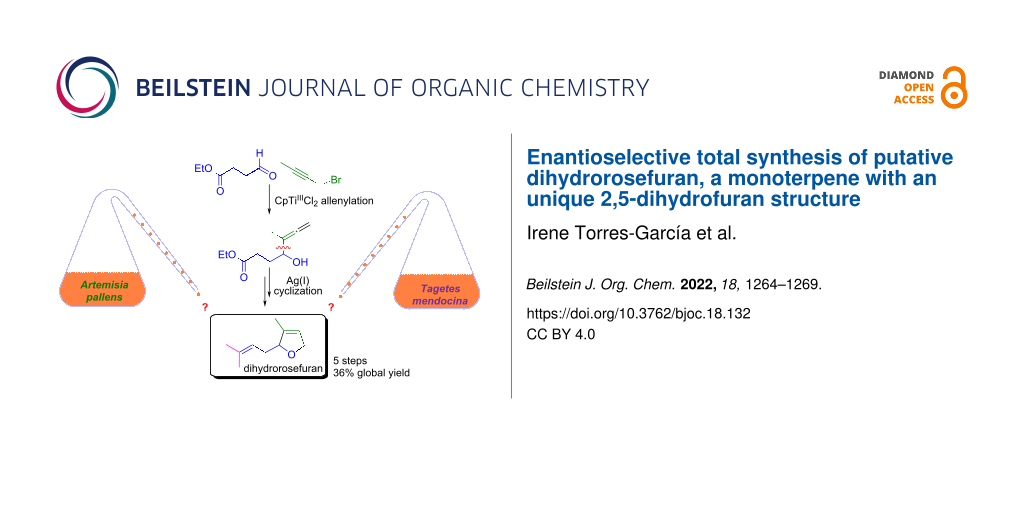
Abstract
An original synthesis of the structure of dihydrorosefuran, a compound allegedly identified in Artemisia pallens and Tagetes mendocina, has been developed. The key steps in the five-step 36% overall yield synthesis are a CpTiIIICl2 mediated Barbier-type allenylation of a linear aldehyde and the formation of a 2,5-dihydrofuran scaffold through a Ag(I)-mediated cyclization. Neither of the reported spectral data for dihydrorosefuran match those of the synthetic product, suggesting that the isolated compound from Tagetes mendocina is in fact the natural product rosiridol, while the real structure of the product from Artemisia pallens remains unknown.

Graphical Abstract
Introduction
Artemisia pallens is an aromatic plant from southern India whose essential oil, known as Davana oil, has shown increasing interest mainly for its use in some beverages, cakes, pastries, etc., as well as in the perfumery industry [1]. In addition, A. pallens has been used in Indian traditional medicine (Ayurveda) for the treatment of measles, cough, cold, depression, diabetes, and high blood pressure [2]. More recently other biological activities have been reported, such as the blood glucose lowering effect of A. pallens [3,4], and its anti-asthmatic potential [5].
A component responsible for the fresh and floral odor of the essential oil was isolated from Davana oil and was assigned as a 2,5-dihydrofuranic monoterpenoid (compound 1 in Scheme 1) and named dihydrorosefuran [6-8]. Furthermore, the same structure was attributed to an isolated substance from the Argentinean herb Tagetes mendocina [9], although not all of its spectroscopic features did match point by point with those previously reported. This made us think that this could be a case of misassigned natural product [10], hence we decided to perform its total synthesis.
Results and Discussion
Our synthetic strategy is based on two metal-mediated steps (Scheme 1). In this way, we thought that the 2,5-dihydrofuran structural motif that is found in the target molecule 1 could be prepared through a Ag(I)-induced intramolecular addition of the hydroxy group to the terminal double bond of the allene in compound 3. Another key step is the Ti(III)-mediated straightforward synthesis of this α-hydroxyallene, which could be achieved through a regioselective Barbier-type coupling of a propargylic halide (1-bromo-2-butyne) with the aldehyde 4 mediated by the organometallic half-sandwich complex [CpTiIIICl2] [11,12].
