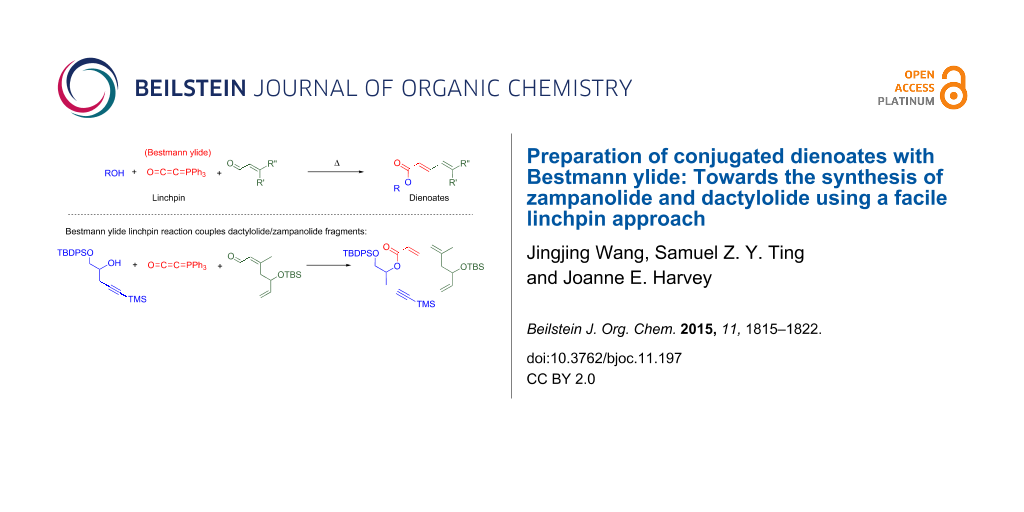
Abstract
Bestmann ylide [(triphenylphosphoranylidene)ketene] acts as a chemical linchpin that links nucleophilic entities, such as alcohols or amines, with carbonyl moieties to produce unsaturated esters and amides, respectively. In this work, the formation of α,β,γ,δ-unsaturated esters (dienoates) is achieved through the coupling of Bestmann ylide, an alcohol and an α,β-unsaturated aldehyde. Primary and secondary alcohols, including allylic alcohols, are suitable substrates; the newly formed alkene has an E-geometry. Strategically, this represents a highly efficient route to unsaturated polyketide derivatives. A linchpin approach to the synthesis of a major fragment of the natural products zampanolide and dactylolide is investigated using Bestmann ylide to link the C16–C20 alcohol with the C3–C8 aldehyde fragment.

Graphical Abstract
Introduction
(Triphenylphosphoranylidene)ketene, Ph3P=C=C=O (1), was first reported in 1966 [1,2]. It initially attracted attention due to its unique structure, namely the 145.5° angle of the C=C=P moiety and the unusually short C=C bond (1.210 Å). Its utility was subsequently explored, with pioneering work by Bestmann and co-workers [3-5] lending the name Bestmann ylide to this versatile and readily obtained reagent [6-8]. Studies revealed that the ylide readily reacts with alcohols and amines to form α-phosphoranylidene esters or amides, providing diverse isolable Wittig reagents that can be used in subsequent transformations [4,5,9-11]. Furthermore, if the α-phosphoranylidene ester or amide is formed in the presence of an aldehyde, ketone or ester, an additional in situ Wittig reaction can occur [12-18]. In this way, amides, esters and thioesters can be obtained through three-component couplings [14,16]. Intramolecular couplings with a Bestmann ylide linchpin have enabled direct lactone and lactam synthesis [12-15], including the preparation of macrolactones [16-18]. An extension of this methodology to γ-hydroxyenone substrates allows the preparation of α-alkylidene-γ-butyrolactones through tandem acylation and Michael addition, followed by a Wittig reaction [19,20].
Although the utility of the Bestmann ylide in the synthesis of acyclic α,β-unsaturated esters and dienamides has already been reported [11,13,16], its application to the synthesis of α,β,γ,δ-unsaturated esters (i.e., dienoates) remains uncharted. In this paper, a three-component reaction between α,β-unsaturated aldehydes, alcohols and the Bestmann ylide is described. The scope of this esterification–Wittig reaction sequence in the synthesis of α,β,γ,δ-unsaturated esters is studied, and the method is applied in an approach towards the structurally related marine natural products, zampanolide (2) and dactylolide (3, Figure 1), wherein the Bestmann ylide represents a C1–C2 linchpin that connects two segments of the macrocylic ring.
![[1860-5397-11-197-1]](/bjoc/content/figures/1860-5397-11-197-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of (−)-zampanolide (2) and (+)-dactylolide (3).
Figure 1: Structures of (−)-zampanolide (2) and (+)-dactylolide (3).
(−)-Zampanolide (2, Figure 1) was first isolated from the marine sponge Fasciospongia rimosa found at Cape Zampa, Japan [21], and subsequently from a Tongan sponge Cacospongia mycofijiensis [22]. It was found to exhibit potent anticancer activities, with IC50 values in the nM scale against a number of cell lines [22,23]. The structurally related compound, (+)-dactylolide, was discovered in the marine sponge Dactylospongia [24] and has significantly lower cytotoxicity. The absolute configuration of the natural material is not firmly established due to discrepancies in optical rotation values between natural and synthetic samples [25].
Zampanolide and dactylolide have engendered world-wide interest from the synthetic community, culminating in a number of total syntheses [26-41]. Zampanolide is invariably prepared by appending the amide side-chain 4 to the aldehyde moiety of dactylolide, so the synthesis of zampanolide requires the prior generation of dactylolide. Although fragment syntheses vary, the late-stage fragment assembly of the dactylolide macrocycle has centred mostly around construction of the C1–C5 dienoate by Wittig-type olefination reactions followed by ester hydrolysis and esterification with the C19 hydroxy group, combined with metathesis to form the alkene at C8–C9. In our synthesis, the doubly unsaturated ester moiety will be formed through an efficient linchpin reaction between the C16–C20 fragment (alcohol 7) [42-45], the C3–C8 fragment (α,β-unsaturated aldehyde 8) and Bestmann ylide (1, Scheme 1). We plan to then attach the C9–C15 aldehyde fragment 6 by asymmetric alkynylation, and form the pyran using an oxa-Michael addition, in a manner reminiscent of that employed by Uenishi and co-workers [34]. Finally, macrocyclisation will be achieved through the well-established strategy of ring-closing metathesis at C8–C9.
![[1860-5397-11-197-i1]](/bjoc/content/inline/1860-5397-11-197-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of zampanolide involving a Bestmann ylide linchpin strategy.
Scheme 1: Retrosynthesis of zampanolide involving a Bestmann ylide linchpin strategy.
Results and Discussion
Evaluation of the three-component reaction for the synthesis of α,β,γ,δ-unsaturated esters
The study of the reactions between Bestmann ylide, alcohols and α,β-unsaturated aldehydes began with the investigation of the coupling between linchpin 1 [46], E-hex-2-en-1-ol (9a) and E-cinnamaldehyde (10a) (Table 1, entry 1). Typically, Bestmann ylide reactions are performed at elevated temperature in a high boiling and/or ether solvent, such as toluene, 1,4-dioxane or THF [11,14-17,19,20]. To investigate the necessity for high temperature, the primary reaction in this study was initiated at room temperature (19 °C) in toluene and then progressively warmed to reflux (110 °C) while monitoring the progress by TLC. It was noted that the Bestmann ylide reagent was insoluble up to 80 °C and no reaction was observed until the reaction mixture was heated at reflux. Under these conditions, incomplete consumption of the starting materials was seen after 18 h and the poor conversion was attributed to the instability of Bestmann ylide over long periods at elevated temperatures and in the presence of any adventitious nucleophilic source. Nonetheless, the product 11a was obtained in a modest yield (Table 1, entry 1). In response to these observations, the reaction was attempted in THF, a solvent in which Bestmann ylide readily dissolved at room temperature. This provided better conversion, although the isolated yield was only marginally improved (Table 1, entry 2). Gratifyingly, the reaction of oct-2-en-1-ol (9b) with cinnamaldehyde (10a) was efficient and high yielding (Table 1, entry 3). Use of a Z-allylic alcohol 9c, likewise produced excellent amounts of the product dienoate (Table 1, entry 4), although a longer reaction time was required to achieve this. The Z-geometry of the allylic alcohol was retained, as expected. After this, the secondary allylic alcohol 9d was investigated and a reasonable yield of the product was obtained when the reaction was carried out in THF (Table 1, entry 5). A comparative reaction in toluene was also performed and found to deliver a better yield of the product (Table 1, entry 6). The saturated secondary alcohol menthol (9e), with additional steric encumbrance and stereogenic centres, provided a good yield of the product 11e [47] in THF, despite incomplete conversion (Table 1, entry 7). Full conversion but a poor isolated yield of the product was achieved in toluene after reaction for 23 hours (Table 1, entry 8). Decreasing the reaction time provided better results (Table 1, entry 9), indicating that the product may decompose upon prolonged periods at elevated temperature. The Bestmann ylide coupling of menthol (9e) and octa-2,4-dienal (10b) delivered the trienoate product 11f in good yields at both 0.1 and 0.3 mmol scale (Table 1, entries 10 and 11). Taken together, these results indicate that both primary and secondary alcohol substrates react effectively with conjugated unsaturated aldehydes and the Bestmann ylide linchpin in either THF or toluene, although prolonged reflux in toluene may cause degradation of the products. Small quantities (<10%) of isomeric products, presumed to be the corresponding 2Z,4E-dienes of 11a, 11b and 11c, were observed in the reaction mixtures derived from the primary alcohols. Only barely trace amounts of Z-alkenes were ever observed with secondary alcohols. As the minor isomers were not able to be isolated, their identities are unconfirmed.
Table 1: Coupling reactions of alcohols 9 and aldehydes 10 with Bestmann ylide (1)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-197-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Alcohol | Aldehyde | Conditionsb | Product | Yieldc |
|---|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-197-i5.svg?max-width=637&scale=1.0)
9a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-197-i6.svg?max-width=637&scale=1.0)
10a |
toluene, rt to 110 °C, 18 hd |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-197-i7.svg?max-width=637&scale=1.0)
11a |
53% (60%) |
| 2 | 9a | 10ae | THF, 66 °C, 2 hf | 11a | 55% (94%) |
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-197-i8.svg?max-width=637&scale=1.0)
9b |
10a | THF, 66 °C, 1.5 h |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-197-i9.svg?max-width=637&scale=1.0)
11b |
93% (100%) |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-197-i10.svg?max-width=637&scale=1.0)
9c |
10a | THF, 66 °C, 6 hg |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-197-i11.svg?max-width=637&scale=1.0)
11c |
91% (100%) |
| 5 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-197-i12.svg?max-width=637&scale=1.0)
9d |
10a | THF, 66 °C, 4.5 h |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-197-i13.svg?max-width=637&scale=1.0)
11d |
61% (100%) |
| 6 | 9d | 10a | toluene, 110 °C, 5.5 h | 11d | 71% (94%) |
| 7 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-197-i14.svg?max-width=637&scale=1.0)
9e |
10a | THF, 66 °C, 22 h |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-197-i15.svg?max-width=637&scale=1.0)
11e |
77% (85%) |
| 8 | 9e | 10a | toluene, 110 °C, 23 h | 11e | 29% (100%) |
| 9 | 9e | 10a | toluene, 110 °C, 9.5 hg | 11e | 53% (100%) |
| 10 | 9e |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-197-i16.svg?max-width=637&scale=1.0)
10b |
toluene, 110 °C, 3 h |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-197-i17.svg?max-width=637&scale=1.0)
11f |
67% (90%) |
| 11 | 9e | 10b | toluene, 110 °C, 4.5 hg | 11f | 66% (100%) |
| 12 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-197-i18.svg?max-width=637&scale=1.0)
7a |
10a | THF, 66 °C, 3.5 h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-197-i19.svg?max-width=637&scale=1.0)
11g |
<36% (43%)h |
| 13 | 7a | 10a | toluene, 110 °C, 2 h | 11g | <49% (73%)h |
| 14 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-197-i20.svg?max-width=637&scale=1.0)
7b |
10a | THF, 66 °C, 8 h |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-197-i21.svg?max-width=637&scale=1.0)
11h |
42% (69%) |
| 15 | 7b | 10a | toluene, 110 °C, 4 hg | 11h | 70% (100%) |
aUnless otherwise stated, reactions were performed on a 0.1 mmol scale using approximately 1:1:1 ratio of alcohol/Bestmann ylide/aldehyde. bSolvent, temperature, reaction time. cIsolated yield. Conversion (given in brackets) was calculated based on relative integrations of peaks assigned to the limiting reagent (aldehyde) and product in 1H NMR spectra of the crude reaction mixture after work-up. dReaction was carried out on 0.8 mmol scale. e0.57 equiv of aldehyde 10a were used. fReaction was carried out on 0.2 mmol scale. gReaction was carried out on 0.3 mmol scale. hProduct 11g was contaminated with the regioisomer resulting from silyl migration and esterification of the primary hydroxy group (3:2 ratio 11g:isomeric C20 ester).
