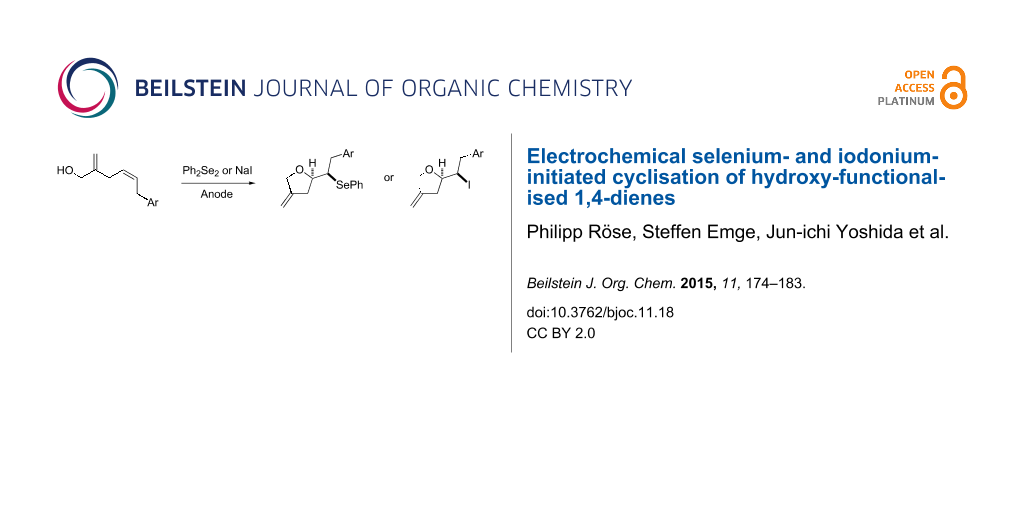
Abstract
The cobalt(I)-catalysed 1,4-hydrovinylation reaction of allyloxytrimethylsilane and allyl alcohol with substituted 1,3-dienes leads to hydroxy-functionalised 1,4-dienes in excellent regio- and diastereoselective fashion. Those 1,4-dienols can be converted into tetrahydrofuran and pyran derivatives under indirect electrochemical conditions generating selenium or iodonium cations. The reactions proceed in good yields and regioselectivities for the formation of single diastereomers.

Graphical Abstract
Introduction
The reaction of terminal alkenes with 1,3-dienes under cobalt catalysis results in 1,4-dienes in a 1,4-hydrovinylation reaction. Besides cobalt, also other transition metals were described to undergo such transformations [1-4]. However, only for the cobalt-catalysed reactions a regiodiverse reaction has been described where the carbon–carbon bond formation, either at the terminal carbon of the double bond (C1) or on C2 was formed, depending on the ligand system applied [5,6]. Besides the ozonolysis of the 1,4-dienes for the generation of 1,3-dicarbonyl derivatives [7-9], these 1,4-dienes are in turn potential substrates for the synthesis of functionalised heterocycles. Particularly, we were interested in the synthesis of tetrahydrofuran and pyran derivatives. Those heterocycles are prevalent substructures in many natural compounds, pesticides and drugs with antifungal and antibacterial properties [10-13]. For this purpose, we investigated a protocol for the straight forward synthesis of 1,4-dienols which should be cyclised into the corresponding tetrahydrofuran or pyran derivatives. With our sight set on efficient and atom economic organic reactions electrochemistry seems to be a powerful tool for the transformation of those 1,4-dienols. Although it seems that all possible functional groups have been investigated in organic electrochemistry, reports on electrochemical transformations of 1,4-dienes are rare [14-17]. First attempts of a direct electrochemical conversion were not very successful, so that we turned our attention towards indirect electrochemical methods [18,19]. Among these we became interested in the electrochemical generation of reactive cations inducing a transformation of the 1,4-diene moiety. As a starting point we put our interest in electrochemical reactions using selenium cations. Several methods applying electrochemically generated selenium cations with alkenes or alkynes including seleno-etherification and lactonisation, epoxidation and oxoselenylation sequences have been reported in the last decades [20-27]. The reactions often proceed in a regio- and stereoselective fashion and tolerate a wide range of functionalities.
Next to selenium cation induced reactions we put our focus on using halonium cations. The advantage of this type of reaction is its lower toxicity and the easy access towards the halonium source. Several reactions using bromonium- and iodonium cations such as iodo-etherification, lactonisation or Friedel–Crafts alkylation reactions can be found in literature. However, these procedures often use expensive or toxic halonium sources like molecular bromine [10,28,29] or organic trihalide salts [30], N-bromosuccinimide or N-iodosuccinimide and its derivatives [31-36], or more specialised reagents such as bis(pyridinium)iodonium(I) tetrafluoroborate [37-39]. Next to those, the in situ oxidation of halogenide ions with strong oxidants such as oxone, Pb(IV), mCPBA, FeCl3 or H2O2 have been reported [40-45]. A more efficient and versatile method is the electrochemical generation of halonium ions. Thereby, it is possible to accumulate the halonium ions in solution and to add those to a substrate in a separated process (“pool” method) [46-51] or to consume the halonium ions in situ in follow-up reactions inside the cell [52].
Accordingly, we envisaged the generation of suitable starting materials via a cobalt-catalysed hydrovinylation reaction and investigated their in situ conversion via electrochemically generated selenium- or iodonium cations.
Results and Discussion
Cobalt-catalysed 1,4-hydrovinylation of allylic alcohols
For the successful application of 1,4-dienes in the electrochemical reactions, 1,4-dienes with additional internal nucleophiles, such as an alcohol group, were envisaged and those 1,4-dienols could be generated from simple 1,3-dienes, such as 1,3-butadiene or 1-aryl-substituted 1,3-dienes 1, and TMS-protected allylic alcohol (Scheme 1) for the synthesis of 1,4-dienols of type 2.