![[1860-5397-18-132-i1]](/bjoc/content/inline/1860-5397-18-132-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic scheme of the target molecule 1.
Scheme 1: Retrosynthetic scheme of the target molecule 1.
Following this retrosynthetic proposal, our route starts from ethyl 4-oxobutanoate (4) [13] which was prepared by ozonolysis of commercially available ethyl pent-4-enoate (Scheme 2). Coupling of the aldehyde 4 with 1-bromobut-2-yne in the presence of CpTiIIICl2 (generated in situ by reduction of CpTiCl3 with Mn) afforded α-hydroxyallene 3. We have recently described that this Barbier-type reaction affords α-hydroxyallenes as major products, mixed with smaller amounts of homopropargylic alcohols, either if the reaction is performed with stoichiometric amounts of CpTiCl3 or if catalytic amounts are used [12]. However, using the particular substrates in this approach, the allenic compound 3 was exclusively formed, in a satisfactory 81% yield [14]. It is also important to control the pH during the reaction workup, as some contamination of the product with lactone 5 can arise at low pH values, which goes in detriment of the yield. The 2,5-dihydrofuran ring in target compound 1 was obtained through a Ag(I)-mediated intramolecular addition of the hydroxy to the allene group, a process that transformed allene 3 into compound 2. The isopropenyl residue of the target compound 1 was assembled through a two-step sequence. The first one was the addition of an excess of methylmagnesium bromide to the ester 2, that completed the carbon skeleton. The second step was the pH-controlled regioselective dehydration of the tertiary alcohol 6 with amberlyst-15® leading to the monoterpene 1. Other systems tested for the elimination of the hydroxy group in 6 were pyridinium p-toluenesulfonate (PPTS) and camphorsulfonic acid (CSA), that gave poorer results, failing to afford a single product. On the other hand, lactone 5 could also be transformed into alcohol 6 through a simple change in the order of the reactions: addition of methylmagnesium bromide to 5 afforded 7, which was then transformed into 6 by the Ag(I)-mediated cyclization (Scheme 2).
![[1860-5397-18-132-i2]](/bjoc/content/inline/1860-5397-18-132-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of dihydrofuran-monoterpenoid 1. a) i. O3, −78 °C; ii. PPh3, rt, 76%; b) 1-bromobut-2-yne, CpTiCl3, Mn and THF (3: 71–81%, 5: 0–10%); c) AgNO3, Me2CO, 88%; d) MeMgBr, Et2O, 90%; e) Amberlyst®-15, DCM, 74%; f) MeMgBr, Et2O, 83%; g) AgNO3, Me2CO, 75%.
Scheme 2: Synthesis of dihydrofuran-monoterpenoid 1. a) i. O3, −78 °C; ii. PPh3, rt, 76%; b) 1-bromobut-2-yne...
Once we had synthesized racemic compound 1, we designed a chiral version using a stereoselective kinetic resolution of allenol 3 via lipase AK-catalyzed acetylation [15]. In this way, unaltered, (−)-hydroxyallene 3 could be separated from (+)-acetyl derivative 9 through standard column chromatography (Scheme 3). Enantiomeric excesses of (−)-3 and (+)-9 were determined by chiral HPLC analyses. Analysis of the NMR data of the Mosher's derivatives of 8 suggested (S) configuration for the alcohol (−)-3 [16].
![[1860-5397-18-132-i3]](/bjoc/content/inline/1860-5397-18-132-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Racemic resolution of allenol 3 and synthesis of derivatives. a) Lipase AK, vinyl acetate, t-BuOMe, 30 °C, ((−)-(S)-3: 46%, 90% ee, (+)-(R)-9: 39%, 95% ee); b) N,N’-dicyclohexylcarbodiimide (DCC), dimethylaminopyridine (DMAP), (S) or (R)-(−)-α-methoxy-α-(trifluoromethyl)phenylacetic acid, 66% [17], c) MeMgBr (5 equiv), Et2O, 67%.
