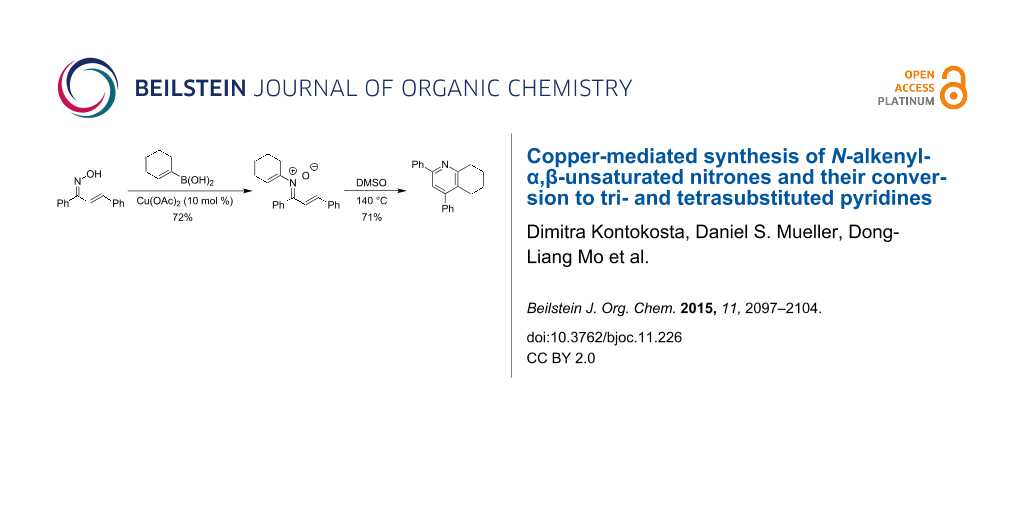
Abstract
A Chan–Lam reaction has been used to prepare N-alkenyl-α,β-unsaturated nitrones, which undergo a subsequent thermal rearrangement to the corresponding tri- and tetrasubstituted pyridines. The optimization and scope of these transformations is discussed. Initial mechanistic experiments suggest a reaction pathway involving oxygen transfer followed by cyclization.

Graphical Abstract
Introduction
While most applications of the Chan–Lam reaction are focused on the synthesis of aryl ethers and aryl amines, our group has been interested in the use of the Chan–Lam reaction for the synthesis of O-alkenyl oximes and hydroxylamines, as well as N-alkenyl and N-arylnitrones [1-5]. We have discovered that when this transformation is performed with oxime and hydroxamic acid substrates, these reactive intermediates can be accessed and subsequently rearrange to a variety of challenging organic fragments and heterocyclic products [6-13]. Specifically, we reported that N-arylnitrones 3 can be prepared by a Chan–Lam coupling of 1 and 2 and that these compounds undergo a copper-catalyzed rearrangement to α,β-epoxyimines such as 4 [8]. Reduction of these products in the presence of a Lewis acid gave tetrahydroquinolines such as 5 (Scheme 1A). These studies encouraged us to consider if similar N-alkenylnitrones 8 could be accessed by a Chan–Lam coupling and transformed into the corresponding substituted pyridines 9 (Scheme 1B).
![[1860-5397-11-226-i1]](/bjoc/content/inline/1860-5397-11-226-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Use of a Chan–Lam reaction for the synthesis of tetrahydroquinolines and potential extension to pyridines [8].
Scheme 1: Use of a Chan–Lam reaction for the synthesis of tetrahydroquinolines and potential extension to pyr...
Pyridines are important heterocycles that are often found in biologically active molecules [14-21]. Due to the high demand for these compounds, there are many methods for preparing substituted pyridines through condensation reactions [22-26], cycloadditions [27-30], functionalization of parent pyridine structures [31-38], fragment couplings [39-42], and transition metal-catalyzed C–H bond functionalization of α,β-unsaturated imines and oximes [43-50]. We were inspired by the copper-catalyzed coupling of protected α,β-unsaturated oximes and alkenylboronic acids developed by Liebeskind and coworkers due to its modularity and control of regioselectivity and wondered if a Chan–Lam route to N-alkenylnitrones would allow us to prepare similar intermediates (Scheme 2A) [51]. Nakamura and coworkers have reported that N-allenylnitrones can be accessed through rearrangements of O-propargylic oximes and undergo similar electrocyclizations to form pyridines (Scheme 2B) [52]. Herein, we show that N-alkenylnitrones 8 can be prepared through a Chan–Lam coupling of α,β-unsaturated oximes 6 and an alkenylboronic acids 7 and that these compounds undergo a novel thermal rearrangement to the corresponding tri- and tetrasubstituted pyridines 9 (Scheme 2C). This use of α,β-unsaturated oxime reagents for the synthesis of pyridines is unique from transition metal-catalyzed C–H bond functionalization processes that require a regioselective migratory insertion. This route is appealing due to the modularity of the Chan–Lam coupling process, and proceeds through a pathway that is distinct from the Liebeskind copper-catalyzed C–N bond coupling and electrocyclization (Scheme 2).