![[1860-5397-11-18-i1]](/bjoc/content/inline/1860-5397-11-18-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Cobalt-catalysed 1,4-hydrovinylation.
Scheme 1: Cobalt-catalysed 1,4-hydrovinylation.
The cobalt-catalysed hydrovinylation reaction is highly regiospecific for the carbon–carbon bond formation which takes place exclusively at the internal carbon of the double bond of the terminal alkene (C2) and C4 of the 1-aryl-substituted 1,3-diene. The key intermediate A in the reaction mechanism is proposed to be a cobaltacycle which only allows the double bond generated from the 1,3-diene component to adopt a Z-configuration. Accordingly, the products of type 2 are formed in high selectivity in terms of regio- and diastereomeric control.
The starting materials of type 1 were generated from the aromatic aldehydes and allyltriphenylphosphonium bromide in a Wittig reaction following a known protocol [53]. The synthesis of the 1,4-dienes was then accomplished utilising the cobalt-catalyst precursor and reducing conditions in the presence of zinc iodide for abstracting the bromide anions at room temperature. The TMS-protected allylic alcohol was applied in the cobalt-catalysed 1,4-hydrovinylation process with aryl-substituted 1,3-dienes 1a–k because the use of allylic alcohol itself led to significant lower yields (up to 30%). Only in case of buta-1,3-diene, 2,3-dimethyl-1,3-butadiene and isoprene allyl alcohol could be used directly without decreasing the yield (Table 1, entries 12–14). The results of the 1,4-dienol syntheses are summarised in Table 1.
Table 1: Results of the cobalt-catalysed 1,4-hydrovinylation reaction of TMS-protected allylic alcohol with 1,3-dienes of type 1.
| Entry | 1,3-Diene (1) | 1,4-Dienol (2) | Yielda |
|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-18-i4.svg?max-width=637&scale=1.0)
|
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-18-i5.svg?max-width=637&scale=1.0)
|
60% |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-18-i6.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-18-i7.svg?max-width=637&scale=1.0)
|
71% |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-18-i8.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-18-i9.svg?max-width=637&scale=1.0)
|
81% |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-18-i10.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-18-i11.svg?max-width=637&scale=1.0)
|
51% |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-18-i12.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-18-i13.svg?max-width=637&scale=1.0)
|
69% |
| 6 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-18-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-18-i15.svg?max-width=637&scale=1.0)
|
87% |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-18-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-18-i17.svg?max-width=637&scale=1.0)
|
64% |
| 8 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-18-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-18-i19.svg?max-width=637&scale=1.0)
|
67% |
| 9 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-18-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-18-i21.svg?max-width=637&scale=1.0)
|
60% |
| 10 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-18-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-18-i23.svg?max-width=637&scale=1.0)
|
59% |
| 11 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-18-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-18-i25.svg?max-width=637&scale=1.0)
|
87% |
| 12 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-18-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-18-i27.svg?max-width=637&scale=1.0)
|
92%b |
| 13 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-18-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-18-i29.svg?max-width=637&scale=1.0)
|
90%b |
| 14 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-18-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-18-i31.svg?max-width=637&scale=1.0)
|
89%
(2n:2o = 79:21) |
aReaction conditions: (i) 1,3-diene (1.0 equiv), allyloxytrimethylsilane (1.2 equiv), CoBr2(dppe) (5–10 mol %), Zn powder (10–20 mol %), ZnI2 (10–20 mol %), CH2Cl2 (1 mL/mmol), rt, 12–16 h (ii) TBAF (1.1 equiv), THF, 0 °C, 3 h. bReaction conditions: 1,3-diene (1.0 equiv), allyl alcohol (1.2–2.0 equiv), CoBr2(dppe) (5–10 mol %), Zn powder (10–20 mol %), ZnI2 (10–20 mol %), CH2Cl2 (1 mL/mmol), rt, 12–16 h.
The cobalt-catalysed hydrovinylation tolerates halide, ether, ester, trifluoromethyl and heterocyclic substituents and gave the desired products in acceptable to good yields over a two-step reaction sequence of hydrovinylation and deprotection. Electron-withdrawing and electron-donating groups as well as sterically hindered aryl substituents are also accepted (see Table 1, entries 7 and 9). The use of buta-1,3-diene and 2,3-dimethyl-1,3-butadiene gave the 1,4-dienols in excellent yields (Table 1, entries 12 and 13). When isoprene was used, the regioisomeric products 2n and 2o were formed in good yields and acceptable regioselectivity, with the carbon–carbon bond formation taking place predominantly at the lower substituted end of the 1,3-diene moiety. These results demonstrate that the cobalt-catalysed hydrovinylation reaction is a powerful tool for the straightforward synthesis of various 1,4-dienols of type 2 and the mild reaction conditions made it possible to generate and isolate the products without isomerisation of the double bonds towards undesired side-products.
Transformation of 1,4-dienols via electro-generated selenium cations
The 1,4-dienols were transformed into cyclic phenylselenoethers by intramolecular cyclisation using selenium cations generated by indirect electrolysis. The reaction was carried out by electrolysing a mixture of the 1,4-dienol, diphenyl diselenide and tetraethylammonium bromide in CH3CN at room temperature in an undivided cell, using platinum foil electrodes (constant current 10 mA). In this investigation only diphenyl diselenide was used as selenium source. These reaction conditions led to the formation of products of type 3 as exclusive diastereomers (Scheme 2).
![[1860-5397-11-18-i2]](/bjoc/content/inline/1860-5397-11-18-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Electrochemical selenoalkoxylation of 2.
Scheme 2: Electrochemical selenoalkoxylation of 2.