Scheme 3: Racemic resolution of allenol 3 and synthesis of derivatives. a) Lipase AK, vinyl acetate, t-BuOMe,...
On the other hand, enantiopure acetate (+)-(R)-9 was transformed into diol (+)-(R)-7 by the addition of an excess of MeMgBr. Finally, these enantiopure compounds, α-hydroxyallene (−)-(S)-3 and diol (+)-(R)-7, can be used to prepare both enantiomers of compound 1 following the procedures shown in Scheme 2.
Unfortunately, once the racemic synthesis was successfully completed and the chiral design was fulfilled, it was found that the spectroscopic data of compound 1 did not match neither with those published for the allegedly dihydrorosefuran isolated from Artemisia pallens nor with those reported for the compound from Tagetes mendocina (see Table 1 and Table 2). The 13C NMR data of compound 1 are quite similar to those of the natural product isolated from T. mendocina, except for the signals of the oxygenated carbons (C2 and C5). The same behavior pattern can be observed in the 1H NMR data. This made us think that the natural product of T. mendocina could have an acyclic skeleton instead of a dihydrofuran one. For this reason, we propose this compound should be the diol called rosiridol (Table 1), a substance that has been isolated from other natural sources [18,19], whose structure was also confirmed by total synthesis some years ago [20]. Comparison of NMR data (Table 1) confirmed the initial suspicion. We are still intrigued about the real structure of the natural product isolated from A. pallens. However, it must be considered that this product was elucidated using low frequency NMR machines, which suggests that further research on the chemical composition of this oil is needed.
Table 1: 1H NMR data of isolated and synthetic products.a
| sources of claimed dihydrorosefuran |
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-132-i4.svg?max-width=637&scale=1.0)
1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-132-i5.svg?max-width=637&scale=1.0)
rosiridolb |
||
| A. pallensa,c [7,8] | T. mendocinad [9] | this work synthesise | literature synthesisd [20] | |
| H2 | 4.04 (td, J = 8.0, 1.0 Hz) | 4.03 (t, J = 8 Hz) | 4.67 (br s) | 4.02–3.99 (m) |
| H4 | 5.07 (td, J = 8.0, 1.0 Hz) | 5.66 (t, J = 8 Hz) | 5.51 (br s) | 5.67–5.62 (m) |
| H5 | 4.55 (d, J = 7.0 Hz) | 4.21 (dd) | 4.62–4.53 (m) | 4.24–4.15 (m) |
| H1' | 2.30 (m) | 2.24 (m) | 2.41 (m), 2.20 (ddd, J = 14.0, 7.0, 6.5 Hz) | 2.28–2.19 (m) |
| H2' | 5.32 (t, J = 7.0 Hz) | 5.13 (t, J = 8 Hz) | 5.21 (t, hept, J = 6.9, 1.5 Hz) | 5.13–5.09 (m) |
| H4' | 1.67 (s) | 1.75 (s) | 1.74 (s) | 1.73 (d, J = 1.1 Hz) |
| H5' | 1.60 (s) | 1.66 (s) | 1.66 (s) | 1.64 (s) |
| H1" | 2.05 (s) | 1.69 (s) | 1.72 (s) | 1.67 (s) |
aCDCl3 in all cases; barbitrary numbering for comparison purposes; c80 MHz; d400 MHz; e500 MHz.
Table 2: 13C NMR data of isolated and synthetic productsa.
| sources of claimed dihydrorosefuran |
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-132-i6.svg?max-width=637&scale=1.0)
1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-132-i7.svg?max-width=637&scale=1.0)
rosiridolb |
||
| A. pallensa,c [7,8] | T. mendocinad [9] | this work synthesise | literature synthesisd [20] | |
| C2 | 64.0 | 76.4 | 87.5 | 76.4 |
| C3 | 145.5 | 140.4 | 138.1 | 140.2 |
| C4 | 129.3 | 124.5 | 120.7 | 124.5 |
| C5 | 61.0 | 59.1 | 74.5 | 59.0 |
| C1' | 39.8 | 34.2 | 32.9 | 34.1 |
| C2’ | 118.0 | 119.7 | 119.7 | 119.8 |
| C3' | 142.0 | 135.3 | 133.6 | 135.0 |
| C4' | 26.2 | 25.9 | 25.9 | 25.8 |
| C5' | 24.8 | 18.0 | 18.0 | 18.0 |
| C1" | 17.5 | 12.2 | 12.5 | 12.0 |
aCDCl3 in all cases; barbitrary numbering for comparison purposes; c80 MHz; d100 MHz; e125 MHz.