![[1860-5397-11-226-i2]](/bjoc/content/inline/1860-5397-11-226-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Examples of pyridine synthesis from oxime precursors [51,52].
Scheme 2: Examples of pyridine synthesis from oxime precursors [51,52].
Results and Discussion
A Chan–Lam coupling between chalcone oxime 6a and cyclohexenylboronic acid (7a) was initially tested using reaction conditions that we had previously identified as optimal for analogous N-alkenylnitrone syntheses from fluorenone oxime [7]. Nitrone 8aa was successfully isolated in 40% yield using 2 equiv of Cu(OAc)2 and the reaction conditions indicated in Table 1, entry 1. Only the E-nitrone isomer was observed and isolated. Decreasing the amount of copper reagent to 1 equiv had little influence on the reaction and a screen of other common copper salts only resulted in diminished yields (Table 1, entries 2–7). Further reduction of the copper loading to 10–30 mol % of Cu(OAc)2 was tolerated without a decrease in reaction efficiency (Table 1, entries 8 and 9). The key factor in improving the yield of the transformation was identified as an alkene additive. Alkene and alkyne additives have previously been observed to improve similar copper-catalyzed coupling reactions [53]. As shown in Table 1, entries 10–14, the addition of 1.2 equiv of cyclooctadiene (COD), cyclooctene (COE), norbornadiene (NBD), 1-octene, and dibenzylideneacetone (dba) all improved the yield of the Chan–Lam reaction, but COD was most efficient.
Table 1: Optimization of Chan–Lam coupling for the synthesis of N-cyclohexenyl-α,β-unsaturated nitrones.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-226-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | catalyst | cat. conc | additive | yield (%)b |
|---|---|---|---|---|
| 1 | Cu(OAc)2 | 2 equiv | none | 40 |
| 2 | Cu(OAc)2 | 1 equiv | none | 46 |
| 3 | CuTC | 1 equiv | none | 30 |
| 4 | CuBr | 1 equiv | none | dec |
| 5 | CuI | 1 equiv | none | trace |
| 6 | CuOTf2 | 1 equiv | none | trace |
| 7 | Cu(TFA)2 | 1 equiv | none | trace |
| 8 | Cu(OAc)2 | 10 mol % | none | 45 |
| 9 | Cu(OAc)2 | 30 mol % | none | 45 |
| 10 | Cu(OAc)2 | 10 mol % | COE | 65 |
| 11 | Cu(OAc)2 | 10 mol % | NBD | 58 |
| 12 | Cu(OAc)2 | 10 mol % | COD | 72 |
| 13 | Cu(OAc)2 | 10 mol % | 1-octene | 57 |
| 14 | Cu(OAc)2 | 10 mol % | dba | 51 |
aConditions: 6a (1 equiv), 7a (2 equiv), pyridine (5 equiv), Na2SO4 (8–9 equiv), 0.1 M in DCE, 25 °C, air, 18 h. bDetermined by 1H NMR spectroscopy using CH2Br2 as a reference.
Having identified optimal conditions for the synthesis of N-cyclohexenylnitrone 8aa, the scope of the nitrone synthesis was explored by varying the oxime and alkenylboronic acid reagents. As shown in Table 2, chalcone oximes with both electron-rich and electron-poor aryl substituents, as well as heteroaryl substituents, were tolerated for the transformation with electron-donating substituents providing higher yields (Table 2, entries 1–4). A significant increase in reaction efficiency was also observed for dba oxime (Table 2, entry 5). Evaluation of acyclic alkenylboronic acids further highlighted the differences between dba oxime and chalcone oximes as substrates for the Chan–Lam reaction. When dba oxime was treated with but-2-en-2-ylboronic acid, nitrone 8eb was isolated in good yield; in contrast, treatment of chalcone oxime with but-2-en-2-ylboronic acid, resulted in the isolation of nitrone 8ab in only 15% yield (Table 2, entries 6 and 7). Phenyl-substituted alkenylboronic acid 7c and monosubstituted alkenylboronic acids 7d and 7e, were more efficient reaction partners with chalcone oxime and gave the corresponding nitrones in good to excellent yield (Table 2, entries 8–10). Acyclic alkenylboronic acids were also incompatible with the optimal conditions determined for cyclohexenylboronic acid (7a) and required the use of 1–2 equiv of Cu(OAc)2. The use of both copper-catalyzed and copper-mediated reaction conditions with oximes 6 and alkenylboronic acids 7, allowed for the preparation of a variety of N-alkenyl-α,β-unsaturated nitrones to test for further reactivity.
