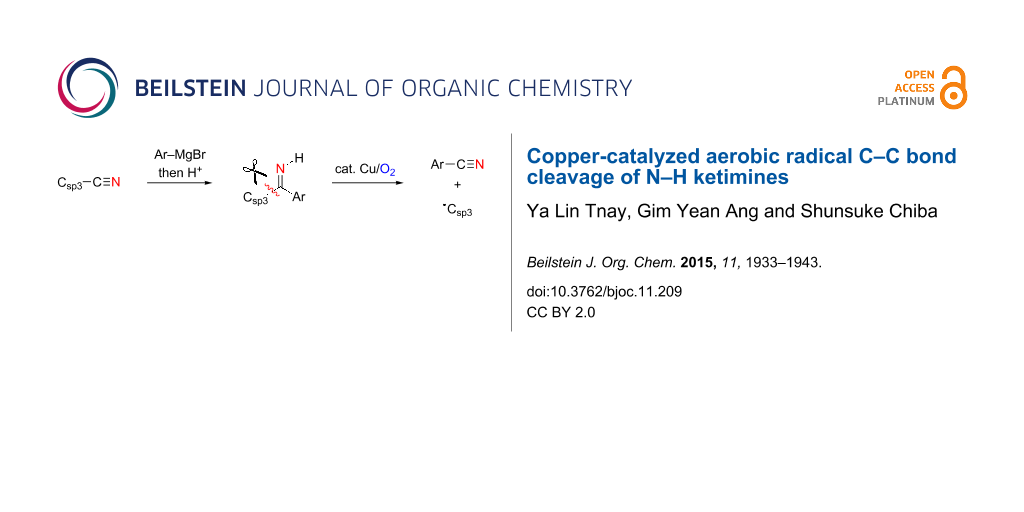
Abstract
We report herein studies on copper-catalyzed aerobic radical C–C bond cleavage of N–H ketimines. Treatment of N–H ketimines having an α-sp3 hybridized carbon under Cu-catalyzed aerobic reaction conditions resulted in a radical fragmentation with C–C bond cleavage to give the corresponding carbonitrile and carbon radical intermediate. This radical process has been applied for the construction of oxaspirocyclohexadienones as well as in the electrophilic cyanation of Grignard reagents with pivalonitrile as a CN source.

Graphical Abstract
Introduction
Alkylideneaminyl radicals (iminyl radicals) have been utilized for the synthesis of azaheterocycles through an intramolecular N–C bond formation with the unsaturated systems [1-6]. As precursors of iminyl radicals, readily available oximes and their derivatives have commonly been utilized. The generation of iminyl radicals involves the homolysis of the N–O bond with radical initiators [7-15] (Scheme 1a) or using thermal [16-20] or photoreaction conditions [21-27] (Scheme 1b). An alternative route to iminyl radicals is the single-electron reduction of oxime derivatives mediated by the appropriate lower valent transition metals [28-32], electron-rich organic electron donors [33-38], or sensitized photolysis [39-42] (Scheme 1c). Although the oxidative generation of iminyl radicals has also been reported, only iminooxyacetic acids have been used as the precursors when we started our studies on the oxidative reactions of N–H ketimines [43-49] (Scheme 1d).
![[1860-5397-11-209-i1]](/bjoc/content/inline/1860-5397-11-209-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Generation of iminyl radicals from oxime derivatives.
Scheme 1: Generation of iminyl radicals from oxime derivatives.
The generation of iminyl radicals by single-electron oxidation of N–H ketimines appears to be the most atom economical method, since only protons (H+) are produced along with the iminyl radicals (Scheme 2). However, the instability of N–H ketimines [50] limits their use as starting materials.
![[1860-5397-11-209-i2]](/bjoc/content/inline/1860-5397-11-209-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Oxidative generation of iminyl radicals from N–H ketimines.
Scheme 2: Oxidative generation of iminyl radicals from N–H ketimines.
Recently, we have studied the chemical reactivity of N–H ketimines towards copper-catalyzed aerobic reaction conditions [51-53]. In these studies, we employed the nucleophilic addition of Grignard reagents to carbonitriles followed by protonation as one of the methods for in situ generation of N–H ketimines, which were directly subjected to Cu-catalyzed aerobic reactions without further purification [54]. In this way, biaryl N–H ketimines generated from biaryl-2-carbonitriles were found to undergo copper-catalyzed aerobic aromatic C–H amination (Scheme 3a) [52] or 1,4-aminooxygenation (spirocyclization) (Scheme 3b) [51], affording phenanthridine derivatives and azaspirocyclohexadienones, respectively, depending on the helical sense of the biaryl axis.
![[1860-5397-11-209-i3]](/bjoc/content/inline/1860-5397-11-209-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Copper-catalyzed aerobic reactions of in situ generated biaryl N–H ketimines.
Scheme 3: Copper-catalyzed aerobic reactions of in situ generated biaryl N–H ketimines.
Herein we report applications of the copper-catalyzed aerobic C–C bond fission of iminyl radical species for the synthesis of oxaspirocyclohexadienones as well as the electrophilic cyanation of Grignard reagents using the readily available pivalonitrile as a CN source.
Results and Discussion
We further explored the reactivity of biaryl N–H ketimines under copper-catalyzed aerobic reaction conditions, aiming at the synthesis of 6-membered azaspirocycles such as 3a’ from carbonitrile 1a having a quaternary sp3-hybridized carbon center at its α-position. The reaction of p-tolylmagnesium bromide (2a) to carbonitrile 1a proceeded smoothly in Et2O at 80 °C in a sealed tube, generating N–H ketimine 1aa after protonation with MeOH. Subsequently, Cu(OAc)2 (20 mol %), 1,10-phen (20 mol %) and DMF (to 0.1 M final concentration) were added and stirred at room temperature under an air atmosphere (Scheme 4). Interestingly, no formation of the desired 6-membered azaspirocycle 3a’ was observed, while oxaspirocyclohexadienone 3a, biaryl alkene 4a, and p-tolunitrile (5a) were isolated in 29%, 32%, and 86% yields, respectively (Scheme 4).
![[1860-5397-11-209-i4]](/bjoc/content/inline/1860-5397-11-209-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Copper-catalyzed aerobic C–C bond cleavage reactions of N–H ketimines.
Scheme 4: Copper-catalyzed aerobic C–C bond cleavage reactions of N–H ketimines.
Oxaspirocyclohexadienone 3a was formed through C–C bond cleavage from N–H ketimine intermediate 1aa most likely via the corresponding iminyl radical, that undergoes radical fragmentation to afford the corresponding C-radical and carbonitrile 5a [55]. Thus, it is deduced that oxaspirocyclohexadienone 3a was formed through oxygenation of the putative C-radical. Moreover, in this transformation, the cyano group of carbonitrile 1a is transferred to Grignard reagent 2a to afford p-tolunitrile (5a). Considering the importance of carbonitriles in organic synthesis [56], we postulated that the cyano group transfer from simple carbonitriles onto Grignard reagents could be realized using this strategy.