Conclusion
In summary, we have proved that the two-step sequence TiIII allenylation–AgI cyclization is a simple and efficient strategy for the preparation of the 2,5-dihydrofuran moiety present in many natural products. In fact, we have achieved the total synthesis of the 2,5-dihydrofuran structure 1. After systematic data analysis of our prepared compound and those in the literature, it can be concluded that the proposed structure of the product isolated from Artemisia pallens oil, dihydrorosefuran, is not correct. In addition, it is clear that the compound isolated from Tagetes mendocina is the acyclic diol named rosiridol.
Experimental
Ti-induced allenylation of ethyl 4-oxobutanoate (4)
Under an Ar atmosphere, dry THF (8 mL) that was deoxygenated prior to use was added to a mixture of CpTiCl3 (329 mg, 1.50 mmol) and Mn dust (165 mg, 3.00 mmol) resulting in a green suspension. Then, a solution of ethyl 4-oxobutanoate (4, 196 mg, 1.50 mmol) and 1-bromobut-2-yne (0.27 mL, 3.00 mmol) in THF (2 mL) was dripped and the mixture was stirred for 2.5 hours. The mixture was filtered, diluted with EtOAc, washed with 3% HCl and brine, and dried (anhydrous MgSO4), and the solvent was removed. The residue was purified by flash chromatography (n-hexane/EtOAc 8:2) to afford ethyl 4-hydroxy-5-methylhepta-5,6-dienoate (3, 225 mg, 81%) isolated as light yellow oil. IR (ATR) ν (cm−1): 3434, 2972, 2928, 1958, 1723, 1436, 1374, 1172, 1028, 925, 853; 1H NMR (300 MHz, CDCl3) δ 4.77 (dq, J = 2.3, 3.2 Hz, 2H), 4.12 (q, J = 7.1 Hz, 2H), 4.05 (m, 1H), 2.42 (t, J = 7.2 Hz, 2H), 2.18 (s, 1H), 2.04–1.77 (m, 2H), 1.70 (td, J = 0.5, 3.2 Hz, 3H), 1.25 (t, J = 7.1 Hz, 3H) ppm; 13C NMR (75 MHz, CDCl3, DEPT) δ 204.8 (C), 174.0 (C), 101.6 (C), 77.0 (CH2), 71.5 (CH), 60.5 (CH2), 30.4 (CH2), 30.0 (CH2), 14.5 (CH3), 14.2 (CH3) ppm; HRMS–ESI (Q-TOF, m/z): [M + H]+ calcd for C10H17O3, 185.1178; found, 185.1158. A lactone is formed as side product (0–10%) when the HCl solution used for the workup has a concentration higher than 3%. Compound 5: IR (ATR) ν (cm−1): 2982, 2927, 1960, 1772, 1427, 1331, 1162, 974, 918, 855; 1H NMR (300 MHz, CDCl3) δ 4.89 (m, 1H), 4.86 (m, 2H), 2.55 (m, 2H), 2.30 (m, 2H), 1.79 (t, J = 3.1 Hz, 3H) ppm; 13C{1H} NMR (75 MHz, CDCl3, DEPT) δ 206.0 (C), 177.0 (C), 98.0 (C), 80.4 (CH), 77.5 (CH2), 28.5 (CH2), 26.1 (CH2), 15.0 (CH3) ppm; HRMS–ESI (Q-TOF, m/z): [M + H]+ calcd for C8H11O2,139.0759; found, 139.0782.