Table 2: Scope of N-alkenyl-α,β-unsaturated nitrone synthesis.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-226-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | 8 | yield (%)b | entry | 8 | yield (%)b |
|---|---|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-226-i7.svg?max-width=637&scale=1.0)
8aa |
72c | 6 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-226-i8.svg?max-width=637&scale=1.0)
8ab |
15d |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-226-i9.svg?max-width=637&scale=1.0)
8ba |
41c | 7 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-226-i10.svg?max-width=637&scale=1.0)
8eb |
68d |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-226-i11.svg?max-width=637&scale=1.0)
8ca |
63c | 8 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-226-i12.svg?max-width=637&scale=1.0)
8ac |
57d |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-226-i13.svg?max-width=637&scale=1.0)
8da |
70e | 9 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-226-i14.svg?max-width=637&scale=1.0)
8ad |
75d |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-226-i15.svg?max-width=637&scale=1.0)
8ea |
84c | 10 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-226-i16.svg?max-width=637&scale=1.0)
8ae |
83d |
aConditions: 6 (1 equiv), 7 (2 equiv), pyridine (5 equiv), COD (1.2 equiv), Na2SO4 (8–9 equiv), 0.1 M in DCE, 25 °C, air, 18 h. bPercent isolated yield. cCu(OAc)2 (10 mol %). dCu(OAc)2 (1–2 equiv), pyridine (10 equiv), no COD. eCu(OAc)2 (10 mol %), no COD.
The preparation of the N-alkenyl-α,β-unsaturated nitrones shown in Table 2, allowed for further study of their conversion to tri- and tetrasubstituted pyridines. The preliminary evaluation of this thermal transformation with 8aa indicated that DMSO was a more efficient reaction medium than PhMe, dioxane, or DMF (Scheme 3). As shown in Table 3, all of the chalcone nitrones were readily converted to the corresponding pyridines in good yield (Table 3, entries 1–4, 6, 8–10). In contrast, the dba nitrones gave the corresponding pyridines in attenuated yields (Table 3, entries 5 and 7). The high density of substituents and regiospecificity of the transformation due to the use of the Chan–Lam reaction for the synthesis of the nitrone precursor are noteworthy and provide advantages over pyridine syntheses that require regioselective insertion reactions or nucleophilic additions.
![[1860-5397-11-226-i3]](/bjoc/content/inline/1860-5397-11-226-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Solvent effect on conversion of N-alkenylnitrones to pyridines.
Scheme 3: Solvent effect on conversion of N-alkenylnitrones to pyridines.
Table 3: Scope of conversion of N-alkenyl-α,β-unsaturated nitrones to pyridines.a
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-226-i17.svg?max-width=637&scale=1.0)
|
|||||
| entry | 9 | yield (%)b | entry | 9 | yield (%)b |
|---|---|---|---|---|---|
| 1 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-226-i18.svg?max-width=637&scale=1.0)
9aa |
71 | 6 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-226-i19.svg?max-width=637&scale=1.0)
9ab |
50 |
| 2 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-226-i20.svg?max-width=637&scale=1.0)
9ba |
68 | 7 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-226-i21.svg?max-width=637&scale=1.0)
9eb |
36 |
| 3 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-226-i22.svg?max-width=637&scale=1.0)
9ca |
64 | 8 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-226-i23.svg?max-width=637&scale=1.0)
9ac |
87 |
| 4 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-226-i24.svg?max-width=637&scale=1.0)
9da |
76 | 9 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-226-i25.svg?max-width=637&scale=1.0)
9ad |
71 |
| 5 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-226-i26.svg?max-width=637&scale=1.0)
9ea |
43 | 10 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-226-i27.svg?max-width=637&scale=1.0)
9ae |
65 |
aConditions: 8 (1 equiv), 4 Å MS, 0.1 M in DMSO, 140 °C, 6–8 h. bPercent isolated yield.