After these promising results, the homopropargylic secondary alcohol 7a [42], representing the C16–C20 fragment of zampanolide, was subjected to Bestmann ylide linchpin reactions with cinnamaldehyde (10a) in THF and toluene (Table 1, entries 12 and 13). In an attempt to avoid degradation, the reactions were terminated prior to full conversion, leading to the dienoate product in modest yields. Unfortunately, it was found that the product was not isolable in pure form but was contaminated with further isomeric material. Careful analysis of the product mixtures led to the realisation that silyl migration from the primary to the secondary hydroxy group was occurring in the reaction, leading to the C20-linked ester isomer in addition to the desired C19 ester 11g. In contrast, reactions of the PMB-protected variant 7b [43-45] with cinnamaldehyde afforded the product 11h in pure form and reasonable-to-good yields in THF and toluene (Table 1, entries 14 and 15, respectively).
Preparation of the C3–C8 fragment of dactylolide/zampanolide
The aldehyde 8, representing the C3–C8 fragment of zampanolide, was synthesised from acrolein (12) (Scheme 2). Firstly, Barbier reaction of acrolein with propargyl bromide followed by silyl protection of the resulting alcohol afforded enyne 13. Treatment of the lithium alkynylide derived from 13 with methyl chloroformate produced the methyl ester 14 [48]. Cuprate-mediated conjugate addition of a methyl nucleophile to the ynoate 14 provided the Z-enoate 15. Our attempts to directly reduce the ester 15 to the aldehyde 8 were unsuccessful due to competitive over-reduction to the corresponding alcohol. Therefore, a two-step reduction–oxidation process was undertaken to afford the aldehyde 8 in a good yield.
![[1860-5397-11-197-i2]](/bjoc/content/inline/1860-5397-11-197-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Three-component coupling of dactylolide/zampanolide fragments with Bestmann ylide
With aldehyde 8 in hand, a reaction with Bestmann ylide (1) was performed in toluene using the TBDPS-protected alcohol 7a. The α,β,γ,δ-unsaturated ester 5a was formed as a mixture of diastereomers (Scheme 3), as expected, but was again contaminated with the regioisomer resulting from silyl migration (2:1 ratio 5a:isomeric C20 ester). Employing the PMB-protected C19 alcohols 7b [43-45] and 7c [43-45,49] led to the desired products 5b and 5c, respectively. While the reaction of 7b with aldehyde 8 in THF took 11 h to go to completion (62% isolated yield of 5b), the equivalent reaction in toluene required only 5 h (68% yield). Alcohol 7c was used to assess the compatibility of an unprotected, terminal alkyne in this linchpin reaction with a view to future synthetic ease [50]. Pleasingly, the reaction of aldehyde 8 with alcohol 7c in toluene provided the desired product 5c in a comparable yield (66%) after 5 h. In these reactions, only the desired E,Z-diene isomer was observed.
![[1860-5397-11-197-i3]](/bjoc/content/inline/1860-5397-11-197-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Bestmann ylide linchpin coupling of the C16–C20 and C3–C8 fragments of zampanolide/dactylolide.
Scheme 3: Bestmann ylide linchpin coupling of the C16–C20 and C3–C8 fragments of zampanolide/dactylolide.
Conclusion
In summary, an efficient three-component reaction between (triphenylphosphoranylidene)ketene (Bestmann ylide, 1), an alcohol and an unsaturated aldehyde delivers α,β,γ,δ-unsaturated esters. This methodology enabled the facile synthesis of E,Z-dienoate products 5b and 5c, which represent two-thirds of the dactylolide/zampanolide macrocycle, suitably functionalised for transformation to the natural products. This highly efficient method for connecting the C3–C8 and C16–C20 fragments through a C1–C2 linchpin in a single step contrasts with other routes that require multiple-step sequences including Wittig-type reaction, ester hydrolysis and coupling to the C19 alcohol. Elaboration of these compounds into the targets – dactylolide, zampanolide and analogues thereof – is currently under development in our lab.
Experimental
General procedure for Bestmann ylide linchpin reaction: To a mixture of alcohol (1 equiv, 0.1–0.3 M) and Bestmann ylide (1 equiv) in solvent (toluene or tetrahydrofuran) heated at reflux, a solution of aldehyde (1 equiv, 1.0 M) was added. The reaction was heated at reflux until full consumption of starting material aldehyde was observed by TLC. After cooling to rt, the reaction was concentrated and purified by silica column chromatography.
(2′E,2E,4E)-Hex-2′-enyl 5-phenylpenta-2,4-dienoate (11a). Rf = 0.23 (20:1 pet. ether:Et2O); 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (complex m, 3H), 7.36 (app. t, J = 7.6 Hz, 2H), 7.30 (t, J = 6.9 Hz, 1H), 6.93–6.82 (complex m, 2H), 6.01 (d, J = 15.6 Hz, 1H), 5.81 (dt, J = 15.4, 6.6 Hz, 1H), 5.62 (dt, J = 15.4, 6.2 Hz, 1H), 4.63 (d, J = 6.6 Hz, 2H), 2.14 (app. q, J = 7.1 Hz, 2H), 1.43 (app. sext, J = 7.4 Hz, 2H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.8, 144.7, 140.4, 136.3, 136.0, 129.0, 128.8, 127.2, 126.2, 124.0, 121.2, 65.2, 34.3, 22.0, 13.6; IR (neat) cm−1: 2958 (m), 2929 (m), 1706 (s), 1625 (s), 1449 (m), 1236 (s), 1172 (s), 997 (m), 689 (m); HRMS (ESI) m/z: [M + H]+ calcd for C17H21O2, 257.1536 found, 257.1529, (Δ = 2.7 ppm).
(1′R,2′S,5′R,2E,4E,6E)-2′-Isopropyl-5′-methylcyclohex-1′-yl deca-2,4,6-trienoate (11f). Rf = 0.14 (40:1 pet. ether:Et2O); 1H NMR (500 MHz, CDCl3) δ 7.28 (dd, J = 15.4, 11.2 Hz, 1H), 6.53 (dd, J = 14.9, 10.7 Hz, 1H), 6.21 (dd, J = 14.9, 11.2 Hz, 1H), 6.13 (dd, J = 15.1, 10.7 Hz, 1H), 5.92 (dt, J = 14.9, 7.2 Hz, 1H), 5.83 (d, J = 15.4 Hz, 1H), 4.75 (app. td, J = 10.9, 4.4 Hz, 1H), 2.12 (app. q, J = 7.3 Hz, 2H), 2.02 (br d, J = 12.2 Hz, 1H), 1.88 (septd, J = 6.8, 2.3 Hz, 1H), 1.71–1.65 (complex m, 2H), 1.50 (partially obs. m, 1H), 1.44 (m, 2H), 1.40 (partially obs. m, 1H), 1.07 (app. qd, J = 12.9, 2.9 Hz, 1H), 0.99 (app. q, J = 11.5 Hz, 1H), 0.91 (t, J = 7.3 Hz, 3H), 0.90 (d, J = 6.1 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H), 0.87 (partially obs. m, 1H), 0.76 (d, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.8, 144.6, 141.0, 140.2, 130.0, 127.8, 120.5, 73.9, 47.2, 41.0, 35.0, 34.3, 31.4, 26.3, 23.6, 22.2, 22.0, 20.7, 16.4, 13.7; IR (neat) cm−1: 2955 (s), 2930 (s), 2869 (s), 1694 (s), 1615 (s), 1456 (m), 1342 (m), 1133 (s), 1007 (s); HRMS (ESI) m/z: [M + H]+ calcd for C20H33O2, 305.2475; found, 305.2486, (Δ = 3.6 ppm). [α]D22 = −26 (c 0.42, CH2Cl2).
(2E,4E)-[1′-(para-Methoxybenzyloxy)-5′-trimethylsilyl]pent-4′-yn-2′-yl 5-phenylpenta-2,4-dienoate (11h). Rf = 0.28 (80% CH2Cl2 in n-hexane); 1H NMR (500 MHz, CDCl3) δ 7.51–7.44 (complex m, 3H), 7.37 (app. t, J = 7.1 Hz, 2H), 7.33 (m, 1H), 7.28 (br d, J = 8.8 Hz, 2H), 6.95–6.84 (complex m, 4H), 6.02 (d, J = 15.4 Hz, 1H), 5.20 (app. dt, J = 10.5, 5.7 Hz, 1H), 4.55 (d, J = 11.5 Hz, 1H), 4.50 (d, J = 11.7 Hz, 1H), 3.80 (s, 3H), 3.72–3.67 (m, 2H), 2.68 (dd, J = 17.1, 7.1 Hz, 1H), 2.63 (dd, J = 16.9, 5.9 Hz, 1H), 0.14 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 166.2, 159.2, 145.2, 140.7, 136.0, 130.1, 129.3, 129.1, 128.8, 127.2, 126.2, 121.0, 113.8, 101.9, 87.1, 73.0, 70.7, 69.4, 55.2, 22.3, 0.00; IR (neat) cm−1: 3028 (w), 2957 (m), 2901 (m), 2178 (m), 1709 (s), 1625 (s), 1512 (m), 1245 (s), 1128 (s), 840 (s), 757 (s); HRMS (ESI) m/z: [M + H]+ calcd for C27H33O4Si, 449.2143; found, 449.2146, (Δ = 0.67 ppm).
(2E,4Z)-1′-(para-Methoxybenzyloxy)-5′-(trimethylsilyloxy)pent-4′-yn-2′-yl 7-(tert-butyldimethylsilyloxy)-5-methylnona-2,4,8-trienoate (5b). Rf = 0.30 (80% CH2Cl2 in n-hexane); 1H NMR (500 MHz, CDCl3) δ 7.57 (dd, J = 14.9, 11.8 Hz, 1H), 7.26 (d, J = 8.3 Hz, 2H), 6.87 (d, J = 8.3 Hz, 2H), 6.07 (d, J = 11.5 Hz, 1H), 5.80 (obs. ddd, J = 16.8, 10.6, 5.7 Hz, 1H), 5.80 (obs. d, J = 15.2 Hz, 1H), 5.23–5.11 (obs. m, 1H), 5.18 (obs. d, J = 17.2 Hz, 1H), 5.05 (d, J = 10.3 Hz, 1H), 4.53 (d, J = 11.5 Hz, 1H), 4.48 (d, J = 11.5 Hz, 1H), 4.25 (app. q, J = 6.2 Hz, 1H), 3.80 (s, 3H), 3.67 (app. d, J = 4.9 Hz, 2H), 2.69–2.54 (complex m, 3H), 2.36 (ddd, J = 13.0, 6.5, 5.5 Hz, 1H), 1.92 (s, 3H), 0.87 (s, 9H), 0.13 (s, 9H), 0.01 (s, 6H); 13C NMR (500 MHz, CDCl3) δ 166.71, 166.69, 159.2, 146.58, 146.56, 141.70, 141.67, 140.9, 130.1, 129.3, 126.09, 129.07, 118.89, 118.87, 114.2, 113.8, 102.0, 86.92, 86.90, 73.00, 72.99, 72.86, 72.82, 70.48, 70.47, 69.45, 69.41, 55.2, 41.7, 25.83, 25.82, 25.64, 25.62, 22.32, 22.29, 18.1, 0.00, −0.01, −4.54, −4.88; IR (neat) cm−1: 2956 (s), 2857 (m), 2180 (w), 1714 (s), 1636 (m), 1513 (m), 1249 (s), 1033 (m), 837 (s), 775 (m), 760 (m); HRMS (ESI) m/z: [M + H]+ calcd for C32H51O5Si2, 571.3270; found, 571.3276, (Δ = 1.1 ppm).
(2E,4Z)-1′-(para-Methoxybenzyloxy)pent-4′-yn-2′-yl 7-(tert-butyldimethylsilyloxy)-5-methylnona-2,4,8-trienoate (5c). Rf = 0.32 (80% CH2Cl2 in n-hexane); 1H NMR (500 MHz, CDCl3) δ 7.58 (dd, J = 14.9, 11.7 Hz, 1H), 7.26 (d, J = 8.6 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 6.07 (d, J = 11.5 Hz, 1H), 5.80 (obs. ddd, J = 17.1, 10.5, 5.9 Hz, 1H), 5.80 (obs. d, J = 14.9 Hz, 1H), 5.23–5.14 (obs. m, 1H), 5.18 (obs. d, J = 16.9 Hz, 1H), 5.05 (d, J = 10.3 Hz, 1H), 4.53 (d, J = 11.5 Hz, 1H), 4.48 (d, J = 11.5 Hz, 1H), 4.25 (app. q, J = 5.6 Hz, 1H), 3.80 (s, 3H), 3.71–3.62 (m, 2H), 2.65 (ddd, J = 16.9, 6.6, 2.7 Hz, 1H), 2.58 (ddd, J = 16.9, 5.4, 2.5 Hz, 1H), 2.61–2.54 (obs. m, 1H), 2.36 (app. dt, J = 13.1, 4.9 Hz, 1H), 1.97 (m, 1H), 1.92 (s, 3H), 0.86 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 166.7, 159.2, 146.81, 146.78, 141.92, 141.89, 140.95, 140.93, 130.1, 129.31, 129.30, 126.07, 126.05, 118.71, 118.68, 114.2, 113.8, 79.6, 73.0, 72.81, 72.77, 70.33, 70.30, 70.18, 70.13, 69.3, 55.2, 41.8, 25.8, 25.66, 25.65, 20.97, 20.95, 18.1, −4.54, −4.88; IR (neat) cm−1: 3308 (w), 2930 (m), 2857 (s), 2214 (w), 1712 (s), 1635 (m), 1612 (m), 1514 (s), 1248 (s), 1033 (m), 836 (s), 776 (s); HRMS (ESI) m/z: [M + H]+ calcd for C29H43O5Si, 499.2874; found, 499.2868, (Δ = 1.2 ppm).
Supporting Information
| Supporting Information File 1: Full experimental methods; spectroscopic data and NMR spectra of all new compounds. | ||
| Format: PDF | Size: 1.7 MB | Download |
Acknowledgements
The authors gratefully acknowledge financial support from Genesis Oncology Trust and the Health Research Council of New Zealand, and helpful discussions with Paul Teesdale-Spittle, Rob Keyzers, Peter Northcote, Mark Bartlett and Kalpani Somarathne (VUW). Sophie Geyrhofer prepared and generously supplied IBX. Ian Vorster, Teresa Gen (VUW) and Yinrong Lu (Callaghan Innovation) are thanked for technical support.
References
-
Daly, J. J.; Wheatley, P. J. J. Chem. Soc. A 1966, 1703–1706. doi:10.1039/j19660001703
Return to citation in text: [1] -
Matthews, C. N.; Birum, G. H. Tetrahedron Lett. 1966, 7, 5707–5710. doi:10.1016/S0040-4039(01)84182-4
Return to citation in text: [1] -
Bestmann, H. J.; Sandmeier, D. Chem. Ber. 1980, 113, 274–277. doi:10.1002/cber.19801130129
Return to citation in text: [1] -
Bestmann, H. J.; Schobert, R. Synthesis 1989, 419–423. doi:10.1055/s-1989-27271
Return to citation in text: [1] [2] -
Bestmann, H. J.; Kellermann, W. Synthesis 1994, 1257–1261. doi:10.1055/s-1994-25676
Return to citation in text: [1] [2] -
Bartlett, M. Synlett 2013, 24, 773–774. doi:10.1055/s-0032-1318264
Return to citation in text: [1] -
Bestmann, H. J. Angew. Chem., Int. Ed. Engl. 1977, 16, 349–364. doi:10.1002/anie.197703491
Return to citation in text: [1] -
Schobert, R. Org. Synth. 2005, 82, 140–146. doi:10.1002/0471264229.os082.22
Return to citation in text: [1] -
Boeckman, R. K., Jr.; Song, X.; Pero, J. E. J. Org. Chem. 2006, 71, 8969–8972. doi:10.1021/jo061732y
Return to citation in text: [1] -
Schlenk, A.; Diestel, R.; Sasse, F.; Schobert, R. Chem. – Eur. J. 2010, 16, 2599–2604. doi:10.1002/chem.200902544
Return to citation in text: [1] -
Pachali, S.; Hofmann, C.; Rapp, G.; Schobert, R.; Baro, A.; Frey, W.; Laschat, S. Eur. J. Org. Chem. 2009, 2828–2835. doi:10.1002/ejoc.200900176
Return to citation in text: [1] [2] [3] -
Jung, M. E.; Yoo, D. Org. Lett. 2011, 13, 2698–2701. doi:10.1021/ol200796r
Return to citation in text: [1] [2] -
Westman, J.; Orrling, K. Comb. Chem. High Throughput Screening 2002, 5, 571–574. doi:10.2174/1386207023330066
Return to citation in text: [1] [2] [3] -
Fedoseyenko, D.; Raghuraman, A.; Ko, E.; Burgess, K. Org. Biomol. Chem. 2012, 10, 921–924. doi:10.1039/C2OB06692K
Return to citation in text: [1] [2] [3] [4] -
Raghuraman, A.; Ko, E.; Perez, L. M.; Ioerger, T. R.; Burgess, K. J. Am. Chem. Soc. 2011, 133, 12350–12353. doi:10.1021/ja2033734
Return to citation in text: [1] [2] [3] -
Schobert, R.; Siegfried, S.; Gordon, G. J. J. Chem. Soc., Perkin Trans. 1 2001, 2393–2397. doi:10.1039/b105745f
Return to citation in text: [1] [2] [3] [4] [5] -
Risi, R. M.; Burke, S. D. Org. Lett. 2012, 14, 1180–1182. doi:10.1021/ol2034299
Return to citation in text: [1] [2] [3] -
Bestmann, H. J.; Kellermann, W.; Pecher, B. Synthesis 1993, 149–152. doi:10.1055/s-1993-25820
Return to citation in text: [1] [2] -
Kitson, R. R. A.; McAllister, G. D.; Taylor, R. J. K. Tetrahedron Lett. 2011, 52, 561–564. doi:10.1016/j.tetlet.2010.11.143
Return to citation in text: [1] [2] -
Kitson, R. R. A.; Taylor, R. J. K.; Wood, J. L. Org. Lett. 2009, 11, 5338–5341. doi:10.1021/ol902191d
Return to citation in text: [1] [2] -
Tanaka, J.; Higa, T. Tetrahedron Lett. 1996, 37, 5535–5538. doi:10.1016/0040-4039(96)01149-5
Return to citation in text: [1] -
Field, J. J.; Singh, A. J.; Kanakkanthara, A.; Halafihi, T.; Northcote, P. T.; Miller, J. H. J. Med. Chem. 2009, 52, 7328–7332. doi:10.1021/jm901249g
Return to citation in text: [1] [2] -
Field, J. J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodriguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefield, S. J.; Singh, A. J.; Jiménez-Barbero, J.; Northcote, P.; Miller, J. H.; López, J. A.; Hamel, E.; Barasoain, I.; Altmann, K.-H.; Díaz, J. F. Chem. Biol. 2012, 19, 686–698. doi:10.1016/j.chembiol.2012.05.008
Return to citation in text: [1] -
Cutignano, A.; Bruno, I.; Bifulco, G.; Casapullo, A.; Debitus, C.; Gomez-Paloma, L.; Riccio, R. Eur. J. Org. Chem. 2001, 775–778. doi:10.1002/1099-0690(200102)2001:4<775::AID-EJOC775>3.0.CO;2-Z
Return to citation in text: [1] -
Chen, Q.-H.; Kingston, D. G. I. Nat. Prod. Rep. 2014, 31, 1202–1226. doi:10.1039/C4NP00024B
Return to citation in text: [1] -
Smith, A. B., III; Safanov, I. G.; Corbett, R. M. J. Am. Chem. Soc. 2001, 123, 12426–12427. doi:10.1021/ja012220y
Return to citation in text: [1] -
Smith, A. B., III; Safonov, I. G.; Corbett, R. M. J. Am. Chem. Soc. 2002, 124, 11102–11113. doi:10.1021/ja020635t
Return to citation in text: [1] -
Hoye, T. R.; Hu, M. J. Am. Chem. Soc. 2003, 125, 9576–9577. doi:10.1021/ja035579q
Return to citation in text: [1] -