Silver(I)-promoted cyclization of ethyl 4-hydroxy-5-methylhepta-5,6-dienoate (3)
A solution of the allenol 3 (65 mg, 0.35 mmol) in acetone (2 mL) was added to a suspension of AgNO3 (120 mg, 0.70 mmol) in acetone (1.5 mL) in the absence of light, and the mixture was stirred at 40 °C overnight. Brine was added and the mixture was extracted with Et2O. The organic phase was dried over anhydrous MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel flash column chromatography (n-hexane/EtOAc 9:1) to afford ethyl 3-(3-methyl-2,5-dihydrofuran-2-yl)propanoate (2, 57 mg, 88%) isolated as colorless oil. IR (ATR) ν (cm−1): 2969, 2927, 2849, 1731, 1442, 1376, 1251, 1160, 1092, 1026, 895, 734. 1H NMR (300 MHz, CDCl3) δ 5.53 (m, 1H), 4.69 (br s, 1H), 4.56 (m, 2H), 4.15 (q, J = 7.1 Hz, 2H), 2.39 (m, 2H), 2.12–2.06 (m, 1H), 1.83–1.74 (m, 1H), 1.72 (br s, 3H), 1.27 (t, J = 7.1 Hz, 3H) ppm; 13C{1H} NMR (75 MHz, CDCl3, DEPT) δ 173.9 (C), 137.2 (C), 121.3 (CH), 86.4 (CH), 74.7 (CH2), 60.3 (CH2), 29.4 (CH2), 28.9 (CH2), 14.2 (CH3), 12.3 (CH3) ppm; HRMS–ESI (Q-TOF, m/z): [M + H]+ calcd for C10H17O3,185.1172; found, 184.1162.
Synthesis of 2-methyl-4-(3-methyl-2,5-dihydrofuran-2-yl)butan-2-ol (6)
Under an N2 atmosphere, methylmagnesium bromide (3 M in Et2O, 0.075 mL, 0.23 mmol) was diluted with dry Et2O (1.5 mL). A solution of ethyl 3-(3-methyl-2,5-dihydrofuran-2-yl)propanoate (2, 16 mg, 0.087 mmol) in Et2O (1 mL) was added dropwise and the reaction mixture was stirred for 3 hours at room temperature. The reaction was quenched with saturated NH4Cl and extracted with EtOAc. The combined organic layer was washed with saturated NaHCO3, brine, and dried over anhydrous MgSO4. The solvent was evaporated in vacuum and the residue was purified using column chromatography (n-hexane/EtOAc 7:3) to give 2-methyl-4-(3-methyl-2,5-dihydrofuran-2-yl)butan-2-ol (6, 169 mg, 90%) isolated as colorless oil. IR (ATR) ν (cm−1): 3425, 2969, 2922, 2852, 1636, 1444, 1382, 1089, 1057, 1025; 1H NMR (300 MHz, CDCl3) δ 5.51 (br s, 1H), 4.68 (br s, 1H), 4.58 (m, 2H), 2.05 (br s, 1H), 1.82 (m, 1H), 1.72 (br s, 3H), 1.57 (m, 3H), 1.24 (s, 6H) ppm; 13C{1H} NMR (75 MHz, CDCl3, DEPT) δ 137.8 (C), 120.7 (CH), 87.7 (CH), 74.5 (CH2), 70.4 (C), 38.5 (CH2), 29.5 (CH3), 29.4 (CH3), 28.3 (CH2), 12.5 (CH3) ppm; HRMS–ESI (Q-TOF, m/z): [M + H]+ calcd for C10H19O2, 171.1385; found, 171.1368.