To better understand the conversion of N-alkenyl-α,β-unsaturated nitrones 8 to pyridines 9, two mechanistic experiments were evaluated (Scheme 4). The conversion of nitrone 8ae to pyridine 9ae was monitored by 1H and 13C NMR spectroscopy. Surprisingly, after heating 8ae for 4 h at 140 °C in DMSO-d6, a 1:1:1 mixture of isoxazoline 15ae, enaminoketone 16ae, and pyridine 9ae was observed [54,55]. Further heating this mixture of intermediates for 4 h resulted in the sole formation of pyridine 9ae. This experiment suggests that the conversion of nitrone 8ae to pyridine 9ae proceeds by oxygen transfer to give 16ae and nucleophilic addition of the enamine to the ketone. This pathway may explain the solvent dependence that was observed for the transformation (Scheme 3). The lack of any observation of dihydropyridine N-oxide intermediate 17ae suggests that the reaction is not proceeding through an electrocyclization and elimination process. A second experiment tested the electronic effect of this oxygen-transfer process. Unsymmetrically substituted dba nitrone 8fa was subjected to the cyclization conditions and a 1:1 mixture of 9fa:9fa' was observed. This experiment indicated a lack of any significant electronic preference for the oxygen-transfer process.
![[1860-5397-11-226-i4]](/bjoc/content/inline/1860-5397-11-226-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
A new method for the preparation of tri- and tetrasubstituted pyridines has been developed that hinges on the use of a Chan–Lam coupling to construct N-alkenyl-α,β-unsaturated nitrone precursors from the corresponding oximes and alkenylboronic acids. This method is tolerant of a variety of chalcone- and dba-derived oxime substrates as well as both mono- and disubstituted alkenylboronic acids. Initial reaction monitoring experiments suggest that the cyclization of the N-alkenyl-α,β-unsaturated nitrone to the pyridine occurs through an oxygen transfer from the nitrone functionality to the β-position of the conjugated olefin followed by nucleophilic attack of the enamine.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 6.6 MB | Download |
References
-
Qiao, J. X.; Lam, P. Y. S. Synthesis 2011, 829–856. doi:10.1055/s-0030-1258379
Return to citation in text: [1] -
Partyka, D. V. Chem. Rev. 2011, 111, 1529–1595. doi:10.1021/cr1002276
Return to citation in text: [1] -
Luo, F.; Pan, C.; Cheng, J. Synlett 2012, 357–366. doi:10.1055/s-0031-1290101
Return to citation in text: [1] -
Rao, K. S.; Wu, T.-S. Tetrahedron 2012, 68, 7735–7754. doi:10.1016/j.tet.2012.06.015
Return to citation in text: [1] -
Petrassi, H. M.; Sharpless, K. B.; Kelly, J. W. Org. Lett. 2001, 3, 139–142. doi:10.1021/ol0003533
Return to citation in text: [1] -
Patil, A. S.; Mo, D.-L.; Wang, H.-Y.; Mueller, D. S.; Anderson, L. L. Angew. Chem., Int. Ed. 2012, 51, 7799–7803. doi:10.1002/anie.201202704
Return to citation in text: [1] -
Mo, D.-L.; Wink, D. A.; Anderson, L. L. Org. Lett. 2012, 14, 5180–5183. doi:10.1021/ol3022885
Return to citation in text: [1] [2] -
Mo, D.-L.; Anderson, L. L. Angew. Chem., Int. Ed. 2013, 52, 6722–6725. doi:10.1002/anie.201301963
Return to citation in text: [1] [2] [3] -