Sanchez, C. C.; Keck, G. E. Org. Lett. 2005, 7, 3053–3056. doi:10.1021/ol051040g
Return to citation in text: [1] -
Aubele, D. L.; Wan, S.; Floreancig, P. E. Angew. Chem., Int. Ed. 2005, 44, 3485–3488. doi:10.1002/anie.200500564
Return to citation in text: [1] -
Ding, F.; Jennings, M. P. Org. Lett. 2005, 7, 2321–2324. doi:10.1021/ol0504897
Return to citation in text: [1] -
Ding, F.; Jennings, M. P. J. Org. Chem. 2008, 73, 5965–5976. doi:10.1021/jo8009853
Return to citation in text: [1] -
Louis, I.; Hungerford, N. L.; Humphries, E. J.; McLeod, M. D. Org. Lett. 2006, 8, 1117–1120. doi:10.1021/ol053092b
Return to citation in text: [1] -
Uenishi, J.; Iwamoto, T.; Tanaka, J. Org. Lett. 2009, 11, 3262–3265. doi:10.1021/ol901167g
Return to citation in text: [1] [2] -
Zurwerra, D.; Gertsch, J.; Altmann, K.-H. Org. Lett. 2010, 12, 2302–2305. doi:10.1021/ol100665m
Return to citation in text: [1] -
Zurwerra, D.; Glaus, F.; Betschart, L.; Schuster, J.; Gertsch, J.; Ganci, W.; Altmann, K.-H. Chem. – Eur. J. 2012, 18, 16868–16883. doi:10.1002/chem.201202553
Return to citation in text: [1] -
Yun, S. Y.; Hansen, E. C.; Volchkov, I.; Cho, E. J.; Lo, W. Y.; Lee, D. Angew. Chem., Int. Ed. 2010, 49, 4261–4263. doi:10.1002/anie.201001681
Return to citation in text: [1] -
Ghosh, A. K.; Cheng, X. Org. Lett. 2011, 13, 4108–4111. doi:10.1021/ol201626h
Return to citation in text: [1] -
Ghosh, A. K.; Cheng, X.; Bai, R.; Hamel, E. Eur. J. Org. Chem. 2012, 4130–4139. doi:10.1002/ejoc.201200286
Return to citation in text: [1] -
Lee, K.; Kim, H.; Hong, J. Angew. Chem., Int. Ed. 2012, 51, 5735–5738. doi:10.1002/anie.201201653
Return to citation in text: [1] -
Larsen, E. M.; Wilson, M. R.; Zajicek, J.; Taylor, R. E. Org. Lett. 2013, 15, 5246–5249. doi:10.1021/ol402462h
and references cited therein.
Return to citation in text: [1] -
Kuntiyong, P.; Lee, T. H.; Kranemann, C. L.; White, J. D. Org. Biomol. Chem. 2012, 10, 7884–7899. doi:10.1039/c2ob25766a
Return to citation in text: [1] [2] -
Trost, B. M.; Machacek, M. R.; Faulk, B. D. J. Am. Chem. Soc. 2006, 128, 6745–6754. doi:10.1021/ja060812g
Return to citation in text: [1] [2] [3] [4] -
Kiyotsuka, Y.; Igarashi, J.; Kobayashi, Y. Tetrahedron Lett. 2002, 43, 2725–2729. doi:10.1016/S0040-4039(02)00403-3
Return to citation in text: [1] [2] [3] [4] -
Kiyotsuka, Y.; Kobayashi, Y. J. Org. Chem. 2009, 74, 7489–7495. doi:10.1021/jo901728b
Return to citation in text: [1] [2] [3] [4] -
Bestmann ylide [1, (triphenylphosphoranylidene)ketene] is commercially available from Sigma–Aldrich as a stable crystalline solid or can be generated by the method of reference [8]. It is essential to thoroughly dry the crystals to remove residual nucleophilic solvent, such as methanol, that would partake, upon dissolution, in reaction with the ylide to afford a methyl ester side-product.
Return to citation in text: [1] -
Larson, G. L.; Quiroz, F.; Suárez, J. Synth. Commun. 1983, 13, 833–844. doi:10.1080/00397918308063719
Return to citation in text: [1] -
Direct formylation of the alkyne 13 was attempted using n-butyllithium and N,N-dimethylformamide but afforded the ynal in only 28% yield. Discouraged by the small quantity of material available from this transformation, it was decided to prepare the enal 8 by a longer but better-established route via the corresponding ester 14.
Return to citation in text: [1] -
Maguire, R. J.; Munt, S. P.; Thomas, E. J. J. Chem. Soc., Perkin Trans. 1 1998, 2853–2864. doi:10.1039/a803422b
Return to citation in text: [1] -
The ester products 5 slowly decompose to methyl dienoate byproducts in the presence of methanol. As the most common acetylenic TMS deprotection methods involve the use of methanol, it is deemed beneficial to carry out the alkyne desilylation prior to formation of the dienoate by the Bestmann ylide linchpin reaction.
Return to citation in text: [1]
| 1. | Daly, J. J.; Wheatley, P. J. J. Chem. Soc. A 1966, 1703–1706. doi:10.1039/j19660001703 |
| 2. | Matthews, C. N.; Birum, G. H. Tetrahedron Lett. 1966, 7, 5707–5710. doi:10.1016/S0040-4039(01)84182-4 |
| 12. | Jung, M. E.; Yoo, D. Org. Lett. 2011, 13, 2698–2701. doi:10.1021/ol200796r |
| 13. | Westman, J.; Orrling, K. Comb. Chem. High Throughput Screening 2002, 5, 571–574. doi:10.2174/1386207023330066 |
| 14. | Fedoseyenko, D.; Raghuraman, A.; Ko, E.; Burgess, K. Org. Biomol. Chem. 2012, 10, 921–924. doi:10.1039/C2OB06692K |
| 15. | Raghuraman, A.; Ko, E.; Perez, L. M.; Ioerger, T. R.; Burgess, K. J. Am. Chem. Soc. 2011, 133, 12350–12353. doi:10.1021/ja2033734 |
| 16. | Schobert, R.; Siegfried, S.; Gordon, G. J. J. Chem. Soc., Perkin Trans. 1 2001, 2393–2397. doi:10.1039/b105745f |
| 17. | Risi, R. M.; Burke, S. D. Org. Lett. 2012, 14, 1180–1182. doi:10.1021/ol2034299 |
| 18. | Bestmann, H. J.; Kellermann, W.; Pecher, B. Synthesis 1993, 149–152. doi:10.1055/s-1993-25820 |