The cyclisation of 2 could lead to a number of products. The PhSe+ cation could interact with the 1,1-disubstituted double bond and nucleophilic attack could lead to oxiran- or oxetan-type products. On the other hand, the interaction of the PhSe+ ion with the 1,2-disubstituted double bond would lead to the furan-type products 3 or alternatively to pyran-type product 4. The results of the electrochemical selenoalkoxylation of the 1,4-dienols are summarised in Table 2.
Table 2: Electrochemical selenoalkoxylation of 1,4-dienols 2a.
| Entry | 1,4-Dienol (2) | Furan (3) or pyran (4) | Yieldb |
|---|---|---|---|
| 1 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-18-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-18-i33.svg?max-width=637&scale=1.0)
|
50% |
| 2 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-18-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-18-i35.svg?max-width=637&scale=1.0)
|
82% |
| 3 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-18-i36.svg?max-width=637&scale=1.0)
|
![[Graphic 34]](/bjoc/content/inline/1860-5397-11-18-i37.svg?max-width=637&scale=1.0)
|
58% |
| 4 |
![[Graphic 35]](/bjoc/content/inline/1860-5397-11-18-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 36]](/bjoc/content/inline/1860-5397-11-18-i39.svg?max-width=637&scale=1.0)
|
68% |
| 5 |
![[Graphic 37]](/bjoc/content/inline/1860-5397-11-18-i40.svg?max-width=637&scale=1.0)
|
![[Graphic 38]](/bjoc/content/inline/1860-5397-11-18-i41.svg?max-width=637&scale=1.0)
|
42% |
| 6 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-11-18-i42.svg?max-width=637&scale=1.0)
|
![[Graphic 40]](/bjoc/content/inline/1860-5397-11-18-i43.svg?max-width=637&scale=1.0)
|
90% |
| 7 |
![[Graphic 41]](/bjoc/content/inline/1860-5397-11-18-i44.svg?max-width=637&scale=1.0)
|
![[Graphic 42]](/bjoc/content/inline/1860-5397-11-18-i45.svg?max-width=637&scale=1.0)
|
62% |
| 8 |
![[Graphic 43]](/bjoc/content/inline/1860-5397-11-18-i46.svg?max-width=637&scale=1.0)
|
![[Graphic 44]](/bjoc/content/inline/1860-5397-11-18-i47.svg?max-width=637&scale=1.0)
|
86% |
| 9 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-11-18-i48.svg?max-width=637&scale=1.0)
|
![[Graphic 46]](/bjoc/content/inline/1860-5397-11-18-i49.svg?max-width=637&scale=1.0)
|
46% (3m)
47% (4m) |
| 10 |
![[Graphic 47]](/bjoc/content/inline/1860-5397-11-18-i50.svg?max-width=637&scale=1.0)
|
![[Graphic 48]](/bjoc/content/inline/1860-5397-11-18-i51.svg?max-width=637&scale=1.0)
|
10% (3o+3o’)
58% (4n) |
| 11 |
![[Graphic 49]](/bjoc/content/inline/1860-5397-11-18-i52.svg?max-width=637&scale=1.0)
|
![[Graphic 50]](/bjoc/content/inline/1860-5397-11-18-i53.svg?max-width=637&scale=1.0)
|
75%
(3p:4p = 1:2.5) |
| 12 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-11-18-i54.svg?max-width=637&scale=1.0)
|
![[Graphic 52]](/bjoc/content/inline/1860-5397-11-18-i55.svg?max-width=637&scale=1.0)
|
53%
(3q:4q = 1:1.4) |
aReaction conditions: 1,4-dienol (0.5 mmol, 1.0 equiv), Ph2Se2 (0.5 equiv), Et4NBr (0.1 equiv), CH3CN (10 mL), rt, undivided cell, Pt/Pt, 10 mA, 6.67 mA/cm2. bOxidation of the alcohol could be observed in all reactions (less than 10%).
The electrochemical selenoalkoxylation of the aryl-substituted derivatives of type 2 (Table 2, entries 1–7) led exclusively to the tetrahydrofuran derivatives 3 via a 5-exo-tet-type cyclisation in moderate to good yields. The products were generated in diastereoselective fashion and the configuration of the formed diastereomer could be identified by 1H NMR experiments comparing a mixture of both diastereomers, synthesised by conventional selenoalkoxylation, with the electrochemically generated selenoether (see Supporting Information File 1). As a side reaction the oxidation of the alcohol could be observed in all reactions (less than 10%). Moreover, under electrochemical conditions the simple methyl-substituted derivative 2l led to the tetrahydrofuran-type product 3l in 86% yield (Table 2, entry 8) while the reaction using PhSeBr under conventional methods gave the product in 76% yield and a diastereoselectivity of 73:27 (threo:erythro). When alkyl-substituted 1,4-dienols are used, the formation of tetrahydrofuran or pyran derivatives can be observed (Table 2, entries 9 and 10). Depending on the substitution grade of the internal double bond, the alcohol functionality attacks at the less-substituted carbon of the internal double bond based on better stabilisation of the cationic intermediate. The triple methyl-substituted starting material 2m gave a 1:1 mixture of tetrahydrofuran and pyran products 3m and 4m, the latter product is formed via a 6-endo-tet-type cyclisation, in an excellent combined yield of 93%. When the mixture of the 1,4-dienols (2n and 2o) was applied, the major regioisomer 2n gave the pyran exclusively in good yields, while the minor 1,4-dienol 2o resulted in the formation of two tetrahydrofuran diastereomers which could be separated easily by column chromatography (Table 2, entry 10). Using higher substituted 1,4-dienols (2p and 2q) a preferred formation of the pyran products was observed (Table 2, entries 11 and 12). However, under no circumstances the previously discussed strained epoxy-derivatives could be detected.
Transformation of 1,4-dienols via electro-generated iodonium cations
In a similar approach we investigated the cyclisation of the 1,4-dienols 2 with in situ electrochemically generated iodonium ions for the desired synthesis of iodoalkoxylated products of type 5 (Scheme 3).
![[1860-5397-11-18-i3]](/bjoc/content/inline/1860-5397-11-18-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Electrochemical iodoalkoxylation of 2.
Scheme 3: Electrochemical iodoalkoxylation of 2.
The electrolysis was carried out in an H-type divided cell (4G glass filter) equipped with carbon fiber electrodes (see Supporting Information File 1). Each chamber was charged with 2,6-lutidine and TBABF4 in CH3CN (0.3 M) and additionally the 1,4-dienol and sodium iodide were placed in the anode chamber. The reaction was performed at constant current electrolysis (10 mA) at 0 °C. It is considerable that the presence of 2,6-lutidine is crucial for a successful reaction. In the absence of 2,6-lutidine only traces of the product can be observed and oxidation of the alcohol functionality takes place. It is mentionable that under the reaction conditions no aromatic iodination could be observed. Next to sodium iodide other iodide sources such as KI, I2 or Bu4NI can be used, whereas applying other halogenides such as NaBr, Et4NBr or Bu4NCl led to a complex mixture of products.
In this series of experiments we focussed our attention on the aryl-substituted 1,4-dienes to avoid undesired mixtures of tetrahydrofuran and pyran derivatives as obtained in the selenoalkoxylation for alkyl-substituted dienols. The results of the electrochemical iodoalkoxylation reactions are summarised in Table 3.
Table 3: Electrochemical iodoalkoxylation of 1,4-dienols 2.
| Entry | 1,4-Dienol 2 | Iodofuran 5 | Yielda |
|---|---|---|---|
| 1 |
![[Graphic 53]](/bjoc/content/inline/1860-5397-11-18-i56.svg?max-width=637&scale=1.0)
|
![[Graphic 54]](/bjoc/content/inline/1860-5397-11-18-i57.svg?max-width=637&scale=1.0)
|
66% |
| 2 |
![[Graphic 55]](/bjoc/content/inline/1860-5397-11-18-i58.svg?max-width=637&scale=1.0)
|
![[Graphic 56]](/bjoc/content/inline/1860-5397-11-18-i59.svg?max-width=637&scale=1.0)
|
60% |
| 3 |
![[Graphic 57]](/bjoc/content/inline/1860-5397-11-18-i60.svg?max-width=637&scale=1.0)
|
![[Graphic 58]](/bjoc/content/inline/1860-5397-11-18-i61.svg?max-width=637&scale=1.0)
|
40% |
| 4 |
![[Graphic 59]](/bjoc/content/inline/1860-5397-11-18-i62.svg?max-width=637&scale=1.0)
|
![[Graphic 60]](/bjoc/content/inline/1860-5397-11-18-i63.svg?max-width=637&scale=1.0)
|
68% |
| 5 |
![[Graphic 61]](/bjoc/content/inline/1860-5397-11-18-i64.svg?max-width=637&scale=1.0)
|
![[Graphic 62]](/bjoc/content/inline/1860-5397-11-18-i65.svg?max-width=637&scale=1.0)
|
46% |
| 6 |
![[Graphic 63]](/bjoc/content/inline/1860-5397-11-18-i66.svg?max-width=637&scale=1.0)
|
![[Graphic 64]](/bjoc/content/inline/1860-5397-11-18-i67.svg?max-width=637&scale=1.0)
|
68% |
| 7 |
![[Graphic 65]](/bjoc/content/inline/1860-5397-11-18-i68.svg?max-width=637&scale=1.0)
|
![[Graphic 66]](/bjoc/content/inline/1860-5397-11-18-i69.svg?max-width=637&scale=1.0)
|
70% |
| 8 |
![[Graphic 67]](/bjoc/content/inline/1860-5397-11-18-i70.svg?max-width=637&scale=1.0)
|
![[Graphic 68]](/bjoc/content/inline/1860-5397-11-18-i71.svg?max-width=637&scale=1.0)
|
67% |
| 9 |
![[Graphic 69]](/bjoc/content/inline/1860-5397-11-18-i72.svg?max-width=637&scale=1.0)
|
![[Graphic 70]](/bjoc/content/inline/1860-5397-11-18-i73.svg?max-width=637&scale=1.0)
|
46% |
| 10 |
![[Graphic 71]](/bjoc/content/inline/1860-5397-11-18-i74.svg?max-width=637&scale=1.0)
|
![[Graphic 72]](/bjoc/content/inline/1860-5397-11-18-i75.svg?max-width=637&scale=1.0)
|
82% |
| 11 |
![[Graphic 73]](/bjoc/content/inline/1860-5397-11-18-i76.svg?max-width=637&scale=1.0)
|
![[Graphic 74]](/bjoc/content/inline/1860-5397-11-18-i77.svg?max-width=637&scale=1.0)
|
66% |
aReaction conditions: H-type cell, anodic chamber: 1,4-dienol (0.5 mmol, 1.0 equiv), NaI (1.1 equiv), 2,6-lutidine (2.0 equiv), TBABF4/CH3CN (0.3 M, 10 mL); cathode chamber: 2,6-lutidine (2.0 equiv), TBABF4/CH3CN (0.3 M, 10 mL); 0 °C, C/C, 10 mA.
The electrochemical iodoalkoxylation of the aryl-substituted 1,4-dienols 2 led to the formation of the tetrahydrofuran derivatives 5 as single regio- and diastereomers. 1,4-Dienols with electron-donating and electron-withdrawing substituents as well as heterocyclic compounds gave the product of type 5 in good yields. It is noteworthy, that under these reaction conditions the electron-rich 1,4-dienols (2g and 2h) gave higher yields than in the selenoalkoxylation (Table 3, entries 7 and 8). For compound 5g the structure could be verified by X-ray analysis (see Supporting Information File 1). It is also considerable that the 1,4-dienols 2c, 2e and 2i which did not gave the selenoether 3 led to a product formation in at least moderate yields of 40% to 46% (Table 3, entries 3, 5 and 9).