Wang, H.-Y.; Anderson, L. L. Org. Lett. 2013, 15, 3362–3365. doi:10.1021/ol401416r
Return to citation in text: [1] -
Kontokosta, D.; Mueller, D. S.; Wang, H.-Y.; Anderson, L. L. Org. Lett. 2013, 15, 4830–4833. doi:10.1021/ol402237w
Return to citation in text: [1] -
Pecak, W. H.; Son, J.; Burnstine, A. J.; Anderson, L. L. Org. Lett. 2014, 16, 3440–3443. doi:10.1021/ol501230e
Return to citation in text: [1] -
Mo, D.-L.; Pecak, W. H.; Zhao, M.; Wink, D. J.; Anderson, L. L. Org. Lett. 2014, 16, 3696–3699. doi:10.1021/ol501503a
Return to citation in text: [1] -
Mo, D.-L.; Wink, D. J.; Anderson, L. L. Chem. – Eur. J. 2014, 20, 13217–13225. doi:10.1002/chem.201403268
Return to citation in text: [1] -
Roughley, S. D.; Jordan, A. M. J. Med. Chem. 2011, 54, 3451–3479. doi:10.1021/jm200187y
Return to citation in text: [1] -
Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337–2347. doi:10.1039/b602413k
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2005, 22, 627–646. doi:10.1039/b413750g
Return to citation in text: [1] -
Hill, M. D. Chem. – Eur. J. 2010, 16, 12052–12062. doi:10.1002/chem.201001100
Return to citation in text: [1] -
Henry, G. D. Tetrahedron 2004, 60, 6043–6061. doi:10.1016/j.tet.2004.04.043
Return to citation in text: [1] -
Varela, J. A.; Saá, C. Chem. Rev. 2003, 103, 3787–3802. doi:10.1021/cr030677f
Return to citation in text: [1] -
Zeni, G.; Larock, R. L. Chem. Rev. 2006, 106, 4644–4680. doi:10.1021/cr0683966
Return to citation in text: [1] -
Bull, J. A.; Mousseau, J. J.; Pelletier, G.; Charette, A. B. Chem. Rev. 2012, 112, 2642–2713. doi:10.1021/cr200251d
Return to citation in text: [1] -
Hantzsch, A. Justus Liebigs Ann. Chem. 1882, 215, 1–82. doi:10.1002/jlac.18822150102
Return to citation in text: [1] -
Barbe, G.; Charette, A. B. J. Am. Chem. Soc. 2008, 130, 18–19. doi:10.1021/ja077463q
Return to citation in text: [1] -
Bagley, M. C.; Glover, C.; Merritt, E. A. Synlett 2007, 2459–2482. doi:10.1055/s-2007-986674
Return to citation in text: [1] -
Kharchenko, V. G.; Markova, L. I.; Fedotova, O. V.; Pchelintseva, N. V. Chem. Heterocycl. Compd. 2003, 39, 1121–1142. doi:10.1023/B:COHC.0000008257.42400.9e
Return to citation in text: [1] -
Sausins, A.; Duburs, G. Heterocycles 1988, 27, 269–289. doi:10.3987/REV-87-370
Return to citation in text: [1] -
Boger, D. L.; Panek, J. S.; Meier, M. M. J. Org. Chem. 1982, 47, 895–897. doi:10.1021/jo00344a031
Return to citation in text: [1] -
Heller, B.; Hapke, M. Chem. Soc. Rev. 2007, 36, 1085–1094. doi:10.1039/b607877j
Return to citation in text: [1] -
McCormick, M. M.; Duong, H. A.; Zuo, G.; Louie, J. J. Am. Chem. Soc. 2005, 127, 5030–5031. doi:10.1021/ja0508931
Return to citation in text: [1] -
Tanaka, R.; Yuza, A.; Watai, Y.; Suzuki, D.; Takayama, Y.; Sato, F.; Urabe, H. J. Am. Chem. Soc. 2005, 127, 7774–7780. doi:10.1021/ja050261e
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C.; Yus, M. Chem. Rev. 2004, 104, 2667–2722. doi:10.1021/cr020101a
Return to citation in text: [1] -
Mongin, F.; Quéguiner, G. Tetrahedron 2001, 57, 4059–4090. doi:10.1016/S0040-4020(01)00100-4
Return to citation in text: [1] -
Fürstner, A.; Leitner, A.; Méndez, M.; Krause, H. J. Am. Chem. Soc. 2002, 124, 13856–13863. doi:10.1021/ja027190t
Return to citation in text: [1] -
Billingsley, K.; Buchwald, S. L. J. Am. Chem. Soc. 2007, 129, 3358–3366. doi:10.1021/ja068577p
Return to citation in text: [1] -
Liu, S.; Sawicki, J.; Driver, T. G. Org. Lett. 2012, 14, 3744–3747. doi:10.1021/ol301606y
Return to citation in text: [1] -
Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. J. Org. Chem. 2014, 79, 230–239. doi:10.1021/jo4024123
Return to citation in text: [1] -
Colombe, J. R.; Bernhardt, S.; Stathakis, C.; Buchwald, S. L.; Knochel, P. Org. Lett. 2013, 15, 5754–5757. doi:10.1021/ol402798z
Return to citation in text: [1] -
Luzung, M. R.; Patel, J. S.; Yin, J. J. Org. Chem. 2010, 75, 8330–8332. doi:10.1021/jo1018798
Return to citation in text: [1] -
Movassaghi, M.; Hill, M. D. J. Am. Chem. Soc. 2006, 128, 4592–4593. doi:10.1021/ja060626a
Return to citation in text: [1] -
Movassaghi, M.; Hill, M. D.; Ahmad, O. K. J. Am. Chem. Soc. 2007, 129, 10096–10097. doi:10.1021/ja073912a
Return to citation in text: [1] -
Manning, J. R.; Davies, H. M. L. J. Am. Chem. Soc. 2008, 130, 8602–8603. doi:10.1021/ja803139k
Return to citation in text: [1] -
Chen, M. Z.; Micalizio, G. C. J. Am. Chem. Soc. 2012, 134, 1352–1356. doi:10.1021/ja2105703
Return to citation in text: [1] -
Colby, D. A.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 3645–3651. doi:10.1021/ja7104784
Return to citation in text: [1] -
Martin, R. M.; Bergman, R. G.; Ellman, J. A. J. Org. Chem. 2012, 77, 2501–2507. doi:10.1021/jo202280e
Return to citation in text: [1] -
Parthasarathy, K.; Jeganmohan, M.; Cheng, C.-H. Org. Lett. 2008, 10, 325–328. doi:10.1021/ol7028367
Return to citation in text: [1] -
Parthasarathy, K.; Cheng, C.-H. Synthesis 2009, 1400–1402. doi:10.1055/s-0028-1088009
Return to citation in text: [1] -
Too, P. C.; Noji, T.; Lim, Y. J.; Li, X.; Chiba, S. Synlett 2011, 2789–2794. doi:10.1055/s-0031-1289556
Return to citation in text: [1] -
Hyster, T. K.; Rovis, T. Chem. Commun. 2011, 47, 11846–11848. doi:10.1039/C1CC15248C
Return to citation in text: [1] -
Neely, J. M.; Rovis, T. J. Am. Chem. Soc. 2013, 135, 66–69. doi:10.1021/ja3104389
Return to citation in text: [1] -
Neely, J. M.; Rovis, T. J. Am. Chem. Soc. 2014, 136, 2735–2738. doi:10.1021/ja412444d
Return to citation in text: [1] -
Liu, S.; Liebeskind, L. S. J. Am. Chem. Soc. 2008, 130, 6918–6919. doi:10.1021/ja8013743
Return to citation in text: [1] [2] -
Nakamura, I.; Zhang, D.; Terada, M. J. Am. Chem. Soc. 2010, 132, 7884–7886. doi:10.1021/ja102436z
Return to citation in text: [1] [2] -
Winternheimer, D. J.; Merlic, C. A. Org. Lett. 2010, 12, 2508–2510. doi:10.1021/ol100707s
Return to citation in text: [1] -
Katritzky, A. R.; Hayden, A. E.; Kirichenko, K.; Pelphrey, P.; Ji, Y. J. Org. Chem. 2004, 69, 5108–5111. doi:10.1021/jo0496594
Return to citation in text: [1] -
Zhang, Z.-H.; Yin, L.; Wang, Y.-M. Adv. Synth. Catal. 2006, 348, 184–190. doi:10.1002/adsc.200505268
Return to citation in text: [1]
| 1. | Qiao, J. X.; Lam, P. Y. S. Synthesis 2011, 829–856. doi:10.1055/s-0030-1258379 |
| 2. | Partyka, D. V. Chem. Rev. 2011, 111, 1529–1595. doi:10.1021/cr1002276 |