Based on these preliminary results (Scheme 4), we next started to investigate the synthesis of oxaspirocylohexadienones as the target product. Using carbonitrile 1a and p-tolylmagnesium bromide (2a), an optimization of the reaction conditions was conducted (Table 1). Increasing the amount of the additive 1,10-phenanthroline to 40 mol % slightly improved the yield, giving 3a in 40% yield (Table 1, entry 1). The use of 2,2’-bipyridine (bpy) provided a comparable result (Table 1, entry 2), while performing the reaction in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO) led to lower yields of spirodienone 3a (Table 1, entry 3). We therefore decided to proceed with 1,10-phenanthroline as the optimal ligand and subsequently tested different Cu(II) and Cu(I) salts (Table 1, entries 4–7). The best results were obtained using 40 mol % CuI which provided 3a in 46% isolated yield (Table 1, entry 7). A reduction of the catalyst loading to 20 mol % slightly lowered the yield of 3a (Table 1, entry 9) and a stoichiometric amount of catalyst did not significantly improve the yield of 3a (Table 1, entry 10). Performing the reaction under an O2 atmosphere did also not increase the yield of 3a (Table 1, entry 8).
Table 1: Optimization of reaction conditions: oxaspirocyclohexadienone synthesis.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-209-i9.svg?max-width=637&scale=1.0)
|
||||||
| Entry |
Cu salt
[mol %] |
Additive
[mol %] |
Atmosphere | Yield [%]b | ||
|---|---|---|---|---|---|---|
| 3a | 4a | 5a | ||||
| 1 | Cu(OAc)2 [20] | 1,10-phen [40] | air | 35 (40) | 32 (31) | 85 (86) |
| 2 | Cu(OAc)2 [20] | bpy [40] | air | 32 | 36 | 80 |
| 3 | Cu(OAc)2 [20] | DABCO [40] | air | 10 | 21 | 87 |
| 4 | CuCl2 [20] | 1,10-phen [40] | air | 27 | 22 | 78 |
| 5 | CuCl [20] | 1,10-phen [40] | air | 37 | 28 | 96 |
| 6 | [20] | 1,10-phen [40] | air | 40 (40)c | 34 (27)c | 95 (90)c |
| 7 | CuI [20] | 1,10-phen [40] | air | 42 (46)c | 34 (28)c | 92 (78)c |
| 8 | CuI [20] | 1,10-phen [40] | O2 | 43 | 37 | 99 |
| 9 | CuI [10] | 1,10-phen [20] | air | 37 | 34 | 95 |
| 10 | CuI [100] | 1,10-phen [100] | air | 46 | 30 | 93 |
aAll reactions were carried out using 0.5 mmol of biaryl carbonitrile 1a with 1.3 equiv of Grignard reagent 2a in Et2O (0.5 mL) at 80 °C (sealed tube) for 4 h followed by the addition of MeOH (60 μL, 3.0 equiv), DMF (5 mL), Cu catalyst and additive and subsequent stirring for 4 h at rt: 1,10-phen = 1,10-phenanthroline; bpy = 2,2’-bipyridine; DABCO = 1,4-diazabicyclo[2.2.2]octane. bCrude yields determined by 1H NMR based on 1,1,2,2-tetrachloroethane as an internal standard. cIsolated yields are given in parentheses.
A proposed reaction mechanism for the formation of oxaspirocyclohexadienone 3a, alkene 4a, and p-tolunitrile (5a) are depicted in Scheme 5. Single-electron oxidation of N–H ketimine 1aa with higher valent Cu(II) species generated under the aerobic reaction conditions forms iminyl radical species A, that undergoes β-carbon fragmentation to give p-tolunitrile (5a) and biaryl-2-isopropyl radical B (Scheme 5a). The aerobic oxygenation of C-radical B affords peroxy radical C, that is presumably reduced by Cu(I) species through the Fenton-type mechanism [57] to give alkoxy radical D [58]. Subsequent spirocyclization of the alkoxy radical D onto the benzene ring affords cyclohexadienyl radical F, oxygenation of which followed by C=O bond formation finally provides the oxaspirocyclohexadienone product 3a. Whereas, the oxidation of the benzylic radical B by the existing Cu(II) species to carbocation G and subsequent E1-type elimination of a proton provides biaryl alkene 4a (Scheme 5b). The presence of alkoxy radical D in the reaction process could be further supported by the reaction of biaryl hydroperoxide 6, which could be converted into the alkoxy radical D under copper-catalyzed aerobic reaction conditions [58]. Indeed treatment of hydroperoxide 6 under the standard reaction conditions afforded 3a in 24% yield along with biaryl alcohol 7 in 25% yield (Scheme 5c).
![[1860-5397-11-209-i5]](/bjoc/content/inline/1860-5397-11-209-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed reaction mechanisms for the formation of 3a, 4a and 5a, and the reaction of hydroperoxide 6.
Scheme 5: Proposed reaction mechanisms for the formation of 3a, 4a and 5a, and the reaction of hydroperoxide 6...
While formation of biaryl alkene 4a could not be avoided at this moment, it is still appealing to probe the potential utility of the current transformation toward the synthesis of oxaspirocyclohexadienones 3. Therefore, the reactions of carbonitriles 1b–d were examined under the current best reaction conditions (Table 1, entry 8) using p-tolyl Grignard reagent 2a (Table 2). Carbonitrile 1b, having methyl groups in ortho-positions on both aryl rings of the biaryl moiety, underwent the C–C bond fission process smoothly to afford oxaspirocyclized product 3b and biaryl alkene 4b in 48% and 35% yields, respectively, along with p-tolunitrile (5a) in 90% yield (Table 2, entry 1). The reactions of N–H ketimines having cyclopentyl and tetrahydropyranyl rings derived from nitriles 1c and 1d, respectively, afforded tricyclic oxaspirocyclohexadienones 3c and 3d in moderate yields along with the corresponding alkenes 4c and 4d as well as p-tolunitrile (5a) (Table 2, entries 2 and 3).