The products of the selenoalkoxylation as well as of the iodoalkoxylation are interesting building blocks for further transformations which are under current investigation.
Conclusion
In conclusion, we have developed the alkoxylation of 1,4-dienols by electrochemically generated selenium and iodonium cations. First, the synthesis of 1,4-dienols via a cobalt-catalysed 1,4-hydrovinylation of substituted 1,3-dienes with allyloxy-trimethylsilane or allyl alcohol has been elaborated. Those 1,4-dienols have been transformed into tetrahydrofuran or pyran derivatives by constant current electrolysis of suitable selenium and iodonium precursors. The reactions proceed in acceptable to good yields in regio- and diasterioselective fashion and tolerate a range of functionalities.
Experimental
General procedure for the cobalt-catalysed 1,4-hydrovinylation of aryl-substituted buta-1,3-dienes with allyloxytrimethylsilane and subsequent desilylation with TBAF
Cobalt dibromo(1,3-bis(diphenylphosphino)ethane) (5–10 mol %), zinc powder (10–20 mol %) and zinc iodide (10–20 mol %) were suspended in dichloromethane and stirred at room temperature for 20 min. Then the 1,3-butadiene (1.0 equiv) and the allyloxytrimethylsilane (1.2–2.0 equiv) were added and the mixture was stirred at room temperature until complete conversion was detected by TLC and GC–MS analysis. n-Pentane was added, the mixture was filtered through a short pad of silica and concentrated under reduced pressure. The crude material was dissolved in 5 mL tetrahydrofuran, TBAF (1 M in THF, 1.1 equiv) was added and the mixture was stirred at 0 °C for 3 h. Upon completion of the reaction 15 mL water were added, the mixture was extracted with diethyl ether (three times 15 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The product was obtained after column chromatography (n-pentane/diethyl ether).
General procedure for the cobalt-catalysed 1,4-hydrovinylation of buta-1,3-dienes with allyl alcohol
Cobalt dibromo(1,3-bis(diphenylphosphino)ethane) (5 mol %), zinc powder (10 mol %) and zinc iodide (10 mol %) were suspended in dichloromethane and stirred at room temperature for 20 min. Then the 1,3-butadiene (1.0 equiv) and allyl alcohol (1.2–1.5 equiv) were added and stirred at room temperature until complete conversion was detected by TLC and GC–MS analysis. Pentane was added and the mixture was filtered through a short pad of silica. The solvent was evaporated and the crude product was purified by column chromatography to give the desired 1,4-diene.
General procedure for the electrochemical seleno-alkoxylation of 1,4-dienols
An undivided electrolysis cell was charged with diphenyl diselenide (78 mg, 0.25 mmol, 0.5 equiv), tetraethylammonium bromide (11 mg, 0.05 mmol, 0.1 equiv), the 1,4-diene (0.5 mmol, 1.0 equiv) and 10 mL acetonitrile. Then, the cell was equipped with a platinum plate anode and cathode and electrolysed under a constant current (10 mA, 6.67 mA/cm2) at 20 °C until completion was detected by TLC and GC–MS analysis. Then, 15 mL water were added, the mixture was extracted with diethyl ether (three times 15 mL), dried over Na2SO4 and the solvent was removed under reduced pressure. The product was obtained after column chromatography (n-pentane/diethyl ether).
General procedure for the electrochemical iodonium-induced alkoxylation of 1,4-dienols
An H-type divided cell (4G glass filter) was equipped with a carbon fiber anode and carbon fiber cathode. Each chamber was charged with 10 mL TBABF4 solution (0.3 M in acetonitrile) and 2,6-lutidine (2.0 equiv). The anodic chamber was charged with the 1,4-diene (1.0 equiv) and sodium iodide (1.1 equiv). Constant current electrolysis (10 mA) was carried out at 0 °C until completion was detected by TLC and GC–MS analysis. Then, 15 mL Na2S2O3 solution (10% in water) were added to both chambers, the mixture was extracted with diethyl ether (three times 15 mL), dried over MgSO4 and the solvent was removed under reduced pressure. The product was obtained after column chromatography (n-pentane/diethyl ether).
Supporting Information
Experimental details and detailed spectroscopic data of all compounds are available as Supporting Information. Single crystal data for compound 5g (CCDC 1030546) has been deposited in the Cambridge Crystallographic Data Center.
| Supporting Information File 1: Experimental details and detailed spectroscopic data. | ||
| Format: PDF | Size: 6.4 MB | Download |
References
-
Hilt, G. Eur. J. Org. Chem. 2012, 4441–4451. doi:10.1002/ejoc.201200212
Return to citation in text: [1] -
Hilt, G. Synlett 2011, 1654–1659. doi:10.1055/s-0030-1260800
Return to citation in text: [1] -
RajanBabu, T. V. Synlett 2009, 853–885. doi:10.1055/s-0028-1088213
Return to citation in text: [1] -
RajanBabu, T. V. Chem. Rev. 2003, 103, 2845–2860. doi:10.1021/cr020040g
Return to citation in text: [1] -
Arndt, M.; Dindaroğlu, M.; Schmalz, H.-G.; Hilt, G. Org. Lett. 2011, 13, 6236–6239. doi:10.1021/ol202696n
Return to citation in text: [1] -
Arndt, M.; Dindaroğlu, M.; Schmalz, H.-G.; Hilt, G. Synthesis 2012, 3534–3542. doi:10.1055/s-0032-1316796
Return to citation in text: [1] -
Hilt, G.; Arndt, M.; Weske, D. F. Synthesis 2010, 1321–1324. doi:10.1055/s-0029-1219278
Return to citation in text: [1] -
Erver, F.; Kuttner, J. R.; Hilt, G. J. Org. Chem. 2012, 77, 8375–8385. doi:10.1021/jo301028b
Return to citation in text: [1] -
Kersten, L.; Roesner, S.; Hilt, G. Org. Lett. 2010, 12, 4920–4923. doi:10.1021/ol102083v
Return to citation in text: [1] -
Harmange, J.-C.; Figadère, B. Tetrahedron: Asymmetry 1993, 4, 1711–1754. doi:10.1016/S0957-4166(00)80408-5
Return to citation in text: [1] [2] -
Faulkner, D. J. Nat. Prod. Rep. 1998, 15, 113–158. doi:10.1039/A815113Y
Return to citation in text: [1] -
Faul, M. M.; Huff, B. E. Chem. Rev. 2000, 100, 2407–2474. doi:10.1021/cr940210s
Return to citation in text: [1] -
Lorente, A.; Lamariano-Merketegi, J.; Albericio, F.; Álvarez, M. Chem. Rev. 2013, 113, 4567–4610. doi:10.1021/cr3004778
Return to citation in text: [1] -
Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker, Inc.: New York, 2001.