| 3. | Luo, F.; Pan, C.; Cheng, J. Synlett 2012, 357–366. doi:10.1055/s-0031-1290101 |
| 4. | Rao, K. S.; Wu, T.-S. Tetrahedron 2012, 68, 7735–7754. doi:10.1016/j.tet.2012.06.015 |
| 5. | Petrassi, H. M.; Sharpless, K. B.; Kelly, J. W. Org. Lett. 2001, 3, 139–142. doi:10.1021/ol0003533 |
| 14. | Roughley, S. D.; Jordan, A. M. J. Med. Chem. 2011, 54, 3451–3479. doi:10.1021/jm200187y |
| 15. | Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337–2347. doi:10.1039/b602413k |
| 16. | Michael, J. P. Nat. Prod. Rep. 2005, 22, 627–646. doi:10.1039/b413750g |
| 17. | Hill, M. D. Chem. – Eur. J. 2010, 16, 12052–12062. doi:10.1002/chem.201001100 |
| 18. | Henry, G. D. Tetrahedron 2004, 60, 6043–6061. doi:10.1016/j.tet.2004.04.043 |
| 19. | Varela, J. A.; Saá, C. Chem. Rev. 2003, 103, 3787–3802. doi:10.1021/cr030677f |
| 20. | Zeni, G.; Larock, R. L. Chem. Rev. 2006, 106, 4644–4680. doi:10.1021/cr0683966 |
| 21. | Bull, J. A.; Mousseau, J. J.; Pelletier, G.; Charette, A. B. Chem. Rev. 2012, 112, 2642–2713. doi:10.1021/cr200251d |
| 53. | Winternheimer, D. J.; Merlic, C. A. Org. Lett. 2010, 12, 2508–2510. doi:10.1021/ol100707s |
| 8. | Mo, D.-L.; Anderson, L. L. Angew. Chem., Int. Ed. 2013, 52, 6722–6725. doi:10.1002/anie.201301963 |
| 54. | Katritzky, A. R.; Hayden, A. E.; Kirichenko, K.; Pelphrey, P.; Ji, Y. J. Org. Chem. 2004, 69, 5108–5111. doi:10.1021/jo0496594 |
| 55. | Zhang, Z.-H.; Yin, L.; Wang, Y.-M. Adv. Synth. Catal. 2006, 348, 184–190. doi:10.1002/adsc.200505268 |
| 8. | Mo, D.-L.; Anderson, L. L. Angew. Chem., Int. Ed. 2013, 52, 6722–6725. doi:10.1002/anie.201301963 |
| 51. | Liu, S.; Liebeskind, L. S. J. Am. Chem. Soc. 2008, 130, 6918–6919. doi:10.1021/ja8013743 |
| 52. | Nakamura, I.; Zhang, D.; Terada, M. J. Am. Chem. Soc. 2010, 132, 7884–7886. doi:10.1021/ja102436z |
| 6. | Patil, A. S.; Mo, D.-L.; Wang, H.-Y.; Mueller, D. S.; Anderson, L. L. Angew. Chem., Int. Ed. 2012, 51, 7799–7803. doi:10.1002/anie.201202704 |
| 7. | Mo, D.-L.; Wink, D. A.; Anderson, L. L. Org. Lett. 2012, 14, 5180–5183. doi:10.1021/ol3022885 |
| 8. | Mo, D.-L.; Anderson, L. L. Angew. Chem., Int. Ed. 2013, 52, 6722–6725. doi:10.1002/anie.201301963 |
| 9. | Wang, H.-Y.; Anderson, L. L. Org. Lett. 2013, 15, 3362–3365. doi:10.1021/ol401416r |
| 10. | Kontokosta, D.; Mueller, D. S.; Wang, H.-Y.; Anderson, L. L. Org. Lett. 2013, 15, 4830–4833. doi:10.1021/ol402237w |
| 11. | Pecak, W. H.; Son, J.; Burnstine, A. J.; Anderson, L. L. Org. Lett. 2014, 16, 3440–3443. doi:10.1021/ol501230e |
| 12. | Mo, D.-L.; Pecak, W. H.; Zhao, M.; Wink, D. J.; Anderson, L. L. Org. Lett. 2014, 16, 3696–3699. doi:10.1021/ol501503a |
| 13. | Mo, D.-L.; Wink, D. J.; Anderson, L. L. Chem. – Eur. J. 2014, 20, 13217–13225. doi:10.1002/chem.201403268 |
| 7. | Mo, D.-L.; Wink, D. A.; Anderson, L. L. Org. Lett. 2012, 14, 5180–5183. doi:10.1021/ol3022885 |
| 39. | Movassaghi, M.; Hill, M. D. J. Am. Chem. Soc. 2006, 128, 4592–4593. doi:10.1021/ja060626a |
| 40. | Movassaghi, M.; Hill, M. D.; Ahmad, O. K. J. Am. Chem. Soc. 2007, 129, 10096–10097. doi:10.1021/ja073912a |
| 41. | Manning, J. R.; Davies, H. M. L. J. Am. Chem. Soc. 2008, 130, 8602–8603. doi:10.1021/ja803139k |
