Abstract
Asymmetric allylic alkylation is a powerful reaction that allows the enantioselective formation of C–C bonds. Here we describe the asymmetric alkylation of alkylzirconium species to racemic 3,6-dihydro-2H-pyrans. Two systems were examined: 3-chloro-3,6-dihydro-2H-pyran using linear optimization (45–93% ee, up to 33% yield, 5 examples) and 3,6-dihydro-2H-pyran-3-yl diethyl phosphate with the assistance of a design of experiments statistical approach (83% ee, 12% yield). 1H NMR spectroscopy was used to gain insight into the reaction mechanisms.

Graphical Abstract
Introduction
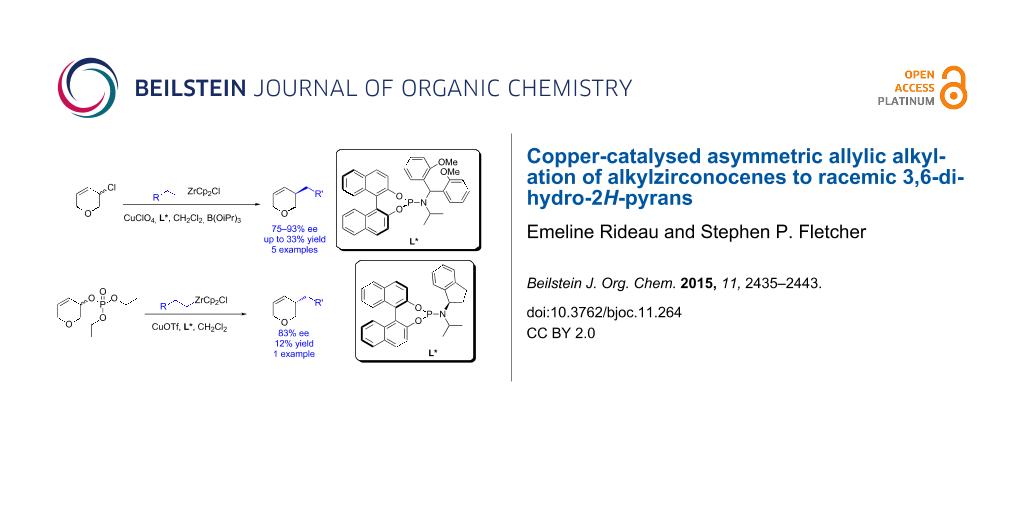
Asymmetry is found in many natural products and biologically active molecules. Using racemic starting materials to synthesize enantiomerically enriched products is a powerful and underdeveloped strategy [1-4]. In some cases transition metal-catalysed asymmetric allylic alkylation (AAA) reactions [5-7] can be used in dynamic kinetic asymmetric transformations (DYKAT) [8-15] to provide single enantiomer products from racemic starting materials. Mechanistically some of these have been shown to occur by direct enantio-convergent transformations [16-18]. We have developed Cu-catalysed asymmetric conjugate additions of alkylzirconium reagents generated in situ by hydrometallation of terminal alkenes [19-25], and recently demonstrated that zirconium nucleophiles may undergo highly enantioselective copper-catalysed AAAs to racemic cyclic allyl halides, such as 1 (Scheme 1a) [26,27].
![[1860-5397-11-264-i1]](/bjoc/content/inline/1860-5397-11-264-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previously reported Cu-AAA of alkylzirconium reagents to racemic allyl chlorides [26] and this work.
Scheme 1: Previously reported Cu-AAA of alkylzirconium reagents to racemic allyl chlorides [26] and this work.
Tetrahydropyrans are a common motif in natural products and pharmaceuticals and are useful synthetic intermediates. However, the direct asymmetric derivatization of pyrans is rare [28] and enantiomerically enriched tetrahydropyrans are often obtained by ring-closing methods [29,30]. To extend our previously reported DYKATs beyond all-carbon electrophiles we decided to examine 3-chloro-3,6-dihydro-2H-pyran (2a, Scheme 1b). This was envisaged to be a challenging substrate. The presence of oxygen in the ring would modify the electronics, and likely the reactivity, of the starting material. The oxygen lone pairs on 2a could potentially interact with the copper-catalyst or alkyl metal nucleophiles.
Results and Discussion
We first examined the in situ hydrometallation/AAA of 4-phenyl-1-butene (4) to racemic 3-chloro-3,6-dihydro-2H-pyran (2a, Table 1). Interestingly, no product was formed using our previously reported conditions for AAA to racemic carbocyclic substrates (CuI, ligand A, CHCl3, Table 1, entry 1) [26] and only unreacted starting material was recovered. Different Cu salts were examined (Table 1, entries 2–7) and more strongly electron withdrawing counterions were found to provide the desired product, with CuClO4 giving the best ee (70% ee, Table 1, entry 3). A solvent screen lead us to the conclusion that chlorinated solvents are best (CH2Cl2 (70% ee) and CHCl3 (67% ee), Table 1, entries 7 and 10, respectively). Extensive examination of phosphoramidite ligands (for example, Table 1, entries 2 and 11–13) did not improve the ee. We then tested many different additives (TMSCl, AgOTf, borates, ZrCl4, Si(OEt)4, etc, for example Table 1, entries 14–18). Using B(OiPr)3, which presumably acts as a Lewis acid, improved the ee to 80% (Table 1, entry 18) and so we re-examined different ligands using CuClO4 in CH2Cl2 with B(OiPr)3 (Table 1, entries 19–21). Derivatives of ligand B were tested and ligand F gave 83% ee (Table 1, entry 21), while electronically similar E was much less selective (47% ee, Table 1, entry 20). The effects of concentration, temperature and catalyst loading were also investigated (not shown) with no improvement on the enantioselectivity.
Table 1: Asymmetric alkylation to 3-chloro-3,6-dihydro-2H-pyran (2a)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-264-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Copper | L* | Solvent | Additive | eeb |
|---|---|---|---|---|---|
| 1 | CuI | A | CHCl3 | NP | |
| 2 | CuClO4 | A | CH2Cl2 | 68% | |
| 3 | CuClO4 | B | CH2Cl2 | 70% | |
| 4 | CuOTf | B | CH2Cl2 | 64% | |
| 5 | CuNTf2 | B | CH2Cl2 | 52% | |
| 6 | CuTC | B | CH2Cl2 | 12% | |
| 7 | CuSbF6 | B | CH2Cl2 | NP | |
| 8 | CuClO4 | B | Et2O | 55% | |
| 9 | CuClO4 | B | Me-THF | 38% | |
| 10 | CuClO4 | B | CHCl3 | 67% | |
| 11 | CuClO4 | C | CH2Cl2 | 53% | |
| 12 | CuClO4 | D | CH2Cl2 | 36% | |
| 13 | CuClO4 | E | CH2Cl2 | 12% | |
| 14 | CuClO4 | B | CH2Cl2 | TMSCl | 73% |
| 15 | CuClO4 | B | CH2Cl2 | Si(OEt)4 | 63% |
| 16 | CuClO4 | B | CH2Cl2 | Ti(OiPr)4 | 25% |
| 17 | CuClO4 | B | CH2Cl2 | AlCl3 | 15% |
| 18 | CuClO4 | B | CH2Cl2 | B(OiPr)3 | 80% |
| 19 | CuClO4 | C | CH2Cl2 | B(OiPr)3 | 78% |
| 20 | CuClO4 | E | CH2Cl2 | B(OiPr)3 | 47% |
| 21 | CuClO4 | F | CH2Cl2 | B(OiPr)3 | 83% |
aConditions: 4-phenyl-1-butene (2.5 equiv), Cp2ZrHCl (2.0 equiv), 2a (1.0 equiv), CuL* as specified (0.1 equiv), additive as specified (1.0 equiv), in specified solvent (2.0 mL), room temperature. bee determined by HPLC. NP = no product. For more information on procedures see Supporting Information File 1.
After extensive optimization, the highest enantiomeric excess obtained was only 83% ee and so we decided to examine other leaving groups (Table 2). Like allyl chloride 2a, allyl bromide 2b gave no desired product when using our previously reported conditions [26] (Table 2, entry 2). The use of 2b also only gave low ee when using the conditions optimized above (38% ee, Table 2, entry 1). Allyl acetate 2c did not give the desired product under any conditions examined, however, allyl phosphate 2d was found to provide 5 with good selectivity (77% ee, Table 2, entry 5). 3,6-Dihydro-2H-pyran-3-yl diethyl phosphate (2d) was also the only substrate to react using our previously reported AAA conditions (CuI, ligand A, CHCl3) [26], albeit with poor enantioselectivity (29% ee, Table 2, entry 6).
Table 2: Effect of leaving groups.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-264-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | X | Copper | L* | Solvent | Additive | eeb |
|---|---|---|---|---|---|---|
| 1 | Br | CuClO4 | F | CH2Cl2 | B(OiPr)3 | 38% |
| 2 | Br | CuI | A | CHCl3 | NP | |
| 3 | OAc | CuClO4 | F | CH2Cl2 | B(OiPr)3 | NP |
| 4 | OAc | CuI | A | CHCl3 | NP | |
| 5 | OPO(OEt)2 | CuClO4 | F | CH2Cl2 | B(OiPr)3 | 77% |
| 6 | OPO(OEt)2 | CuI | A | CHCl3 | 29% | |
aConditions: 4-phenyl-1-butene (2.5 equiv), Cp2ZrHCl (2.0 equiv), 2 (1.0 equiv), CuL*(0.1 equiv), additive (1.0 equiv), in solvent (2.0 mL), room temperature. bee determined by HPLC. NP = no product. For more information on procedures see Supporting Information File 1.
Design of experiments (DoE) [31-37] is a powerful tool for efficient screening and is commonly used in industry, since traditional one-factor-at-a-time optimization poorly covers the available parameter space and may not locate the most optimal conditions. As DoE rapidly explores the response space efficiently and can reveal interdependence of factors at no extra experimental cost, we decided to briefly examine DoE in this complex asymmetric transformation. We note that there are important limits to this investigation. Understanding what interactions give rise to asymmetric induction (particularly in transformations where mechanisms are not understood) is extremely challenging, and it is not obvious how to parameterize the multiple variables present in key factors such as ligand structure [38].
Nevertheless, a Principal Component Analysis using JMP® 12.1.0 (SAS) in 3 waves was carried out using 2d as the starting material. In each experiment, the most promising variables were chosen based on results from previously published methods, the procedure optimised for 2a (above), and the results of previous waves. The first wave was as a third factorial design with 3 categories: Ligand (A, B, C, F and G), counter-ion (ClO4−, I− and OTf−) and solvent (CH2Cl2, Et2O and TBME, Table 1, entries 1–17; ● Figure 1, for more details see Supplorting Information File 1).
![[1860-5397-11-264-1]](/bjoc/content/figures/1860-5397-11-264-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: DoE from 3,6-dihydro-2H-pyran-3-yl diethyl phosphate (2d). Conditions: 4-phenyl-1-butene (2.5 equiv), Cp2ZrHCl (2.0 equiv), 2d (1.0 equiv), CuL* as specified (0.1 equiv), additive as specified (1.0 equiv or 5.0 equiv), in specified solvent (2.0 mL), room temperature. ee determined by HPLC. For more information on the procedures see Supporting Information File 1. ● (wave 1, entries 1–17), ♦ (wave 2, entries 18–30), ▼ (wave 3, entries 31–38).
Figure 1: DoE from 3,6-dihydro-2H-pyran-3-yl diethyl phosphate (2d). Conditions: 4-phenyl-1-butene (2.5 equiv...
This first DoE study suggested that neither CuI nor TBME were good fits for the reaction (both consistently giving low ee). The combination of CuOTf in CH2Cl2 gave the best enantioselectivity (up to 83% ee, Table S1 entry 3, Supporting Information File 1) with ligand G. Unlike with 2a, CuClO4 did not give high ee with 2d; the highest value obtained was 43% ee (Table S1, entry 14). Interestingly Et2O gave mixed results with some low (e.g., 1% ee, Table S1, entry 1) and moderate (e.g., 56% ee, Table S1, entry 6) ee values obtained.
Based on those results, a second wave of DoE was designed as a 6th factorial design with 4 factors: Ligand (G, H, I, J), counter-ion (OTf and NTf2), solvent (CH2Cl2, Et2O and CHCl3) and TMSCl equivalent (0, 1 and 5) (Table S1, entries 18–30, ♦). As mixed results were obtained with Et2O, we decided to investigate it more thoroughly. This second study emphasizes the intrinsic challenge of finding optimum conditions in complex asymmetric reactions. Whereas CuOTf seems to work best with CH2Cl2 as a solvent, CuNTf2 gave better enantioselectivity in Et2O. CHCl3 consistently provided lower enantioselectivity than CH2Cl2. In the small selection of ligands examined, G generally gave better results.
We designed a final study to investigate the role of various equivalents of additive (TMSCl and B(OiPr)3) with CuOTf and CuNTf2 in their respective favoured solvents (CH2Cl2 and Et2O) (Table S1, entries 31–38, ▼). B(OiPr)3 significantly lowered the ee (44% ee, Table S1, entry 33). The influence of TMSCl on the reaction was highly dependent on the other reaction parameters; CuNTf2 in Et2O with no additive gave 67% ee (Table S1, entry 34), while adding 1 equiv of TMSCl gave a slight improvement (73% ee, Table S1, entry 35) but no further improvement was observed by adding more TMSCl (5 equiv, 74% ee, Table S1, entry 36). On the other hand, using 1 equiv of TMSCl with CuOTf in CH2Cl2 did not modify the ee (81% ee, Table S1, entry 32), while adding 5 equiv of TMSCl was detrimental to enantioselectivity (60% ee, Table S1, entry 31).
Despite our efforts to optimise this second system, the highest enantioselectivity obtained was 83% ee, which is the same as for allyl chloride 2a. It became clear that when using alkylzirconocene nucleophiles and Cu catalysis, derivatised 3,6-dihydro-2H-pyrans are difficult to obtain in high enantiomeric excess. Moreover, both optimised systems gave poor yield; 25% yield with 100% conversion from allyl chloride 2a and 17% yield with 31% conversion from allyl phosphate 2d.
Various alkenes were examined using the allyl chloride 2a system (Scheme 2). The reaction showed tolerance in functional groups such as CF3 (6, 75% ee) Cl (7, 77% ee), and cyclohexane (8, 88% ee). Electron rich allyl silane could also be used to introduce a TMS group (9, 93% ee), but all the yields were poor.
![[1860-5397-11-264-i2]](/bjoc/content/inline/1860-5397-11-264-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of nucleophiles. Conditions: alkene (2.5 equiv), Cp2ZrHCl (2.0 equiv), 3-chloro-3,6-dihydro-2H-pyran 2a (1.0 equiv), CuCl (10 mol %), D (10 mol %), AgClO4 (10 mol %), B(OiPr)3 (1.0 equiv), in CH2Cl2 (2.0 mL), room temperature. Isolated yield. ee determined by HPLC or GC. For more information see Supporting Information File 1.
Scheme 2: Scope of nucleophiles. Conditions: alkene (2.5 equiv), Cp2ZrHCl (2.0 equiv), 3-chloro-3,6-dihydro-2H...
To investigate why we obtained such poor yields, and possibly shed light onto the reaction mechanism, we decided to follow both reactions in time using in situ NMR spectroscopy (Figure 2 and Figure 3). Reactions were carried out as normal, but in deuterated solvents and mixed in an NMR tube (see Supporting Information File 1). Ethylene was used as the alkylzirconium precursor as it greatly simplifies the NMR spectra. Spectra were recorded at regular intervals over time where relative concentrations are based on integration of the best resolved 1H signal for each species and calibrated accordingly.
Through these kinetic studies, it is clear that the allyl chloride 2a system fails because the starting material dimerises to give 11 as the major reaction product (60% isolated yield – 30 mol % by NMR) (Figure 2). This is consistent with the observed ≈100% conversion but low product yields. Presumably 11 arises from the homocoupling of allyl chloride 2a, possibly through a π-allyl-Cu intermediate [39-43]. Although both the conditions and leaving groups differ in the two reactions it is not clear why 2a, but not 2d, dimerizes. 11 can exists as 3 isomers, a meso compound and two enantiomers. Upon comparison to literature data [44], we concluded that we form a mixture of all three, as a 1:1 mixture of the meso and racemic material. Our samples did not rotate plane polarized light, emphasizing the racemic nature of the sample and suggesting that 11 is formed in a completely non-selective pathway.
![[1860-5397-11-264-2]](/bjoc/content/figures/1860-5397-11-264-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reaction kinetics as monitored by in situ 1H NMR spectroscopy from 3-chloro-3,6-dihydro-2H-pyran (2a).
Figure 2: Reaction kinetics as monitored by in situ 1H NMR spectroscopy from 3-chloro-3,6-dihydro-2H-pyran (2a...
In the case of allyl phosphate 2d, the system appears to lack reactivity and the reaction quickly dies, so that 10 (Figure 3) is formed with poor conversion, and we speculate that the phosphate leaving group inhibits the catalyst which would explain why only ~10% of product is formed.
![[1860-5397-11-264-3]](/bjoc/content/figures/1860-5397-11-264-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Reaction kinetics as monitored by in situ 1H NMR spectroscopy from 3,6-dihydro-2H-pyran-3-yl diethyl phosphate (2d).
Figure 3: Reaction kinetics as monitored by in situ 1H NMR spectroscopy from 3,6-dihydro-2H-pyran-3-yl diethy...
To obtain further mechanistic information we followed the ee of these reactions in time (Figure 4 and Figure 5). In the system using chloride 2a, the ee of product 5 remains constant throughout the reaction (~75% ee, Figure 4). Starting chloride 2a was found to be quite robust so that we could also determine its enantiomeric excess during the course of the reaction. Initially 2a is racemic but it becomes scalemic to slowly reach 34% ee when the reaction is complete (~12 hours). From these observations and our experimental demonstration that 2a is much more stable than all-carbocyclic 1, it appears that 2a undergoes kinetic resolution. However, this system is clearly complicated by the fact that the majority of 2a is consumed during byproduct 11's formation.
![[1860-5397-11-264-4]](/bjoc/content/figures/1860-5397-11-264-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Kinetic ee analysis using 2a. ee of reaction with 3-chloro-3,6-dihydro-2H-pyran (2a) as measured by removing aliquots in time.
Figure 4: Kinetic ee analysis using 2a. ee of reaction with 3-chloro-3,6-dihydro-2H-pyran (2a) as measured by...
![[1860-5397-11-264-5]](/bjoc/content/figures/1860-5397-11-264-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Kinetic ee analysis using 2d. ee of reaction with 3,6-dihydro-2H-pyran-3-yl diethyl phosphate (2d) as measured by removing aliquots in time.
Figure 5: Kinetic ee analysis using 2d. ee of reaction with 3,6-dihydro-2H-pyran-3-yl diethyl phosphate (2d) ...
In the phosphate system based on 2d, the ee of product 5 was found to increase during the course of the reaction (Figure 5) so that 5 was ~26% ee after a few minutes, and increased to ~82% ee after 5 hours. