J. Am. Chem. Soc. 2002, 124, 390–391. doi:10.1021/ja0173019 |
| 54. | Ishiyama, T.; Takagi, J.; Hartwig, J. F.; Miyaura, N. Angew. Chem., Int. Ed. 2002, 41, 3056–3058. doi:10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-# |
| 55. | Preshlock, S. M.; Plattner, D. L.; Maligres, P. E.; Krska, S. W.; Maleczka, R. E., Jr.; Smith, M. R., III. Angew. Chem., Int. Ed. 2013, 52, 12915–12919. doi:10.1002/anie.201306511 |
| 56. | Dai, H.-X.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 134–137. doi:10.1021/ja2097095 |
| 57. | Kawamorita, S.; Miyazaki, T.; Ohmiya, H.; Iwai, T.; Sawamura, M. J. Am. Chem. Soc. 2011, 133, 19310–19313. doi:10.1021/ja208364a |
| 58. | Ros, A.; Estepa, B.; López-Rodríguez, R.; Álvarez, E.; Fernández, R.; Lassaletta, J. M. Angew. Chem., Int. Ed. 2011, 50, 11724–11728. doi:10.1002/anie.201104544 |
| 14. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8 |
| 7. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 8. | Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003 |
| 9. | Vida, N.; Pastýříková, T.; Klepetářová, B.; Beier, P. J. Org. Chem. 2014, 79, 8906–8911. doi:10.1021/jo501562z |
| 43. | Brown, H. C.; Cole, T. E. Organometallics 1983, 2, 1316–1319. doi:10.1021/om50004a009 |
| 44. | Brown, H. C.; Srebnik, M.; Cole, T. E. Organometallics 1986, 5, 2300–2303. doi:10.1021/om00142a020 |
| 72. | Joliton, A.; Carreira, E. M. Org. Lett. 2013, 15, 5147–5149. doi:10.1021/ol4025666 |
| 5. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 6. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 45. | Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508–7510. doi:10.1021/jo00128a024 |
| 46. | Murata, M.; Watanabe, S.; Masuda, Y. J. Org. Chem. 1997, 62, 6458–6459. doi:10.1021/jo970963p |
| 47. | Murata, M.; Oyama, T.; Watanabe, S.; Masuda, Y. J. Org. Chem. 2000, 65, 164–168. doi:10.1021/jo991337q |
| 48. | Fürstner, A.; Seidel, G. Org. Lett. 2002, 4, 541–543. doi:10.1021/ol0171463 |
| 49. | Zhu, W.; Ma, D. Org. Lett. 2006, 8, 261–263. doi:10.1021/ol052633u |
| 50. | Kleeberg, C.; Dang, L.; Lin, Z.; Marder, T. B. Angew. Chem., Int. Ed. 2009, 48, 5350–5354. doi:10.1002/anie.200901879 |
| 68. | Erb, W.; Hellal, A.; Albini, M.; Rouden, J.; Blanchet, J. Chem. – Eur. J. 2014, 20, 6608–6612. doi:10.1002/chem.201402487 |
| 6. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 18. | Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. |
| 33. | Chia, P. W.; Brennan, S. C.; Slawin, A. M. Z.; Riccardi, D.; O'Hagan, D. Org. Biomol. Chem. 2012, 10, 7922–7927. doi:10.1039/c2ob26402a |
| 34. | Sun, L.; Li, J.; Bera, H.; Dolzhenko, A. V.; Chiu, G. N. C.; Chui, W. K. Eur. J. Med. Chem. 2013, 70, 400–410. doi:10.1016/j.ejmech.2013.10.022 |
| 35. | Stump, B.; Eberle, C.; Schweizer, W. B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R. L.; Lentz, D.; Diederich, F. ChemBioChem 2009, 10, 79–83. doi:10.1002/cbic.200800565 |
| 36. | Altomonte, S.; Baillie, G. L.; Ross, R. A.; Riley, J.; Zanda, M. RSC Adv. 2014, 4, 20164–20176. doi:10.1039/c4ra01212g |
| 37. | Yang, Y.-D.; Tokunaga, E.; Akiyama, H.; Saito, N.; Shibata, N. ChemMedChem 2014, 9, 913–917. doi:10.1002/cmdc.201400059 |
| 38. | Nakayama, H.; Nishida, J.-i.; Takada, N.; Sato, H.; Yamashita, Y. Chem. Mater. 2012, 24, 671–676. doi:10.1021/cm202650u |
| 40. | Hall, D. G., Ed. Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley-VCH: Weinheim, Germany, 2005. |
| 81. | Sakakura, T.; Hara, M.; Tanaka, M. J. Chem. Soc., Perkin Trans. 1 1994, 283–288. doi:10.1039/P19940000283 |
| 31. | Lee, J.-W.; List, B. J. Am. Chem. Soc. 2012, 134, 18245–18248. doi:10.1021/ja3096202 |
| 32. | Yang, Y.-D.; Lu, X.; Tokunaga, E.; Shibata, N. J. Fluorine Chem. 2012, 143, 204–209. doi:10.1016/j.jfluchem.2012.06.007 |
| 41. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 42. | Miyaura, N. Top. Curr. Chem. 2002, 219, 11–59. doi:10.1007/3-540-45313-X |
| 82. | Köster, R.; Bellut, H.; Ziegler, E. Angew. Chem., Int. Ed. 1967, 6, 255. doi:10.1002/anie.196702551 |
| 28. | Sergeeva, T. A.; Dolbier, W. R., Jr. Org. Lett. 2004, 6, 2417–2419. doi:10.1021/ol0491991 |
| 29. | Dolbier, W. R., Jr.; Zheng, Z. J. Fluorine Chem. 2011, 132, 389–393. doi:10.1016/j.jfluchem.2011.03.017 |
| 30. | Dolbier, W. R., Jr.; Zheng, Z. J. Org. Chem. 2009, 74, 5626–5628. doi:10.1021/jo9007699 |
| 78. | Zhao, C.-J.; Xue, D.; Jia, Z.-H.; Wang, C.; Xiao, J. Synlett 2014, 25, 1577–1584. doi:10.1055/s-0033-1339118 |
| 14. | Bowden, R. D.; Comina, P. J.; Greenhall, M. P.; Kariuki, B. M.; Loveday, A.; Philp, D. Tetrahedron 2000, 56, 3399–3408. doi:10.1016/s0040-4020(00)00184-8 |
| 17. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006 |
| 18. | Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. |
| 19. | Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<1989::AID-ANIE1989>3.0.CO;2-K |
| 20. | Frischmuth, A.; Unsinn, A.; Groll, K.; Stadtmüller, H.; Knochel, P. Chem. – Eur. J. 2012, 18, 10234–10238. doi:10.1002/chem.201201485 |
| 21. | Beier, P.; Pastýříková, T.; Vida, N.; Iakobson, G. Org. Lett. 2011, 13, 1466–1469. doi:10.1021/ol2001478 |
| 22. | Beier, P.; Pastýříková, T.; Iakobson, G. J. Org. Chem. 2011, 76, 4781–4786. doi:10.1021/jo200618p |
| 23. | Beier, P.; Pastýříková, T. Tetrahedron Lett. 2011, 52, 4392–4394. doi:10.1016/j.tetlet.2011.06.011 |
| 24. | Pastýříková, T.; Iakobson, G.; Vida, N.; Pohl, R.; Beier, P. Eur. J. Org. Chem. 2012, 2123–2126. doi:10.1002/ejoc.201200127 |
| 25. | Vida, N.; Beier, P. J. Fluorine Chem. 2012, 143, 130–134. doi:10.1016/j.jfluchem.2012.04.001 |
| 26. | Iakobson, G.; Pošta, M.; Beier, P. Synlett 2013, 24, 855–859. doi:10.1055/s-0032-1318452 |
| 27. | Wang, C.; Yu, Y.-B.; Fan, S.; Zhang, X. Org. Lett. 2013, 15, 5004–5007. doi:10.1021/ol4023326 |
| 5. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 19. | Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(SICI)1521-3773(19990712)38:13/14<1989::AID-ANIE1989>3.0.CO;2-K |
| 38. | Nakayama, H.; Nishida, J.-i.; Takada, N.; Sato, H.; Yamashita, Y. Chem. Mater. 2012, 24, 671–676. doi:10.1021/cm202650u |
| 39. | Winter, R.; Nixon, P. G.; Gard, G. L.; Graham, D. J.; Castner, D. G.; Holcomb, N. R.; Grainger, D. W. Langmuir 2004, 20, 5776–5781. doi:10.1021/la040011w |
| 79. | Abramovitch, R. A.; Saha, J. G. Tetrahedron 1965, 21, 3297–3303. doi:10.1016/S0040-4020(01)96951-0 |
| 80. | Loewenschuss, H.; Wahl, G. H., Jr.; Zollinger, H. Helv. Chim. Acta 1976, 59, 1438–1448. doi:10.1002/hlca.19760590505 |
| 68. | Erb, W.; Hellal, A.; Albini, M.; Rouden, J.; Blanchet, J. Chem. – Eur. J. 2014, 20, 6608–6612. doi:10.1002/chem.201402487 |
| 59. | Ingleson, M. J. Synlett 2012, 23, 1411–1415. doi:10.1055/s-0031-1291147 |
| 60. | De Vries, T. S.; Prokofjevs, A.; Vedejs, E. Chem. Rev. 2012, 112, 4246–4282. doi:10.1021/cr200133c |
| 61. | Del Grosso, A.; Singleton, P. J.; Muryn, C. A.; Ingleson, M. J. Angew. Chem., Int. Ed. 2011, 50, 2102–2106. doi:10.1002/anie.201006196 |
| 62. | Del Grosso, A.; Helm, M. D.; Solomon, S. A.; Caras-Quintero, D.; Ingleson, M. J. Chem. Commun. 2011, 47, 12459–12461. doi:10.1039/c1cc14226g |
| 85. | Barluenga, J.; González, J. M.; Campos, P. J.; Asensio, G. Angew. Chem., Int. Ed. 1985, 24, 319–320. doi:10.1002/anie.198503191 |
| 63. | Ma, Y.; Song, C.; Jiang, W.; Xue, G.; Cannon, J. F.; Wang, X.; Andrus, M. B. Org. Lett. 2003, 5, 4635–4638. doi:10.1021/ol035857q |
| 64. | Mo, F.; Jiang, Y.; Qiu, D.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 1846–1849. doi:10.1002/anie.200905824 |
| 65. | Qiu, D.; Jin, L.; Zheng, Z.; Meng, H.; Mo, F.; Wang, X.; Zhang, Y.; Wang, J. J. Org. Chem. 2013, 78, 1923–1933. doi:10.1021/jo3018878 |
| 66. | Yu, H.; Zhang, J.; Wang, X.; Ye, J. Synlett 2012, 23, 1394–1396. doi:10.1055/s-0031-1290960 |
| 67. | Yu, J.; Zhang, L.; Yan, G. Adv. Synth. Catal. 2012, 354, 2625–2628. doi:10.1002/adsc.201200416 |
| 85. | Barluenga, J.; González, J. M.; Campos, P. J.; Asensio, G. Angew. Chem., Int. Ed. 1985, 24, 319–320. doi:10.1002/anie.198503191 |
| 86. | Barluenga, J.; Rodriguez, M. A.; Campos, P. J. J. Org. Chem. 1990, 55, 3104–3106. doi:10.1021/jo00297a027 |
| 87. | Barluenga, J.; Gonzalez, J. M.; Garcia-Martin, M. A.; Campos, P. J.; Asensio, G. J. Org. Chem. 1993, 58, 2058–2060. doi:10.1021/jo00060a020 |
| 88. | Wanderheiden, S.; Bulat, B.; Zevaco, T.; Jung, N.; Bräse, S. Chem. Commun. 2011, 47, 9063–9065. doi:10.1039/c1cc12950c |
| 74. | Okazaki, T.; Laali, K. K.; Bunge, S. D.; Adas, S. K. Eur. J. Org. Chem. 2014, 1630–1644. doi:10.1002/ejoc.201301538 |
| 75. | Okazaki, T.; Laali, K. K.; Reddy, A. S. J. Fluorine Chem. 2014, 165, 91–95. doi:10.1016/j.jfluchem.2014.06.021 |
| 63. | Ma, Y.; Song, C.; Jiang, W.; Xue, G.; Cannon, J. F.; Wang, X.; Andrus, M. B. Org. Lett. 2003, 5, 4635–4638. doi:10.1021/ol035857q |
| 64. | Mo, F.; Jiang, Y.; Qiu, D.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2010, 49, 1846–1849. doi:10.1002/anie.200905824 |
| 65. | Qiu, D.; Jin, L.; Zheng, Z.; Meng, H.; Mo, F.; Wang, X.; Zhang, Y.; Wang, J. J. Org. Chem. 2013, 78, 1923–1933. doi:10.1021/jo3018878 |
| 73. | Doyle, M. P.; Bryker, W. J. J. Org. Chem. 1979, 44, 1572–1574. doi:10.1021/jo01323a048 |
| 32. | Yang, Y.-D.; Lu, X.; Tokunaga, E.; Shibata, N. J. Fluorine Chem. 2012, 143, 204–209. doi:10.1016/j.jfluchem.2012.06.007 |
| 72. | Joliton, A.; Carreira, E. M. Org. Lett. 2013, 15, 5147–5149. doi:10.1021/ol4025666 |
| 69. | Marciasini, L. D.; Richy, N.; Vaultier, M.; Pucheault, M. Adv. Synth. Catal. 2013, 355, 1083–1088. doi:10.1002/adsc.201200942 |
| 89. | Simmons, E. M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 3066–3072. doi:10.1002/anie.201107334 |
| 70. | Sherrington, J. Organoboron compound comprising sulphur pentafluoride. WO 123749 A1, Dec 29, 2005. |
| 71. | Stamford, A. W.; Cumming, J. N. Pentafluorosulfur imino heterocyclic compounds as BACE-1 inhibitors, composition, and their use. WO 044184 A1, April 14, 2011. |
© 2015 Iakobson et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)