Synthesis of 3-methyl-2-(3-methylbut-2-en-1-yl)-2,5-dihydrofuran (1)
The reaction of amberlyst®-15 (dry, 97 mg) and 2-methyl-4-(3-methyl-2,5-dihydrofuran-2-yl)butan-2-ol (6, 97 mg, 0.57 mmol), based on the previously reported literature procedure [21], afforded 3-methyl-2-(3-methylbut-2-en-1-yl)-2,5-dihydrofuran (1, 64 mg, 74%) isolated as colorless oil. IR (ATR) ν (cm−1): 3066, 2965, 2918, 2843, 1668, 1445, 1377, 1080, 975, 850, 778; 1H NMR (500 MHz, CDCl3) δ 5.51 (br s, 1H, H4), 5.21 (t, hept, J = 7.0, 1.5 Hz, 1H, H2'), 4.67 (br s, 1H, H2), 4.62–4.53 (m, 2H, H5), 2.41 (m, 1H, H1'a), 2.20 (ddd, J = 14.0, 7.0, 6.5 Hz, 1H, H1'b), 1.74 (d, J = 1.5 Hz, 3H, H4'*), 1.72 (m, 3H, H1''), 1.66 (br s, 3H, H5'*) ppm; 13C{1H} NMR (125 MHz, CDCl3, DEPT) δ 138.1 (C, C3), 133.6 (C, C3'), 120.7 (CH, C4), 119.7 (CH, C2'), 87.5 (CH, C2), 74.5 (CH2, C5), 32.9 (CH2, C1'), 25.9 (CH3, C4'*), 18.0 (CH3, C5'*), 12.5 (CH3, C1'') ppm (*may be interchanged); HRMS–ESI (Q-TOF, m/z): [M + H]+ calcd for C10H17O, 153.1274; found, 153.1262.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization of other substances, and copies of IR, NMR spectra and HPLC chromatograms. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Hellivan, P.-J. Perfum. Flavor. 2011, 36, 38–43.
Return to citation in text: [1] -
Kulkarni, R. N. Artemisia pallens. In Artemisia; Wright, C. W., Ed.; Taylor & Francis: London, UK, 2002; Vol. 18, pp 119–137.
Return to citation in text: [1] -
Subramoniam, A.; Pushpangadan, P.; Rajasekharan, S.; Evans, D. A.; Latha, P. G.; Valsaraj, R. J. Ethnopharmacol. 1996, 50, 13–17. doi:10.1016/0378-8741(95)01329-6
Return to citation in text: [1] -
Pavithra, K. S.; Annadurai, J.; Ragunathan, R. J. Pharmacogn. Phytochem. 2018, 7, 664–675.
Return to citation in text: [1] -
Mukherjee, A. A.; Kandhare, A. D.; Rojatkar, S. R.; Bodhankar, S. L. Biomed. Pharmacother. 2017, 94, 880–889. doi:10.1016/j.biopha.2017.08.017
Return to citation in text: [1] -
Chandra, A.; Misra, L. N.; Thakur, R. S. Tetrahedron Lett. 1987, 28, 6377–6380. doi:10.1016/s0040-4039(01)91378-4
Return to citation in text: [1] -
Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 549–552. doi:10.1016/0031-9422(91)83725-z
Return to citation in text: [1] [2] [3] -
Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 4212–4213. doi:10.1016/0031-9422(91)83512-j
Return to citation in text: [1] [2] [3] -
Lima, B.; Agüero, M. B.; Zygadlo, J.; Tapia, A.; Solis, C.; Rojas De Arias, A.; Yaluff, G.; Zacchino, S.; Feresin, G. E.; Schmeda-Hirschmann, G. J. Chil. Chem. Soc. 2009, 54, 68–72. doi:10.4067/s0717-97072009000100016
Return to citation in text: [1] [2] [3] -
Nicolaou, K. C.; Snyder, S. A. Angew. Chem., Int. Ed. 2005, 44, 1012–1044. doi:10.1002/anie.200460864
Return to citation in text: [1] -
López-Martínez, J. L.; Torres-García, I.; Rodríguez-García, I.; Muñoz-Dorado, M.; Álvarez-Corral, M. J. Org. Chem. 2019, 84, 806–816. doi:10.1021/acs.joc.8b02643
Return to citation in text: [1] -
Torres‐García, I.; López‐Martínez, J. L.; Martínez‐Martínez, R.; Oltra, J. E.; Muñoz‐Dorado, M.; Rodríguez‐García, I.; Álvarez‐Corral, M. Appl. Organomet. Chem. 2020, 34, 10.1002/aoc.5244. doi:10.1002/aoc.5244
Return to citation in text: [1] [2] -
Smith, A. B., III; Fukui, M.; Vaccaro, H. A.; Empfield, J. R. J. Am. Chem. Soc. 1991, 113, 2071–2092. doi:10.1021/ja00006a029
Return to citation in text: [1] -
Reaction performed with stoichiometric amounts of CpTiCl2.
Return to citation in text: [1] -
Li, W.; Lin, Z.; Chen, L.; Tian, X.; Wang, Y.; Huang, S.-H.; Hong, R. Tetrahedron Lett. 2016, 57, 603–606. doi:10.1016/j.tetlet.2015.12.098
Return to citation in text: [1] -
Hoye, T. R.; Jeffrey, C. S.; Shao, F. Nat. Protoc. 2007, 2, 2451–2458. doi:10.1038/nprot.2007.354
Return to citation in text: [1] -
Under this standard esterification conditions, some lactonization leading to compound 5 is observed.
Return to citation in text: [1] -
Ali, Z.; Fronczek, F. R.; Khan, I. A. Planta Med. 2008, 74, 178–181. doi:10.1055/s-2008-1034288
Return to citation in text: [1] -
Yoshikawa, M.; Nakamura, S.; Li, X.; Matsuda, H. Chem. Pharm. Bull. 2008, 56, 695–700. doi:10.1248/cpb.56.695
Return to citation in text: [1] -
Schöttner, E.; Simon, K.; Friedel, M.; Jones, P. G.; Lindel, T. Tetrahedron Lett. 2008, 49, 5580–5582. doi:10.1016/j.tetlet.2008.07.018
Return to citation in text: [1] [2] [3] -
Frija, L. M. T.; Afonso, C. A. M. Tetrahedron 2012, 68, 7414–7421. doi:10.1016/j.tet.2012.06.083
Return to citation in text: [1]
| 20. | Schöttner, E.; Simon, K.; Friedel, M.; Jones, P. G.; Lindel, T. Tetrahedron Lett. 2008, 49, 5580–5582. doi:10.1016/j.tetlet.2008.07.018 |
| 7. | Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 549–552. doi:10.1016/0031-9422(91)83725-z |
| 8. | Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 4212–4213. doi:10.1016/0031-9422(91)83512-j |
| 9. | Lima, B.; Agüero, M. B.; Zygadlo, J.; Tapia, A.; Solis, C.; Rojas De Arias, A.; Yaluff, G.; Zacchino, S.; Feresin, G. E.; Schmeda-Hirschmann, G. J. Chil. Chem. Soc. 2009, 54, 68–72. doi:10.4067/s0717-97072009000100016 |
| 6. | Chandra, A.; Misra, L. N.; Thakur, R. S. Tetrahedron Lett. 1987, 28, 6377–6380. doi:10.1016/s0040-4039(01)91378-4 |
| 7. | Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 549–552. doi:10.1016/0031-9422(91)83725-z |
| 8. | Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 4212–4213. doi:10.1016/0031-9422(91)83512-j |
| 18. | Ali, Z.; Fronczek, F. R.; Khan, I. A. Planta Med. 2008, 74, 178–181. doi:10.1055/s-2008-1034288 |
| 19. | Yoshikawa, M.; Nakamura, S.; Li, X.; Matsuda, H. Chem. Pharm. Bull. 2008, 56, 695–700. doi:10.1248/cpb.56.695 |
| 5. | Mukherjee, A. A.; Kandhare, A. D.; Rojatkar, S. R.; Bodhankar, S. L. Biomed. Pharmacother. 2017, 94, 880–889. doi:10.1016/j.biopha.2017.08.017 |
| 20. | Schöttner, E.; Simon, K.; Friedel, M.; Jones, P. G.; Lindel, T. Tetrahedron Lett. 2008, 49, 5580–5582. doi:10.1016/j.tetlet.2008.07.018 |