ISBN:0-8247-0430-4.
Return to citation in text: [1] -
Moeller, K. D. Tetrahedron 2000, 56, 9527–9554. doi:10.1016/S0040-4020(00)00840-1
Return to citation in text: [1] -
Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/B512308A
Return to citation in text: [1] -
Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843
Return to citation in text: [1] -
Steckhan, E. Angew. Chem., Int. Ed. Engl. 1986, 25, 683–701. doi:10.1002/anie.198606831
Return to citation in text: [1] -
Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J. J. Am. Chem. Soc. 2006, 128, 7710–7711. doi:10.1021/ja0625778
Return to citation in text: [1] -
Uneyama, K.; Fujibayashi, S.; Torii, S. Tetrahedron Lett. 1985, 26, 4637–4638. doi:10.1016/S0040-4039(00)98772-0
Return to citation in text: [1] -
Vukićević, R.; Konstantinović, S.; Mihailović, M. L. Tetrahedron 1991, 47, 859–865. doi:10.1016/S0040-4020(01)87074-5
Return to citation in text: [1] -
Konstantinović, S.; Vukićević, R.; Mihailović, M. L. Tetrahedron Lett. 1987, 28, 6511–6512. doi:10.1016/S0040-4039(00)96902-8
Return to citation in text: [1] -
Torii, S.; Uneyama, K.; Ono, M.; Tazana, H.; Matsunami, S. Tetrahedron Lett. 1979, 20, 4661–4662. doi:10.1016/S0040-4039(01)86676-4
Return to citation in text: [1] -
Torii, S.; Uneyama, K.; Ono, M. Tetrahedron Lett. 1980, 21, 2653–2654. doi:10.1016/S0040-4039(00)92830-2
Return to citation in text: [1] -
Torii, S.; Uneyama, K.; Ono, M. Tetrahedron Lett. 1980, 21, 2741–2744. doi:10.1016/S0040-4039(00)78594-7
Return to citation in text: [1] -
Torii, S.; Uneyama, K.; Ono, M.; Bannou, T. J. Am. Chem. Soc. 1981, 103, 4606–4608. doi:10.1021/ja00405a064
Return to citation in text: [1] -
Uneyama, K.; Takano, K.; Torii, S. Tetrahedron Lett. 1982, 23, 1161–1164. doi:10.1016/S0040-4039(00)87049-5
Return to citation in text: [1] -
Chamberlin, A. R.; Dezube, M.; Dussault, P.; McMills, M. C. J. Am. Chem. Soc. 1983, 105, 5819–5825. doi:10.1021/ja00356a020
Return to citation in text: [1] -
Labelle, M.; Guindon, Y. J. Am. Chem. Soc. 1989, 111, 2204–2210. doi:10.1021/ja00188a039
Return to citation in text: [1] -
Still, W. C.; Schneider, M. J. J. Am. Chem. Soc. 1977, 99, 948–950. doi:10.1021/ja00445a050
Return to citation in text: [1] -
Hajra, S.; Maji, B.; Karmakar, A. Tetrahedron Lett. 2005, 46, 8599–8603. doi:10.1016/j.tetlet.2005.09.170
Return to citation in text: [1] -
Hartung, J.; Kneuer, R.; Laug, S.; Schmidt, P.; Špehar, K.; Svoboda, I.; Fuess, H. Eur. J. Org. Chem. 2003, 4033–4052. doi:10.1002/ejoc.200300107
Return to citation in text: [1] -
Denmark, S. E.; Burk, M. T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20655–20660. doi:10.1073/pnas.1005296107
Return to citation in text: [1] -
Fujioka, H.; Maehata, R.; Wakamatsu, S.; Nakahara, K.; Hayashi, T.; Oki, T. Org. Lett. 2012, 14, 1054–1057. doi:10.1021/ol203425p
Return to citation in text: [1] -
Fang, C.; Paull, D. H.; Hethcox, J. C.; Shugrue, C. R.; Martin, S. F. Org. Lett. 2012, 14, 6290–6293. doi:10.1021/ol3030555
Return to citation in text: [1] -
Hennecke, U.; Müller, C. H.; Fröhlich, R. Org. Lett. 2011, 13, 860–863. doi:10.1021/ol1028805
Return to citation in text: [1] -
Barluenga, J.; González, J. M.; Campos, P. J. Angew. Chem., Int. Ed. Engl. 1985, 24, 319–320. doi:10.1002/anie.198503191
Return to citation in text: [1] -
Takaku, K.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 1996, 37, 6781–6784. doi:10.1016/S0040-4039(96)01499-2
Return to citation in text: [1] -
Zhang, H.; Mootoo, D. R. J. Org. Chem. 1995, 60, 8134–8135. doi:10.1021/jo00130a009
Return to citation in text: [1] -
Curini, M.; Epifano, F.; Marcotullio, M. C.; Montanari, F. Synthesis 2004, 368–370. doi:10.1055/s-2003-45009
Return to citation in text: [1] -
Bailey, A. D.; Cherney, S. M.; Anzalone, P. W.; Anderson, E. D.; Ernat, J. J.; Mohan, R. S. Synlett 2006, 215–218. doi:10.1055/s-2005-923586
Return to citation in text: [1] -
Miller, L. L.; Watkins, B. F. Tetrahedron Lett. 1974, 15, 4495–4496. doi:10.1016/S0040-4039(01)92201-4
Return to citation in text: [1] -
Srebnik, M.; Mechoulam, R. J. Chem. Soc., Chem. Commun. 1984, 1070–1071. doi:10.1039/C39840001070
Return to citation in text: [1] -
Chavan, S. P.; Sharma, A. K. Tetrahedron Lett. 2001, 42, 4923–4924. doi:10.1016/S0040-4039(01)00876-0
Return to citation in text: [1] -
Jones, C. C. Application of Hydrogen Peroxide and Derivatives. Clark, J. H., Ed.; RSC Clean Technology Monographs; The Royal Society of Chemistry: Cambridge, U.K., 1999.