| 42. | Chen, M. Z.; Micalizio, G. C. J. Am. Chem. Soc. 2012, 134, 1352–1356. doi:10.1021/ja2105703 |
| 51. | Liu, S.; Liebeskind, L. S. J. Am. Chem. Soc. 2008, 130, 6918–6919. doi:10.1021/ja8013743 |
| 31. | Chinchilla, R.; Nájera, C.; Yus, M. Chem. Rev. 2004, 104, 2667–2722. doi:10.1021/cr020101a |
| 32. | Mongin, F.; Quéguiner, G. Tetrahedron 2001, 57, 4059–4090. doi:10.1016/S0040-4020(01)00100-4 |
| 33. | Fürstner, A.; Leitner, A.; Méndez, M.; Krause, H. J. Am. Chem. Soc. 2002, 124, 13856–13863. doi:10.1021/ja027190t |
| 34. | Billingsley, K.; Buchwald, S. L. J. Am. Chem. Soc. 2007, 129, 3358–3366. doi:10.1021/ja068577p |
| 35. | Liu, S.; Sawicki, J.; Driver, T. G. Org. Lett. 2012, 14, 3744–3747. doi:10.1021/ol301606y |
| 36. | Chen, X.; Zhou, L.; Li, Y.; Xie, T.; Zhou, S. J. Org. Chem. 2014, 79, 230–239. doi:10.1021/jo4024123 |
| 37. | Colombe, J. R.; Bernhardt, S.; Stathakis, C.; Buchwald, S. L.; Knochel, P. Org. Lett. 2013, 15, 5754–5757. doi:10.1021/ol402798z |
| 38. | Luzung, M. R.; Patel, J. S.; Yin, J. J. Org. Chem. 2010, 75, 8330–8332. doi:10.1021/jo1018798 |
| 52. | Nakamura, I.; Zhang, D.; Terada, M. J. Am. Chem. Soc. 2010, 132, 7884–7886. doi:10.1021/ja102436z |
| 27. | Boger, D. L.; Panek, J. S.; Meier, M. M. J. Org. Chem. 1982, 47, 895–897. doi:10.1021/jo00344a031 |
| 28. | Heller, B.; Hapke, M. Chem. Soc. Rev. 2007, 36, 1085–1094. doi:10.1039/b607877j |
| 29. | McCormick, M. M.; Duong, H. A.; Zuo, G.; Louie, J. J. Am. Chem. Soc. 2005, 127, 5030–5031. doi:10.1021/ja0508931 |
| 30. | Tanaka, R.; Yuza, A.; Watai, Y.; Suzuki, D.; Takayama, Y.; Sato, F.; Urabe, H. J. Am. Chem. Soc. 2005, 127, 7774–7780. doi:10.1021/ja050261e |
| 22. | Hantzsch, A. Justus Liebigs Ann. Chem. 1882, 215, 1–82. doi:10.1002/jlac.18822150102 |
| 23. | Barbe, G.; Charette, A. B. J. Am. Chem. Soc. 2008, 130, 18–19. doi:10.1021/ja077463q |
| 24. | Bagley, M. C.; Glover, C.; Merritt, E. A. Synlett 2007, 2459–2482. doi:10.1055/s-2007-986674 |
| 25. | Kharchenko, V. G.; Markova, L. I.; Fedotova, O. V.; Pchelintseva, N. V. Chem. Heterocycl. Compd. 2003, 39, 1121–1142. doi:10.1023/B:COHC.0000008257.42400.9e |
| 26. | Sausins, A.; Duburs, G. Heterocycles 1988, 27, 269–289. doi:10.3987/REV-87-370 |
| 43. | Colby, D. A.; Bergman, R. G.; Ellman, J. A. J. Am. Chem. Soc. 2008, 130, 3645–3651. doi:10.1021/ja7104784 |
| 44. | Martin, R. M.; Bergman, R. G.; Ellman, J. A. J. Org. Chem. 2012, 77, 2501–2507. doi:10.1021/jo202280e |
| 45. | Parthasarathy, K.; Jeganmohan, M.; Cheng, C.-H. Org. Lett. 2008, 10, 325–328. doi:10.1021/ol7028367 |
| 46. | Parthasarathy, K.; Cheng, C.-H. Synthesis 2009, 1400–1402. doi:10.1055/s-0028-1088009 |
| 47. | Too, P. C.; Noji, T.; Lim, Y. J.; Li, X.; Chiba, S. Synlett 2011, 2789–2794. doi:10.1055/s-0031-1289556 |
| 48. | Hyster, T. K.; Rovis, T. Chem. Commun. 2011, 47, 11846–11848. doi:10.1039/C1CC15248C |
| 49. | Neely, J. M.; Rovis, T. J. Am. Chem. Soc. 2013, 135, 66–69. doi:10.1021/ja3104389 |
| 50. | Neely, J. M.; Rovis, T. J. Am. Chem. Soc. 2014, 136, 2735–2738. doi:10.1021/ja412444d |
© 2015 Kontokosta et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)