| 3. | Subramoniam, A.; Pushpangadan, P.; Rajasekharan, S.; Evans, D. A.; Latha, P. G.; Valsaraj, R. J. Ethnopharmacol. 1996, 50, 13–17. doi:10.1016/0378-8741(95)01329-6 |
| 4. | Pavithra, K. S.; Annadurai, J.; Ragunathan, R. J. Pharmacogn. Phytochem. 2018, 7, 664–675. |
| 16. | Hoye, T. R.; Jeffrey, C. S.; Shao, F. Nat. Protoc. 2007, 2, 2451–2458. doi:10.1038/nprot.2007.354 |
| 2. | Kulkarni, R. N. Artemisia pallens. In Artemisia; Wright, C. W., Ed.; Taylor & Francis: London, UK, 2002; Vol. 18, pp 119–137. |
| 17. | Under this standard esterification conditions, some lactonization leading to compound 5 is observed. |
| 13. | Smith, A. B., III; Fukui, M.; Vaccaro, H. A.; Empfield, J. R. J. Am. Chem. Soc. 1991, 113, 2071–2092. doi:10.1021/ja00006a029 |
| 20. | Schöttner, E.; Simon, K.; Friedel, M.; Jones, P. G.; Lindel, T. Tetrahedron Lett. 2008, 49, 5580–5582. doi:10.1016/j.tetlet.2008.07.018 |
| 11. | López-Martínez, J. L.; Torres-García, I.; Rodríguez-García, I.; Muñoz-Dorado, M.; Álvarez-Corral, M. J. Org. Chem. 2019, 84, 806–816. doi:10.1021/acs.joc.8b02643 |
| 12. | Torres‐García, I.; López‐Martínez, J. L.; Martínez‐Martínez, R.; Oltra, J. E.; Muñoz‐Dorado, M.; Rodríguez‐García, I.; Álvarez‐Corral, M. Appl. Organomet. Chem. 2020, 34, 10.1002/aoc.5244. doi:10.1002/aoc.5244 |
| 15. | Li, W.; Lin, Z.; Chen, L.; Tian, X.; Wang, Y.; Huang, S.-H.; Hong, R. Tetrahedron Lett. 2016, 57, 603–606. doi:10.1016/j.tetlet.2015.12.098 |
| 21. | Frija, L. M. T.; Afonso, C. A. M. Tetrahedron 2012, 68, 7414–7421. doi:10.1016/j.tet.2012.06.083 |
| 10. | Nicolaou, K. C.; Snyder, S. A. Angew. Chem., Int. Ed. 2005, 44, 1012–1044. doi:10.1002/anie.200460864 |
| 7. | Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 549–552. doi:10.1016/0031-9422(91)83725-z |
| 8. | Misra, L. N.; Chandra, A.; Thakur, R. S. Phytochemistry 1991, 30, 4212–4213. doi:10.1016/0031-9422(91)83512-j |
| 9. | Lima, B.; Agüero, M. B.; Zygadlo, J.; Tapia, A.; Solis, C.; Rojas De Arias, A.; Yaluff, G.; Zacchino, S.; Feresin, G. E.; Schmeda-Hirschmann, G. J. Chil. Chem. Soc. 2009, 54, 68–72. doi:10.4067/s0717-97072009000100016 |
| 12. | Torres‐García, I.; López‐Martínez, J. L.; Martínez‐Martínez, R.; Oltra, J. E.; Muñoz‐Dorado, M.; Rodríguez‐García, I.; Álvarez‐Corral, M. Appl. Organomet. Chem. 2020, 34, 10.1002/aoc.5244. doi:10.1002/aoc.5244 |
| 9. | Lima, B.; Agüero, M. B.; Zygadlo, J.; Tapia, A.; Solis, C.; Rojas De Arias, A.; Yaluff, G.; Zacchino, S.; Feresin, G. E.; Schmeda-Hirschmann, G. J. Chil. Chem. Soc. 2009, 54, 68–72. doi:10.4067/s0717-97072009000100016 |
© 2022 Torres-García et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.