| 25. | Chen, Q.-H.; Kingston, D. G. I. Nat. Prod. Rep. 2014, 31, 1202–1226. doi:10.1039/C4NP00024B |
| 4. | Bestmann, H. J.; Schobert, R. Synthesis 1989, 419–423. doi:10.1055/s-1989-27271 |
| 5. | Bestmann, H. J.; Kellermann, W. Synthesis 1994, 1257–1261. doi:10.1055/s-1994-25676 |
| 9. | Boeckman, R. K., Jr.; Song, X.; Pero, J. E. J. Org. Chem. 2006, 71, 8969–8972. doi:10.1021/jo061732y |
| 10. | Schlenk, A.; Diestel, R.; Sasse, F.; Schobert, R. Chem. – Eur. J. 2010, 16, 2599–2604. doi:10.1002/chem.200902544 |
| 11. | Pachali, S.; Hofmann, C.; Rapp, G.; Schobert, R.; Baro, A.; Frey, W.; Laschat, S. Eur. J. Org. Chem. 2009, 2828–2835. doi:10.1002/ejoc.200900176 |
| 26. | Smith, A. B., III; Safanov, I. G.; Corbett, R. M. J. Am. Chem. Soc. 2001, 123, 12426–12427. doi:10.1021/ja012220y |
| 27. | Smith, A. B., III; Safonov, I. G.; Corbett, R. M. J. Am. Chem. Soc. 2002, 124, 11102–11113. doi:10.1021/ja020635t |
| 28. | Hoye, T. R.; Hu, M. J. Am. Chem. Soc. 2003, 125, 9576–9577. doi:10.1021/ja035579q |
| 29. | Sanchez, C. C.; Keck, G. E. Org. Lett. 2005, 7, 3053–3056. doi:10.1021/ol051040g |
| 30. | Aubele, D. L.; Wan, S.; Floreancig, P. E. Angew. Chem., Int. Ed. 2005, 44, 3485–3488. doi:10.1002/anie.200500564 |
| 31. | Ding, F.; Jennings, M. P. Org. Lett. 2005, 7, 2321–2324. doi:10.1021/ol0504897 |
| 32. | Ding, F.; Jennings, M. P. J. Org. Chem. 2008, 73, 5965–5976. doi:10.1021/jo8009853 |
| 33. | Louis, I.; Hungerford, N. L.; Humphries, E. J.; McLeod, M. D. Org. Lett. 2006, 8, 1117–1120. doi:10.1021/ol053092b |
| 34. | Uenishi, J.; Iwamoto, T.; Tanaka, J. Org. Lett. 2009, 11, 3262–3265. doi:10.1021/ol901167g |
| 35. | Zurwerra, D.; Gertsch, J.; Altmann, K.-H. Org. Lett. 2010, 12, 2302–2305. doi:10.1021/ol100665m |
| 36. | Zurwerra, D.; Glaus, F.; Betschart, L.; Schuster, J.; Gertsch, J.; Ganci, W.; Altmann, K.-H. Chem. – Eur. J. 2012, 18, 16868–16883. doi:10.1002/chem.201202553 |
| 37. | Yun, S. Y.; Hansen, E. C.; Volchkov, I.; Cho, E. J.; Lo, W. Y.; Lee, D. Angew. Chem., Int. Ed. 2010, 49, 4261–4263. doi:10.1002/anie.201001681 |
| 38. | Ghosh, A. K.; Cheng, X. Org. Lett. 2011, 13, 4108–4111. doi:10.1021/ol201626h |
| 39. | Ghosh, A. K.; Cheng, X.; Bai, R.; Hamel, E. Eur. J. Org. Chem. 2012, 4130–4139. doi:10.1002/ejoc.201200286 |
| 40. | Lee, K.; Kim, H.; Hong, J. Angew. Chem., Int. Ed. 2012, 51, 5735–5738. doi:10.1002/anie.201201653 |
| 41. |
Larsen, E. M.; Wilson, M. R.; Zajicek, J.; Taylor, R. E. Org. Lett. 2013, 15, 5246–5249. doi:10.1021/ol402462h
and references cited therein. |
| 6. | Bartlett, M. Synlett 2013, 24, 773–774. doi:10.1055/s-0032-1318264 |
| 7. | Bestmann, H. J. Angew. Chem., Int. Ed. Engl. 1977, 16, 349–364. doi:10.1002/anie.197703491 |
| 8. | Schobert, R. Org. Synth. 2005, 82, 140–146. doi:10.1002/0471264229.os082.22 |
| 22. | Field, J. J.; Singh, A. J.; Kanakkanthara, A.; Halafihi, T.; Northcote, P. T.; Miller, J. H. J. Med. Chem. 2009, 52, 7328–7332. doi:10.1021/jm901249g |
| 23. | Field, J. J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodriguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefield, S. J.; Singh, A. J.; Jiménez-Barbero, J.; Northcote, P.; Miller, J. H.; López, J. A.; Hamel, E.; Barasoain, I.; Altmann, K.-H.; Díaz, J. F. Chem. Biol. 2012, 19, 686–698. doi:10.1016/j.chembiol.2012.05.008 |
| 3. | Bestmann, H. J.; Sandmeier, D. Chem. Ber. 1980, 113, 274–277. doi:10.1002/cber.19801130129 |
| 4. | Bestmann, H. J.; Schobert, R. Synthesis 1989, 419–423. doi:10.1055/s-1989-27271 |
| 5. | Bestmann, H. J.; Kellermann, W. Synthesis 1994, 1257–1261. doi:10.1055/s-1994-25676 |
| 24. | Cutignano, A.; Bruno, I.; Bifulco, G.; Casapullo, A.; Debitus, C.; Gomez-Paloma, L.; Riccio, R. Eur. J. Org. Chem. 2001, 775–778. doi:10.1002/1099-0690(200102)2001:4<775::AID-EJOC775>3.0.CO;2-Z |
| 19. | Kitson, R. R. A.; McAllister, G. D.; Taylor, R. J. K. Tetrahedron Lett. 2011, 52, 561–564. doi:10.1016/j.tetlet.2010.11.143 |
| 20. | Kitson, R. R. A.; Taylor, R. J. K.; Wood, J. L. Org. Lett. 2009, 11, 5338–5341. doi:10.1021/ol902191d |
| 21. | Tanaka, J.; Higa, T. Tetrahedron Lett. 1996, 37, 5535–5538. doi:10.1016/0040-4039(96)01149-5 |
| 16. | Schobert, R.; Siegfried, S.; Gordon, G. J. J. Chem. Soc., Perkin Trans. 1 2001, 2393–2397. doi:10.1039/b105745f |
| 17. | Risi, R. M.; Burke, S. D. Org. Lett. 2012, 14, 1180–1182. doi:10.1021/ol2034299 |
| 18. | Bestmann, H. J.; Kellermann, W.; Pecher, B. Synthesis 1993, 149–152. doi:10.1055/s-1993-25820 |
| 22. | Field, J. J.; Singh, A. J.; Kanakkanthara, A.; Halafihi, T.; Northcote, P. T.; Miller, J. H. J. Med. Chem. 2009, 52, 7328–7332. doi:10.1021/jm901249g |
| 12. | Jung, M. E.; Yoo, D. Org. Lett. 2011, 13, 2698–2701. doi:10.1021/ol200796r |
| 13. | Westman, J.; Orrling, K. Comb. Chem. High Throughput Screening 2002, 5, 571–574. doi:10.2174/1386207023330066 |