Table 2: Substrate scope: oxaspirocyclohexadienone synthesis.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-209-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate | Productsb | ||
|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-209-i11.svg?max-width=637&scale=1.0)
1b |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-209-i12.svg?max-width=637&scale=1.0)
3b 48% |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-209-i13.svg?max-width=637&scale=1.0)
4b 35% |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-209-i14.svg?max-width=637&scale=1.0)
5a 90%c |
| 2 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-209-i15.svg?max-width=637&scale=1.0)
1c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-209-i16.svg?max-width=637&scale=1.0)
3c 36% |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-209-i17.svg?max-width=637&scale=1.0)
4c 27% |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-209-i18.svg?max-width=637&scale=1.0)
5a 76%c |
| 3 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-209-i19.svg?max-width=637&scale=1.0)
1d |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-209-i20.svg?max-width=637&scale=1.0)
3d 48% |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-209-i21.svg?max-width=637&scale=1.0)
4d 40% |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-209-i22.svg?max-width=637&scale=1.0)
5a 90%c |
aAll reactions were carried out using 0.5 mmol of biaryl carbonitrile 1 with 1.3 equiv of Grignard reagents 2a in Et2O (0.5 mL) at 80 °C (sealed tube) for 4 h followed by the addition of MeOH (60 μL , 3.0 equiv), DMF (5 mL), CuI (20 mol %) and 1,10-phen (40 mol %) under ambient air at rt: 1,10-phen = 1,10-phenanthroline. bIsolated yields. c1H NMR crude yields based on 1,1,2,2-tetrachloroethane as an internal standard.
On the other hand, the reaction starting from carbonitrile 1e having a strained cyclobutane ring did not form the desired oxaspirocyclohexadienone (Scheme 6). Instead, γ-bromoketone 8e was isolated in 44% yield along with nitrile 5a in 80% yield. The formation of γ-bromoketone 8e is most likely caused by radical ring opening from the transient cyclobutoxy radical I, which is driven by releasing ring strain of the cyclobutyl ring. The resulting γ-keto radical J subsequently undergoes radical bromination to form 8e [59]. This result unambiguously supports the presence of the alkoxy radical intermediate during oxaspirocyclohexadienone formation in our mechanistic proposal.
![[1860-5397-11-209-i6]](/bjoc/content/inline/1860-5397-11-209-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, we turned our attention to apply the present copper-catalyzed aerobic C–C bond fission process in the electrophilic cyanation of Grignard reagents. As carbonitriles are omnipresent components in various natural products, dyes and potent pharmaceutical drugs [60-62], new and versatile routes towards this substance class are always desirable. Conventional methods to install the cyano group on aryl rings such as the Rosenmund–von Braun reaction [63] or the Sandmeyer reaction [64] require the use of stoichiometric amounts of toxic metal cyanides (such as CuCN) as the “CN” anion source [65]. Therefore, an employment of aliphatic carbonitriles which are less toxic and easier to handle (such as acetonitrile [66-70], benzyl cyanide [71-76], and malononitrile [77,78]) for the “CN” surrogates has been developed more recently [79]. We envisioned that the readily available pivalonitrile (1f) could be a potential CN source for the electrophilic cyanation [80-86] of Grignard reagents using the present protocol (Scheme 7).
![[1860-5397-11-209-i7]](/bjoc/content/inline/1860-5397-11-209-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Electrophilic cyanation of Grignard reagents with pivalonitrile (1f).
Scheme 7: Electrophilic cyanation of Grignard reagents with pivalonitrile (1f).
Thus pivalonitrile (1f) was reacted with 2-naphthylmagnesium bromide (2b) for formation of the corresponding N–H ketimine 1fb, which was subsequently treated with 10 mol % of Cu(OAc)2 under an O2 atmosphere (Scheme 8). As expected, formation of 2-naphthonitrile (5b) was observed in 79% yield. On the other hand, the reaction in the absence of O2 (under an Ar atmosphere) provided only 2-pivaloylnaphthalene (9b) in 83% yield formed through hydrolysis of unreacted N–H imine 1fb during the aqueous work-up. Therefore molecular oxygen is indispensable to achieve the present cyanation through the C–C bond cleavage of the N–H ketimine. It was found that use of CuBr2 as the catalyst resulted in formation of 5b in higher yield (86%).
![[1860-5397-11-209-i8]](/bjoc/content/inline/1860-5397-11-209-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Electrophilic cyanation with pivalonitrile (1e).
Scheme 8: Electrophilic cyanation with pivalonitrile (1e).
We next examined the substrate scope using different Grignard reagents (Table 3). The electrophilic cyanation proceeded smoothly even with sterically bulky Grignard reagents such as 1-naphthyl- and 2,4,6-trimethylphenyl Grignard reagents (for 5c and 5d) (Table 3, entries 1 and 2). Electron-rich aryl Grignard reagents could also be used to give the corresponding benzonitriles 5e, 5f and 5g in good yields (Table 3, entries 3–5). The reaction also proceeded with chlorinated substrates leaving the C–Cl bond intact (for 5h) (Table 3, entry 6). Thiophen-2-carbonitrile (5i) was also prepared in 63% yield. The present method could also be applied for the cyanation of the primary alkyl Grignard reagent, phenethylmagnesium bromide (for 5j), albeit the product yield was moderate.
Table 3: Scope of the reaction using different Grignard reagents.a
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-209-i23.svg?max-width=637&scale=1.0)
|
|||
| Entry | R–MgBr (2) | Time x/y (h) | Product 5b |
|---|---|---|---|
| 1 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-209-i24.svg?max-width=637&scale=1.0)
2c |
12/18 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-209-i25.svg?max-width=637&scale=1.0)
5c 76% |
| 2c |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-209-i26.svg?max-width=637&scale=1.0)
2d |
56/19 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-209-i27.svg?max-width=637&scale=1.0)
5d 81% |
| 3c |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-209-i28.svg?max-width=637&scale=1.0)
2e |
50/18 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-209-i29.svg?max-width=637&scale=1.0)
5e 67% |
| 4 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-209-i30.svg?max-width=637&scale=1.0)
2f |
22/10 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-209-i31.svg?max-width=637&scale=1.0)
5f 74% |
| 5 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-209-i32.svg?max-width=637&scale=1.0)
2g |
48/24 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-209-i33.svg?max-width=637&scale=1.0)
5g 81% |
| 6 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-209-i34.svg?max-width=637&scale=1.0)
2h |
48/55 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-209-i35.svg?max-width=637&scale=1.0)
5h 70% |
| 7 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-209-i36.svg?max-width=637&scale=1.0)
2i |
48/24 |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-209-i37.svg?max-width=637&scale=1.0)
5i 63%d |
| 8 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-209-i38.svg?max-width=637&scale=1.0)
2j |
48/48 |
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-209-i39.svg?max-width=637&scale=1.0)
5j 47% |
aUnless otherwise noted, the reactions were carried out using 1 mmol of pivalonitrile (1f) with 1.3 equiv of Grignard reagents 2 in Et2O (1 mL) at 60 °C (sealed tube) for the time x followed by the addition of MeOH (120 μL), DMF (10 mL), and CuBr2 (10 mol %), and the mixture was stirred at 80 °C for time y under an O2 atmosphere. bIsolated yields. cThe reaction was conducted using Cu(OAc)2 (10 mol %) as the catalyst. d1H NMR yield.
Conclusion
In summary, we have demonstrated a copper-catalyzed aerobic generation of iminyl radicals from the corresponding N–H ketimines and their radical C–C bond fission. These processes could be applied for synthesis of oxaspirocyclohexadienones through spirocylization of the transient alkoxy radicals generated by aerobic oxygenation of the resulting carbon radicals. With the present protocol, the electrophilic cyanation of Grignard reagents was also established using readily available pivalonitrile as a simple CN source.
Supporting Information
| Supporting Information File 1: Full experimental details and analytical data. | ||
| Format: PDF | Size: 5.9 MB | Download |
References
-
Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2008, 81, 539–547. doi:10.1246/bcsj.81.539
Return to citation in text: [1] -
Narasaka, K.; Kitamura, M. Eur. J. Org. Chem. 2005, 4505–4519. doi:10.1002/ejoc.200500389
Return to citation in text: [1] -
Stella, L. Nitrogen-Centered Radicals. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 2, pp 407–426. doi:10.1002/9783527618293.ch45
Return to citation in text: [1] -
Mikami, T.; Narasaka, K. Generation of radical species by single electron transfer reactions and their application to the development of synthetic reactions. In Advances in Free Radical Chemistry; Zard, S. Z., Ed.; JAI: Stanford, 1999; Vol. 2, pp 45–88. doi:10.1016/S1874-5237(99)80004-X
Return to citation in text: [1] -
Fallis, A. G.; Brinza, I. M. Tetrahedron 1997, 53, 17543–17594. doi:10.1016/S0040-4020(97)10060-6
Return to citation in text: [1] -
Zard, S. Z. Synlett 1996, 12, 1148–1154. doi:10.1055/s-1996-5698
Return to citation in text: [1] -
El Kaim, L.; Meyer, C. J. Org. Chem. 1996, 61, 1556–1557. doi:10.1021/jo952173q
Return to citation in text: [1] -
Callier-Dublanchet, A.-C.; Quiclet-Sire, B.; Zard, S. Z. Tetrahedron Lett. 1995, 36, 8791–8794. doi:10.1016/0040-4039(95)01878-L
Return to citation in text: [1] -
Boivin, J.; Callier-Dublanchet, A.-C.; Quiclet-Sire, B.; Schiano, A.-M.; Zard, S. Z. Tetrahedron 1995, 51, 6517–6528. doi:10.1016/0040-4020(95)00319-4
Return to citation in text: [1] -
Callier, A.-C.; Quiclet-Sire, B.; Zard, S. Z. Tetrahedron Lett. 1994, 35, 6109–6112. doi:10.1016/0040-4039(94)88089-1
Return to citation in text: [1] -
Boivin, J.; Schiano, A.-M.; Zard, S. Z. Tetrahedron Lett. 1994, 35, 249–252. doi:10.1016/S0040-4039(00)76523-3
Return to citation in text: [1] -
Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron 1994, 50, 1757–1768. doi:10.1016/S0040-4020(01)80850-4
Return to citation in text: [1] -
Boivin, J.; Fouquet, E.; Zard, S. Z. J. Am. Chem. Soc. 1991, 113, 1055–1057. doi:10.1021/ja00003a057
Return to citation in text: [1] -
Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron 1994, 50, 1745–1756. doi:10.1016/S0040-4020(01)80849-8
Return to citation in text: [1] -
Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron Lett. 1990, 31, 3545–3548. doi:10.1016/S0040-4039(00)94438-1
Return to citation in text: [1] -
Cai, Y.; Jalan, A.; Kobosumi, A. R.; Castle, S. T. Org. Lett. 2015, 17, 488–491. doi:10.1021/ol5035047
Return to citation in text: [1] -
Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. Chem. Commun. 2007, 4041–4043. doi:10.1039/b712582h
Return to citation in text: [1] -
Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. J. Org. Chem. 2008, 73, 5558–5565. doi:10.1021/jo800847h
Return to citation in text: [1] -
Blake, J. A.; Pratt, D. A.; Lin, S.; Walton, J. C.; Mulder, P.; Ingold, K. U. J. Org. Chem. 2004, 69, 3112–3120. doi:10.1021/jo049927y
Return to citation in text: [1] -
Lin, X.; Stien, D.; Weinreb, S. M. Org. Lett. 1999, 1, 637–640. doi:10.1021/ol990720e
Return to citation in text: [1] -
Faulkner, A.; Race, N. J.; Scott, J. S.; Bower, J. F. Chem. Sci. 2014, 5, 2416–2421. doi:10.1039/c4sc00652f
Return to citation in text: [1] -
Bingham, M.; Moutrille, C.; Zard, S. Z. Heterocycles 2014, 88, 953–960. doi:10.3987/COM-13-S(S)94
Return to citation in text: [1] -
Portela-Cubillo, F.; Lymer, J.; Scanlan, E. M.; Scott, J. S.; Walton, J. C. Tetrahedron 2008, 64, 11908–11916. doi:10.1016/j.tet.2008.08.112
Return to citation in text: [1] -
Alonso, R.; Campos, P. J.; Rodríguez, M. A.; Sampedro, D. J. Org. Chem. 2008, 73, 2234–2239. doi:10.1021/jo7025542
Return to citation in text: [1] -
Alonso, R.; Campos, P. J.; García, B.; Rodríguez, M. A. Org. Lett. 2006, 8, 3521–3523. doi:10.1021/ol061258i
Return to citation in text: [1] -
Gagosz, F.; Zard, S. Z. Synlett 1998, 1978–1980.