ISBN:978-0-85404-536-5.
Return to citation in text: [1] -
Yoshida, J.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546–9549. doi:10.1021/ja9920112
Return to citation in text: [1] -
Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J. J. Am. Chem. Soc. 2000, 122, 10244–10245. doi:10.1021/ja002123p
Return to citation in text: [1] -
Yoshida, J.; Suga, S. Chem. – Eur. J. 2002, 8, 2650–2658. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S
Return to citation in text: [1] -
Nokami, T.; Ohata, K.; Inoue, M.; Tsuyama, H.; Shibuya, A.; Soga, K.; Okajima, M.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2008, 130, 10864–10865. doi:10.1021/ja803487q
Return to citation in text: [1] -
Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648
Return to citation in text: [1] -
Morofuji, T.; Shimizu, A.; Yoshida, J. Angew. Chem., Int. Ed. 2012, 51, 7259–7262. doi:10.1002/anie.201202788
Return to citation in text: [1] -
Torii, S.; Inokuchi, T.; Akahosi, F.; Kubota, M. Synthesis 1987, 242–245. doi:10.1055/s-1987-27901
Return to citation in text: [1] -
Hilt, G.; Danz, M.; Treutwein, J. Org. Lett. 2009, 11, 3322–3325. doi:10.1021/ol901064p
Return to citation in text: [1]
| 1. | Hilt, G. Eur. J. Org. Chem. 2012, 4441–4451. doi:10.1002/ejoc.201200212 |
| 2. | Hilt, G. Synlett 2011, 1654–1659. doi:10.1055/s-0030-1260800 |
| 3. | RajanBabu, T. V. Synlett 2009, 853–885. doi:10.1055/s-0028-1088213 |
| 4. | RajanBabu, T. V. Chem. Rev. 2003, 103, 2845–2860. doi:10.1021/cr020040g |
| 14. |
Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker, Inc.: New York, 2001.
ISBN:0-8247-0430-4. |
| 15. | Moeller, K. D. Tetrahedron 2000, 56, 9527–9554. doi:10.1016/S0040-4020(00)00840-1 |
| 16. | Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/B512308A |
| 17. | Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843 |
| 53. | Hilt, G.; Danz, M.; Treutwein, J. Org. Lett. 2009, 11, 3322–3325. doi:10.1021/ol901064p |
| 10. | Harmange, J.-C.; Figadère, B. Tetrahedron: Asymmetry 1993, 4, 1711–1754. doi:10.1016/S0957-4166(00)80408-5 |
| 11. | Faulkner, D. J. Nat. Prod. Rep. 1998, 15, 113–158. doi:10.1039/A815113Y |
| 12. | Faul, M. M.; Huff, B. E. Chem. Rev. 2000, 100, 2407–2474. doi:10.1021/cr940210s |
| 13. | Lorente, A.; Lamariano-Merketegi, J.; Albericio, F.; Álvarez, M. Chem. Rev. 2013, 113, 4567–4610. doi:10.1021/cr3004778 |
| 7. | Hilt, G.; Arndt, M.; Weske, D. F. Synthesis 2010, 1321–1324. doi:10.1055/s-0029-1219278 |
| 8. | Erver, F.; Kuttner, J. R.; Hilt, G. J. Org. Chem. 2012, 77, 8375–8385. doi:10.1021/jo301028b |
| 9. | Kersten, L.; Roesner, S.; Hilt, G. Org. Lett. 2010, 12, 4920–4923. doi:10.1021/ol102083v |
| 46. | Yoshida, J.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546–9549. doi:10.1021/ja9920112 |
| 47. | Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J. J. Am. Chem. Soc. 2000, 122, 10244–10245. doi:10.1021/ja002123p |
| 48. | Yoshida, J.; Suga, S. Chem. – Eur. J. 2002, 8, 2650–2658. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S |
| 49. | Nokami, T.; Ohata, K.; Inoue, M.; Tsuyama, H.; Shibuya, A.; Soga, K.; Okajima, M.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2008, 130, 10864–10865. doi:10.1021/ja803487q |
| 50. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 51. | Morofuji, T.; Shimizu, A.; Yoshida, J. Angew. Chem., Int. Ed. 2012, 51, 7259–7262. doi:10.1002/anie.201202788 |
| 5. | Arndt, M.; Dindaroğlu, M.; Schmalz, H.-G.; Hilt, G. Org. Lett. 2011, 13, 6236–6239. doi:10.1021/ol202696n |
| 6. | Arndt, M.; Dindaroğlu, M.; Schmalz, H.-G.; Hilt, G. Synthesis 2012, 3534–3542. doi:10.1055/s-0032-1316796 |
| 52. | Torii, S.; Inokuchi, T.; Akahosi, F.; Kubota, M. Synthesis 1987, 242–245. doi:10.1055/s-1987-27901 |
| 30. | Still, W. C.; Schneider, M. J. J. Am. Chem. Soc. 1977, 99, 948–950. doi:10.1021/ja00445a050 |
| 37. | Barluenga, J.; González, J. M.; Campos, P. J. Angew. Chem., Int. Ed. Engl. 1985, 24, 319–320. doi:10.1002/anie.198503191 |
| 38. | Takaku, K.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 1996, 37, 6781–6784. doi:10.1016/S0040-4039(96)01499-2 |
| 39. | Zhang, H.; Mootoo, D. R. J. Org. Chem. 1995, 60, 8134–8135. doi:10.1021/jo00130a009 |
| 10. | Harmange, J.-C.; Figadère, B. Tetrahedron: Asymmetry 1993, 4, 1711–1754. doi:10.1016/S0957-4166(00)80408-5 |
| 28. | Chamberlin, A. R.; Dezube, M.; Dussault, P.; McMills, M. C. J. Am. Chem. Soc. 1983, 105, 5819–5825. doi:10.1021/ja00356a020 |
| 29. | Labelle, M.; Guindon, Y. J. Am. Chem. Soc. 1989, 111, 2204–2210. doi:10.1021/ja00188a039 |
| 40. | Curini, M.; Epifano, F.; Marcotullio, M. C.; Montanari, F. Synthesis 2004, 368–370. doi:10.1055/s-2003-45009 |
| 41. | Bailey, A. D.; Cherney, S. M.; Anzalone, P. W.; Anderson, E. D.; Ernat, J. J.; Mohan, R. S. Synlett 2006, 215–218. doi:10.1055/s-2005-923586 |
| 42. | Miller, L. L.; Watkins, B. F. Tetrahedron Lett. 1974, 15, 4495–4496. doi:10.1016/S0040-4039(01)92201-4 |
| 43. | Srebnik, M.; Mechoulam, R. J. Chem. Soc., Chem. Commun. 1984, 1070–1071. doi:10.1039/C39840001070 |
| 44. | Chavan, S. P.; Sharma, A. K. Tetrahedron Lett. 2001, 42, 4923–4924. doi:10.1016/S0040-4039(01)00876-0 |
| 45. |
Jones, C. C. Application of Hydrogen Peroxide and Derivatives. Clark, J. H., Ed.; RSC Clean Technology Monographs; The Royal Society of Chemistry: Cambridge, U.K., 1999.
ISBN:978-0-85404-536-5. |
| 20. | Uneyama, K.; Fujibayashi, S.; Torii, S. Tetrahedron Lett. 1985, 26, 4637–4638. doi:10.1016/S0040-4039(00)98772-0 |
| 21. | Vukićević, R.; Konstantinović, S.; Mihailović, M. L. Tetrahedron 1991, 47, 859–865. doi:10.1016/S0040-4020(01)87074-5 |
| 22. | Konstantinović, S.; Vukićević, R.; Mihailović, M. L. Tetrahedron Lett. 1987, 28, 6511–6512. doi:10.1016/S0040-4039(00)96902-8 |
| 23. | Torii, S.; Uneyama, K.; Ono, M.; Tazana, H.; Matsunami, S. Tetrahedron Lett. 1979, 20, 4661–4662. doi:10.1016/S0040-4039(01)86676-4 |
| 24. | Torii, S.; Uneyama, K.; Ono, M. Tetrahedron Lett. 1980, 21, 2653–2654. doi:10.1016/S0040-4039(00)92830-2 |
| 25. | Torii, S.; Uneyama, K.; Ono, M. Tetrahedron Lett. 1980, 21, 2741–2744. doi:10.1016/S0040-4039(00)78594-7 |
| 26. | Torii, S.; Uneyama, K.; Ono, M.; Bannou, T. J. Am. Chem. Soc. 1981, 103, 4606–4608. doi:10.1021/ja00405a064 |
| 27. | Uneyama, K.; Takano, K.; Torii, S. Tetrahedron Lett. 1982, 23, 1161–1164. doi:10.1016/S0040-4039(00)87049-5 |
| 18. | Steckhan, E. Angew. Chem., Int. Ed. Engl. 1986, 25, 683–701. doi:10.1002/anie.198606831 |
| 19. | Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J. J. Am. Chem. Soc. 2006, 128, 7710–7711. doi:10.1021/ja0625778 |
| 31. | Hajra, S.; Maji, B.; Karmakar, A. Tetrahedron Lett. 2005, 46, 8599–8603. doi:10.1016/j.tetlet.2005.09.170 |
| 32. | Hartung, J.; Kneuer, R.; Laug, S.; Schmidt, P.; Špehar, K.; Svoboda, I.; Fuess, H. Eur. J. Org. Chem. 2003, 4033–4052. doi:10.1002/ejoc.200300107 |
| 33. | Denmark, S. E.; Burk, M. T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20655–20660. doi:10.1073/pnas.1005296107 |
| 34. | Fujioka, H.; Maehata, R.; Wakamatsu, S.; Nakahara, K.; Hayashi, T.; Oki, T. Org. Lett. 2012, 14, 1054–1057. doi:10.1021/ol203425p |
| 35. | Fang, C.; Paull, D. H.; Hethcox, J. C.; Shugrue, C. R.; Martin, S. F. Org. Lett. 2012, 14, 6290–6293. doi:10.1021/ol3030555 |
| 36. | Hennecke, U.; Müller, C. H.; Fröhlich, R. Org. Lett. 2011, 13, 860–863. doi:10.1021/ol1028805 |
© 2015 Röse et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)