| 14. | Fedoseyenko, D.; Raghuraman, A.; Ko, E.; Burgess, K. Org. Biomol. Chem. 2012, 10, 921–924. doi:10.1039/C2OB06692K |
| 15. | Raghuraman, A.; Ko, E.; Perez, L. M.; Ioerger, T. R.; Burgess, K. J. Am. Chem. Soc. 2011, 133, 12350–12353. doi:10.1021/ja2033734 |
| 14. | Fedoseyenko, D.; Raghuraman, A.; Ko, E.; Burgess, K. Org. Biomol. Chem. 2012, 10, 921–924. doi:10.1039/C2OB06692K |
| 16. | Schobert, R.; Siegfried, S.; Gordon, G. J. J. Chem. Soc., Perkin Trans. 1 2001, 2393–2397. doi:10.1039/b105745f |
| 11. | Pachali, S.; Hofmann, C.; Rapp, G.; Schobert, R.; Baro, A.; Frey, W.; Laschat, S. Eur. J. Org. Chem. 2009, 2828–2835. doi:10.1002/ejoc.200900176 |
| 13. | Westman, J.; Orrling, K. Comb. Chem. High Throughput Screening 2002, 5, 571–574. doi:10.2174/1386207023330066 |
| 16. | Schobert, R.; Siegfried, S.; Gordon, G. J. J. Chem. Soc., Perkin Trans. 1 2001, 2393–2397. doi:10.1039/b105745f |
| 46. | Bestmann ylide [1, (triphenylphosphoranylidene)ketene] is commercially available from Sigma–Aldrich as a stable crystalline solid or can be generated by the method of reference [8]. It is essential to thoroughly dry the crystals to remove residual nucleophilic solvent, such as methanol, that would partake, upon dissolution, in reaction with the ylide to afford a methyl ester side-product. |
| 42. | Kuntiyong, P.; Lee, T. H.; Kranemann, C. L.; White, J. D. Org. Biomol. Chem. 2012, 10, 7884–7899. doi:10.1039/c2ob25766a |
| 43. | Trost, B. M.; Machacek, M. R.; Faulk, B. D. J. Am. Chem. Soc. 2006, 128, 6745–6754. doi:10.1021/ja060812g |
| 44. | Kiyotsuka, Y.; Igarashi, J.; Kobayashi, Y. Tetrahedron Lett. 2002, 43, 2725–2729. doi:10.1016/S0040-4039(02)00403-3 |
| 45. | Kiyotsuka, Y.; Kobayashi, Y. J. Org. Chem. 2009, 74, 7489–7495. doi:10.1021/jo901728b |
| 34. | Uenishi, J.; Iwamoto, T.; Tanaka, J. Org. Lett. 2009, 11, 3262–3265. doi:10.1021/ol901167g |
| 43. | Trost, B. M.; Machacek, M. R.; Faulk, B. D. J. Am. Chem. Soc. 2006, 128, 6745–6754. doi:10.1021/ja060812g |
| 44. | Kiyotsuka, Y.; Igarashi, J.; Kobayashi, Y. Tetrahedron Lett. 2002, 43, 2725–2729. doi:10.1016/S0040-4039(02)00403-3 |
| 45. | Kiyotsuka, Y.; Kobayashi, Y. J. Org. Chem. 2009, 74, 7489–7495. doi:10.1021/jo901728b |
| 49. | Maguire, R. J.; Munt, S. P.; Thomas, E. J. J. Chem. Soc., Perkin Trans. 1 1998, 2853–2864. doi:10.1039/a803422b |
| 50. | The ester products 5 slowly decompose to methyl dienoate byproducts in the presence of methanol. As the most common acetylenic TMS deprotection methods involve the use of methanol, it is deemed beneficial to carry out the alkyne desilylation prior to formation of the dienoate by the Bestmann ylide linchpin reaction. |
| 48. | Direct formylation of the alkyne 13 was attempted using n-butyllithium and N,N-dimethylformamide but afforded the ynal in only 28% yield. Discouraged by the small quantity of material available from this transformation, it was decided to prepare the enal 8 by a longer but better-established route via the corresponding ester 14. |
| 43. | Trost, B. M.; Machacek, M. R.; Faulk, B. D. J. Am. Chem. Soc. 2006, 128, 6745–6754. doi:10.1021/ja060812g |
| 44. | Kiyotsuka, Y.; Igarashi, J.; Kobayashi, Y. Tetrahedron Lett. 2002, 43, 2725–2729. doi:10.1016/S0040-4039(02)00403-3 |
| 45. | Kiyotsuka, Y.; Kobayashi, Y. J. Org. Chem. 2009, 74, 7489–7495. doi:10.1021/jo901728b |
| 42. | Kuntiyong, P.; Lee, T. H.; Kranemann, C. L.; White, J. D. Org. Biomol. Chem. 2012, 10, 7884–7899. doi:10.1039/c2ob25766a |
| 43. | Trost, B. M.; Machacek, M. R.; Faulk, B. D. J. Am. Chem. Soc. 2006, 128, 6745–6754. doi:10.1021/ja060812g |
| 44. | Kiyotsuka, Y.; Igarashi, J.; Kobayashi, Y. Tetrahedron Lett. 2002, 43, 2725–2729. doi:10.1016/S0040-4039(02)00403-3 |
| 45. | Kiyotsuka, Y.; Kobayashi, Y. J. Org. Chem. 2009, 74, 7489–7495. doi:10.1021/jo901728b |
| 11. | Pachali, S.; Hofmann, C.; Rapp, G.; Schobert, R.; Baro, A.; Frey, W.; Laschat, S. Eur. J. Org. Chem. 2009, 2828–2835. doi:10.1002/ejoc.200900176 |
| 14. | Fedoseyenko, D.; Raghuraman, A.; Ko, E.; Burgess, K. Org. Biomol. Chem. 2012, 10, 921–924. doi:10.1039/C2OB06692K |
| 15. | Raghuraman, A.; Ko, E.; Perez, L. M.; Ioerger, T. R.; Burgess, K. J. Am. Chem. Soc. 2011, 133, 12350–12353. doi:10.1021/ja2033734 |
| 16. | Schobert, R.; Siegfried, S.; Gordon, G. J. J. Chem. Soc., Perkin Trans. 1 2001, 2393–2397. doi:10.1039/b105745f |
| 17. | Risi, R. M.; Burke, S. D. Org. Lett. 2012, 14, 1180–1182. doi:10.1021/ol2034299 |
| 19. | Kitson, R. R. A.; McAllister, G. D.; Taylor, R. J. K. Tetrahedron Lett. 2011, 52, 561–564. doi:10.1016/j.tetlet.2010.11.143 |
| 20. | Kitson, R. R. A.; Taylor, R. J. K.; Wood, J. L. Org. Lett. 2009, 11, 5338–5341. doi:10.1021/ol902191d |
| 47. | Larson, G. L.; Quiroz, F.; Suárez, J. Synth. Commun. 1983, 13, 833–844. doi:10.1080/00397918308063719 |
© 2015 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)