Return to citation in text: [1] -
Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron Lett. 1991, 32, 4299–4302. doi:10.1016/S0040-4039(00)92153-1
Return to citation in text: [1] -
Zhao, M.-N.; Liang, H.; Ren, Z.-H.; Guan, Z.-H. Synthesis 2012, 44, 1501–1506. doi:10.1055/s-0031-1290779
Return to citation in text: [1] -
Tanaka, K.; Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2005, 78, 1659–1664. doi:10.1246/bcsj.78.1659
Return to citation in text: [1] -
Koganemaru, Y.; Kitamura, M.; Narasaka, K. Chem. Lett. 2002, 31, 784–785. doi:10.1246/cl.2002.784
Return to citation in text: [1] -
Boivin, J.; Schiano, A.-M.; Zard, S. Z.; Zhang, H. Tetrahedron Lett. 1999, 40, 4531–4534. doi:10.1016/S0040-4039(99)00720-0
Return to citation in text: [1] -
Boivin, J.; Schiano, A.-M.; Zard, S. Z. Tetrahedron Lett. 1992, 33, 7849–7852. doi:10.1016/S0040-4039(00)74760-5
Return to citation in text: [1] -
Kitamura, M.; Yoshida, M.; Kikuchi, T.; Narasaka, K. Synthesis 2003, 15, 2415–2426. doi:10.1055/s-2003-42439
Return to citation in text: [1] -
Yoshida, M.; Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2003, 76, 2003–2008. doi:10.1246/bcsj.76.2003
Return to citation in text: [1] -
Yoshida, M.; Kitamura, M.; Narasaka, K. Chem. Lett. 2002, 31, 144–145. doi:10.1246/cl.2002.144
Return to citation in text: [1] -
Uchiyama, K.; Hayashi, Y.; Narasaka, K. Tetrahedron 1999, 55, 8915–8930. doi:10.1016/S0040-4020(99)00453-6
Return to citation in text: [1] -
Uchiyama, K.; Ono, A.; Hayashi, Y.; Narasaka, K. Bull. Chem. Soc. Jpn. 1998, 71, 2945–2955. doi:10.1246/bcsj.71.2945
Return to citation in text: [1] -
Ono, A.; Uchiyama, K.; Hayashi, Y.; Narasaka, K. Chem. Lett. 1998, 27, 437–438. doi:10.1246/cl.1998.437
Return to citation in text: [1] -
Jiang, H.; An, X.; Tong, K.; Zheng, T.; Zhang, Y.; Yu, S. Angew. Chem., Int. Ed. 2015, 54, 4055–4059. doi:10.1002/anie.201411342
Return to citation in text: [1] -
McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a
Return to citation in text: [1] -
Kitamura, M.; Mori, Y.; Narasaka, K. Tetrahedron Lett. 2005, 46, 2373–2376. doi:10.1016/j.tetlet.2005.02.062
Return to citation in text: [1] -
Mikami, T.; Narasaka, K. Chem. Lett. 2000, 29, 338–339. doi:10.1246/cl.2000.338
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Meyer, C. J.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 606–611. doi:10.1039/p19790000606
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 612–615. doi:10.1039/p19790000612
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 616–620. doi:10.1039/p19790000616
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 621–631. doi:10.1039/p19790000621
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Napier, R. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 632–636. doi:10.1039/p19790000632
Return to citation in text: [1] -
Forrester, A. R.; Gill, M.; Meyer, C. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 637–642. doi:10.1039/p19790000637
Return to citation in text: [1] -
Forrester, A. R.; Napier, R. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1981, 984–987. doi:10.1039/p19810000984
Return to citation in text: [1] -
Griller, D.; Mendenhall, G. D.; Van Hoof, W.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 6068–6070. doi:10.1021/ja00826a018
Return to citation in text: [1] -
Tnay, Y. L.; Chen, C.; Chua, Y. Y.; Zhang, L.; Chiba, S. Org. Lett. 2012, 14, 3550–3553. doi:10.1021/ol301583y
Return to citation in text: [1] [2] -
Zhang, L.; Ang, G. Y.; Chiba, S. Org. Lett. 2010, 12, 3682–3685. doi:10.1021/ol101490n
Return to citation in text: [1] [2] -
Chiba, S.; Zhang, L.; Lee, J.-Y. J. Am. Chem. Soc. 2010, 132, 7266–7267. doi:10.1021/ja1027327
Return to citation in text: [1] -
Pickard, P. L.; Tolbert, T. L. J. Org. Chem. 1961, 26, 4886–4888. doi:10.1021/jo01070a025
Return to citation in text: [1] -
Chiba, S.; Zhang, L.; Ang, G. Y.; Hui, B. W.-Q. Org. Lett. 2010, 12, 2052–2055. doi:10.1021/ol100522z
Return to citation in text: [1] -
Paquette, L. A. Comprehensive Organic Transformations; VCH Publishers, Inc./VCH Verlagsgesellschaft: New York/Weinheim, 1989. doi:10.1002/ange.19901020837
Return to citation in text: [1] -
Rachmilovich-Calis, S.; Masarwa, A.; Meyerstein, N.; Myerstein, D.; van Eldik, R. Chem. – Eur. J. 2009, 15, 8303–8309. doi:10.1002/chem.200802572
Return to citation in text: [1] -
Too, P. C.; Tnay, Y. L.; Chiba, S. Beilstein J. Org. Chem. 2013, 9, 1217–1225. doi:10.3762/bjoc.9.138
Return to citation in text: [1] [2] -
Sanjaya, S.; Chiba, S. Tetrahedron 2011, 67, 590–596. doi:10.1016/j.tet.2010.11.060
Return to citation in text: [1] -
Fatiadi, A. J. Preparation and Synthetic Applications of Cyano Compounds. In Triple-Bonded Functional Groups; Patai, S.; Rappaport, Z., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 1983; Vol. 2. doi:10.1002/9780470771709.ch9
Return to citation in text: [1] -
Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Synthesis, Patents, Applications, 4th ed.; Thieme: Stuttgart, Germany, 2001.
Return to citation in text: [1] -
Miller, J. S.; Manson, J. L. Acc. Chem. Res. 2001, 34, 563–570. doi:10.1021/ar0000354
Return to citation in text: [1] -
Lindley, J. Tetrahedron 1984, 40, 1433–1456. doi:10.1016/S0040-4020(01)91791-0
Return to citation in text: [1] -
Galli, C. Chem. Rev. 1988, 88, 765–792. doi:10.1021/cr00087a004
Return to citation in text: [1] -
Wen, Q.; Jin, J.; Zhang, L.; Luo, Y.; Lu, P.; Wang, Y. Tetrahedron Lett. 2014, 55, 1271–1280. doi:10.1016/j.tetlet.2014.01.032
Return to citation in text: [1] -
Zhu, Y.; Zhao, M.; Lu, W.; Li, L.; Shen, Z. Org. Lett. 2015, 17, 2602–2605. doi:10.1021/acs.orglett.5b00886
Return to citation in text: [1] -
Pan, C.; Jin, H.; Xu, P.; Liu, X.; Cheng, Y.; Zhu, C. J. Org. Chem. 2013, 78, 9494–9498. doi:10.1021/jo4014904
Return to citation in text: [1] -
Kou, X.; Zhao, M.; Qiao, X.; Zhu, Y.; Tong, X.; Shen, Z. Chem. – Eur. J. 2013, 19, 16880–16886. doi:10.1002/chem.201303637
Return to citation in text: [1] -
Song, R.-J.; Wu, J.-C.; Liu, Y.; Deng, G.-B.; Wu, C.-Y.; Wei, W.-T.; Li, J.-H. Synlett 2012, 2491–2496. doi:10.1055/s-0032-1317191
Return to citation in text: [1] -
Luo, F.-H.; Chu, C.-I.; Cheng, C.-H. Organometallics 1998, 17, 1025–1030. doi:10.1021/om970842f
Return to citation in text: [1] -
Wen, Q.; Jin, J.; Mei, Y.; Lu, P.; Wang, Y. Eur. J. Org. Chem. 2013, 2013, 4032–4036. doi:10.1002/ejoc.201300052
Return to citation in text: [1] -
Zhang, L.; Wen, Q.; Jin, J.; Wang, C.; Lu, P.; Wang, Y. Tetrahedron 2013, 69, 4236–4240. doi:10.1016/j.tet.2013.03.089
Return to citation in text: [1] -
Luo, Y.; Wen, Q.; Wu, Z.; Jin, J.; Lu, P.; Wang, Y. Tetrahedron 2013, 69, 8400–8404. doi:10.1016/j.tet.2013.07.063
Return to citation in text: [1] -
Yuen, O. Y.; Choy, P. Y.; Chow, W. K.; Wong, W. T.; Kwong, F. Y. J. Org. Chem. 2013, 78, 3374–3378. doi:10.1021/jo3028278
Return to citation in text: [1] -
Wen, Q.; Jin, J.; Hu, B.; Lu, P.; Wang, Y. RSC Adv. 2012, 2, 6167–6169. doi:10.1039/c2ra20770b
Return to citation in text: [1] -
Jin, J.; Wen, Q.; Lu, P.; Wang, Y. Chem. Commun. 2012, 48, 9933–9935. doi:10.1039/c2cc35046g
Return to citation in text: [1] -
Reeves, J. T.; Malapit, C. A.; Buono, F. G.; Sidhu, K. P.; Marsini, M. A.; Sader, C. A.; Fandrick, K. R.; Busacca, C. A.; Senanayake, C. H. J. Am. Chem. Soc. 2015, 137, 9481–9488. doi:10.1021/jacs.5b06136
Return to citation in text: [1] -
Jiang, Z.; Huang, Q.; Chen, S.; Long, L.; Zhou, X. Adv. Synth. Catal. 2012, 354, 589–592. doi:10.1002/adsc.201100836
Return to citation in text: [1] -
Wen, Q.; Lu, P.; Wang, Y. RSC Adv. 2014, 4, 47806–47826. doi:10.1039/C4RA08675A
Return to citation in text: [1] -
Anbarasan, P.; Neumann, H.; Beller, M. Chem. – Eur. J. 2011, 17, 4217–4222. doi:10.1002/chem.201003388
Return to citation in text: [1] -
Anbarasan, P.; Neumann, H.; Beller, M. Chem. – Eur. J. 2010, 16, 4725–4728. doi:10.1002/chem.201000086
Return to citation in text: [1] -
Hughes, T. V.; Cava, M. P. J. Org. Chem. 1999, 64, 313–315. doi:10.1021/jo981924w
Return to citation in text: [1] -
Koo, J. S.; Lee, J. I. Synth. Commun. 1996, 26, 3709–3713. doi:10.1080/00397919608003787
Return to citation in text: [1] -
Klement, I.; Lennick, K.; Tucker, C. E.; Knochel, P. Tetrahedron Lett. 1993, 34, 4623–4626. doi:10.1016/S0040-4039(00)60640-8
Return to citation in text: [1] -
van Leusen, A. M.; Jagt, J. C. Tetrahedron Lett. 1970, 11, 967–970. doi:10.1016/S0040-4039(01)97880-3
Return to citation in text: [1] -
Foulger, N. J.; Wakefield, B. J. Tetrahedron Lett. 1972, 13, 4169–4170. doi:10.1016/S0040-4039(01)94266-2
Return to citation in text: [1]
| 80. | Anbarasan, P.; Neumann, H.; Beller, M. Chem. – Eur. J. 2011, 17, 4217–4222. doi:10.1002/chem.201003388 |
| 81. | Anbarasan, P.; Neumann, H.; Beller, M. Chem. – Eur. J. 2010, 16, 4725–4728. doi:10.1002/chem.201000086 |
| 82. | Hughes, T. V.; Cava, M. P. J. Org. Chem. 1999, 64, 313–315. doi:10.1021/jo981924w |
| 83. | Koo, J. S.; Lee, J. I. Synth. Commun. 1996, 26, 3709–3713. doi:10.1080/00397919608003787 |
| 84. | Klement, I.; Lennick, K.; Tucker, C. E.; Knochel, P. Tetrahedron Lett. 1993, 34, 4623–4626. doi:10.1016/S0040-4039(00)60640-8 |
| 85. | van Leusen, A. M.; Jagt, J. C. Tetrahedron Lett. 1970, 11, 967–970. doi:10.1016/S0040-4039(01)97880-3 |
| 86. | Foulger, N. J.; Wakefield, B. J. Tetrahedron Lett. 1972, 13, 4169–4170. doi:10.1016/S0040-4039(01)94266-2 |
| 1. | Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2008, 81, 539–547. doi:10.1246/bcsj.81.539 |
| 2. | Narasaka, K.; Kitamura, M. Eur. J. Org. Chem. 2005, 4505–4519. doi:10.1002/ejoc.200500389 |
| 3. | Stella, L. Nitrogen-Centered Radicals. In Radicals in Organic Synthesis; Renaud, P.; Sibi, M. P., Eds.; Wiley-VCH: Weinheim, Germany, 2001; Vol. 2, pp 407–426. doi:10.1002/9783527618293.ch45 |
| 4. | Mikami, T.; Narasaka, K. Generation of radical species by single electron transfer reactions and their application to the development of synthetic reactions. In Advances in Free Radical Chemistry; Zard, S. Z., Ed.; JAI: Stanford, 1999; Vol. 2, pp 45–88. doi:10.1016/S1874-5237(99)80004-X |
| 5. | Fallis, A. G.; Brinza, I. M. Tetrahedron 1997, 53, 17543–17594. doi:10.1016/S0040-4020(97)10060-6 |
| 6. | Zard, S. Z. Synlett 1996, 12, 1148–1154. doi:10.1055/s-1996-5698 |
| 28. | Zhao, M.-N.; Liang, H.; Ren, Z.-H.; Guan, Z.-H. Synthesis 2012, 44, 1501–1506. doi:10.1055/s-0031-1290779 |
| 29. | Tanaka, K.; Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2005, 78, 1659–1664. doi:10.1246/bcsj.78.1659 |
| 30. | Koganemaru, Y.; Kitamura, M.; Narasaka, K. Chem. Lett. 2002, 31, 784–785. doi:10.1246/cl.2002.784 |
| 31. | Boivin, J.; Schiano, A.-M.; Zard, S. Z.; Zhang, H. Tetrahedron Lett. 1999, 40, 4531–4534. doi:10.1016/S0040-4039(99)00720-0 |
| 32. | Boivin, J.; Schiano, A.-M.; Zard, S. Z. Tetrahedron Lett. 1992, 33, 7849–7852. doi:10.1016/S0040-4039(00)74760-5 |
| 56. | Paquette, L. A. Comprehensive Organic Transformations; VCH Publishers, Inc./VCH Verlagsgesellschaft: New York/Weinheim, 1989. doi:10.1002/ange.19901020837 |
| 21. | Faulkner, A.; Race, N. J.; Scott, J. S.; Bower, J. F. Chem. Sci. 2014, 5, 2416–2421. doi:10.1039/c4sc00652f |
| 22. | Bingham, M.; Moutrille, C.; Zard, S. Z. Heterocycles 2014, 88, 953–960. doi:10.3987/COM-13-S(S)94 |
| 23. | Portela-Cubillo, F.; Lymer, J.; Scanlan, E. M.; Scott, J. S.; Walton, J. C. Tetrahedron 2008, 64, 11908–11916. doi:10.1016/j.tet.2008.08.112 |
| 24. | Alonso, R.; Campos, P. J.; Rodríguez, M. A.; Sampedro, D. J. Org. Chem. 2008, 73, 2234–2239. doi:10.1021/jo7025542 |
| 25. | Alonso, R.; Campos, P. J.; García, B.; Rodríguez, M. A. Org. Lett. 2006, 8, 3521–3523. doi:10.1021/ol061258i |
| 26. | Gagosz, F.; Zard, S. Z. Synlett 1998, 1978–1980. |
| 27. | Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron Lett. 1991, 32, 4299–4302. doi:10.1016/S0040-4039(00)92153-1 |
| 57. | Rachmilovich-Calis, S.; Masarwa, A.; Meyerstein, N.; Myerstein, D.; van Eldik, R. Chem. – Eur. J. 2009, 15, 8303–8309. doi:10.1002/chem.200802572 |
| 16. | Cai, Y.; Jalan, A.; Kobosumi, A. R.; Castle, S. T. Org. Lett. 2015, 17, 488–491. doi:10.1021/ol5035047 |
| 17. | Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. Chem. Commun. 2007, 4041–4043. doi:10.1039/b712582h |
| 18. | Portela-Cubillo, F.; Scott, J. S.; Walton, J. C. J. Org. Chem. 2008, 73, 5558–5565. doi:10.1021/jo800847h |
| 19. | Blake, J. A.; Pratt, D. A.; Lin, S.; Walton, J. C.; Mulder, P.; Ingold, K. U. J. Org. Chem. 2004, 69, 3112–3120. doi:10.1021/jo049927y |
| 20. | Lin, X.; Stien, D.; Weinreb, S. M. Org. Lett. 1999, 1, 637–640. doi:10.1021/ol990720e |
| 51. | Tnay, Y. L.; Chen, C.; Chua, Y. Y.; Zhang, L.; Chiba, S. Org. Lett. 2012, 14, 3550–3553. doi:10.1021/ol301583y |
| 7. | El Kaim, L.; Meyer, C. J. Org. Chem. 1996, 61, 1556–1557. doi:10.1021/jo952173q |
| 8. | Callier-Dublanchet, A.-C.; Quiclet-Sire, B.; Zard, S. Z. Tetrahedron Lett. 1995, 36, 8791–8794. doi:10.1016/0040-4039(95)01878-L |
| 9. | Boivin, J.; Callier-Dublanchet, A.-C.; Quiclet-Sire, B.; Schiano, A.-M.; Zard, S. Z. Tetrahedron 1995, 51, 6517–6528. doi:10.1016/0040-4020(95)00319-4 |
| 10. | Callier, A.-C.; Quiclet-Sire, B.; Zard, S. Z. Tetrahedron Lett. 1994, 35, 6109–6112. doi:10.1016/0040-4039(94)88089-1 |
| 11. | Boivin, J.; Schiano, A.-M.; Zard, S. Z. Tetrahedron Lett. 1994, 35, 249–252. doi:10.1016/S0040-4039(00)76523-3 |
| 12. | Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron 1994, 50, 1757–1768. doi:10.1016/S0040-4020(01)80850-4 |
| 13. | Boivin, J.; Fouquet, E.; Zard, S. Z. J. Am. Chem. Soc. 1991, 113, 1055–1057. doi:10.1021/ja00003a057 |
| 14. | Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron 1994, 50, 1745–1756. doi:10.1016/S0040-4020(01)80849-8 |
| 15. | Boivin, J.; Fouquet, E.; Zard, S. Z. Tetrahedron Lett. 1990, 31, 3545–3548. doi:10.1016/S0040-4039(00)94438-1 |
| 55. | Chiba, S.; Zhang, L.; Ang, G. Y.; Hui, B. W.-Q. Org. Lett. 2010, 12, 2052–2055. doi:10.1021/ol100522z |
| 50. | Griller, D.; Mendenhall, G. D.; Van Hoof, W.; Ingold, K. U. J. Am. Chem. Soc. 1974, 96, 6068–6070. doi:10.1021/ja00826a018 |
| 54. | Pickard, P. L.; Tolbert, T. L. J. Org. Chem. 1961, 26, 4886–4888. doi:10.1021/jo01070a025 |
| 43. | Forrester, A. R.; Gill, M.; Meyer, C. J.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 606–611. doi:10.1039/p19790000606 |
| 44. | Forrester, A. R.; Gill, M.; Sadd, J. S.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 612–615. doi:10.1039/p19790000612 |
| 45. | Forrester, A. R.; Gill, M.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 616–620. doi:10.1039/p19790000616 |
| 46. | Forrester, A. R.; Gill, M.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 621–631. doi:10.1039/p19790000621 |
| 47. | Forrester, A. R.; Gill, M.; Napier, R. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 632–636. doi:10.1039/p19790000632 |
| 48. | Forrester, A. R.; Gill, M.; Meyer, C. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1979, 637–642. doi:10.1039/p19790000637 |
| 49. | Forrester, A. R.; Napier, R. J.; Thomson, R. H. J. Chem. Soc., Perkin Trans. 1 1981, 984–987. doi:10.1039/p19810000984 |
| 52. | Zhang, L.; Ang, G. Y.; Chiba, S. Org. Lett. 2010, 12, 3682–3685. doi:10.1021/ol101490n |
| 39. | Jiang, H.; An, X.; Tong, K.; Zheng, T.; Zhang, Y.; Yu, S. Angew. Chem., Int. Ed. 2015, 54, 4055–4059. doi:10.1002/anie.201411342 |
| 40. | McBurney, R. T.; Slawin, A. M. Z.; Smart, L. A.; Yu, Y.; Walton, J. C. Chem. Commun. 2011, 47, 7974–7976. doi:10.1039/c1cc12720a |
| 41. | Kitamura, M.; Mori, Y.; Narasaka, K. Tetrahedron Lett. 2005, 46, 2373–2376. doi:10.1016/j.tetlet.2005.02.062 |
| 42. | Mikami, T.; Narasaka, K. Chem. Lett. 2000, 29, 338–339. doi:10.1246/cl.2000.338 |
| 33. | Kitamura, M.; Yoshida, M.; Kikuchi, T.; Narasaka, K. Synthesis 2003, 15, 2415–2426. doi:10.1055/s-2003-42439 |
| 34. | Yoshida, M.; Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2003, 76, 2003–2008. doi:10.1246/bcsj.76.2003 |
| 35. | Yoshida, M.; Kitamura, M.; Narasaka, K. Chem. Lett. 2002, 31, 144–145. doi:10.1246/cl.2002.144 |
| 36. | Uchiyama, K.; Hayashi, Y.; Narasaka, K. Tetrahedron 1999, 55, 8915–8930. doi:10.1016/S0040-4020(99)00453-6 |
| 37. | Uchiyama, K.; Ono, A.; Hayashi, Y.; Narasaka, K. Bull. Chem. Soc. Jpn. 1998, 71, 2945–2955. doi:10.1246/bcsj.71.2945 |
| 38. | Ono, A.; Uchiyama, K.; Hayashi, Y.; Narasaka, K. Chem. Lett. 1998, 27, 437–438. doi:10.1246/cl.1998.437 |
| 51. | Tnay, Y. L.; Chen, C.; Chua, Y. Y.; Zhang, L.; Chiba, S. Org. Lett. 2012, 14, 3550–3553. doi:10.1021/ol301583y |
| 52. | Zhang, L.; Ang, G. Y.; Chiba, S. Org. Lett. 2010, 12, 3682–3685. doi:10.1021/ol101490n |
| 53. | Chiba, S.; Zhang, L.; Lee, J.-Y. J. Am. Chem. Soc. 2010, 132, 7266–7267. doi:10.1021/ja1027327 |
| 59. | Sanjaya, S.; Chiba, S. Tetrahedron 2011, 67, 590–596. doi:10.1016/j.tet.2010.11.060 |
| 58. | Too, P. C.; Tnay, Y. L.; Chiba, S. Beilstein J. Org. Chem. 2013, 9, 1217–1225. doi:10.3762/bjoc.9.138 |
| 58. | Too, P. C.; Tnay, Y. L.; Chiba, S. Beilstein J. Org. Chem. 2013, 9, 1217–1225. doi:10.3762/bjoc.9.138 |
| 77. | Reeves, J. T.; Malapit, C. A.; Buono, F. G.; Sidhu, K. P.; Marsini, M. A.; Sader, C. A.; Fandrick, K. R.; Busacca, C. A.; Senanayake, C. H. J. Am. Chem. Soc. 2015, 137, 9481–9488. doi:10.1021/jacs.5b06136 |
| 78. | Jiang, Z.; Huang, Q.; Chen, S.; Long, L.; Zhou, X. Adv. Synth. Catal. 2012, 354, 589–592. doi:10.1002/adsc.201100836 |
| 79. | Wen, Q.; Lu, P.; Wang, Y. RSC Adv. 2014, 4, 47806–47826. doi:10.1039/C4RA08675A |
| 66. | Zhu, Y.; Zhao, M.; Lu, W.; Li, L.; Shen, Z. Org. Lett. 2015, 17, 2602–2605. doi:10.1021/acs.orglett.5b00886 |
| 67. | Pan, C.; Jin, H.; Xu, P.; Liu, X.; Cheng, Y.; Zhu, C. J. Org. Chem. 2013, 78, 9494–9498. doi:10.1021/jo4014904 |
| 68. | Kou, X.; Zhao, M.; Qiao, X.; Zhu, Y.; Tong, X.; Shen, Z. Chem. – Eur. J. 2013, 19, 16880–16886. doi:10.1002/chem.201303637 |
| 69. | Song, R.-J.; Wu, J.-C.; Liu, Y.; Deng, G.-B.; Wu, C.-Y.; Wei, W.-T.; Li, J.-H. Synlett 2012, 2491–2496. doi:10.1055/s-0032-1317191 |
| 70. | Luo, F.-H.; Chu, C.-I.; Cheng, C.-H. Organometallics 1998, 17, 1025–1030. doi:10.1021/om970842f |
| 71. | Wen, Q.; Jin, J.; Mei, Y.; Lu, P.; Wang, Y. Eur. J. Org. Chem. 2013, 2013, 4032–4036. doi:10.1002/ejoc.201300052 |
| 72. | Zhang, L.; Wen, Q.; Jin, J.; Wang, C.; Lu, P.; Wang, Y. Tetrahedron 2013, 69, 4236–4240. doi:10.1016/j.tet.2013.03.089 |
| 73. | Luo, Y.; Wen, Q.; Wu, Z.; Jin, J.; Lu, P.; Wang, Y. Tetrahedron 2013, 69, 8400–8404. doi:10.1016/j.tet.2013.07.063 |
| 74. | Yuen, O. Y.; Choy, P. Y.; Chow, W. K.; Wong, W. T.; Kwong, F. Y. J. Org. Chem. 2013, 78, 3374–3378. doi:10.1021/jo3028278 |
| 75. | Wen, Q.; Jin, J.; Hu, B.; Lu, P.; Wang, Y. RSC Adv. 2012, 2, 6167–6169. doi:10.1039/c2ra20770b |
| 76. | Jin, J.; Wen, Q.; Lu, P.; Wang, Y. Chem. Commun. 2012, 48, 9933–9935. doi:10.1039/c2cc35046g |
| 65. | Wen, Q.; Jin, J.; Zhang, L.; Luo, Y.; Lu, P.; Wang, Y. Tetrahedron Lett. 2014, 55, 1271–1280. doi:10.1016/j.tetlet.2014.01.032 |
| 60. | Fatiadi, A. J. Preparation and Synthetic Applications of Cyano Compounds. In Triple-Bonded Functional Groups; Patai, S.; Rappaport, Z., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 1983; Vol. 2. doi:10.1002/9780470771709.ch9 |
| 61. | Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Synthesis, Patents, Applications, 4th ed.; Thieme: Stuttgart, Germany, 2001. |
| 62. | Miller, J. S.; Manson, J. L. Acc. Chem. Res. 2001, 34, 563–570. doi:10.1021/ar0000354 |
| 63. | Lindley, J. Tetrahedron 1984, 40, 1433–1456. doi:10.1016/S0040-4020(01)91791-0 |
© 2015 Tnay et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)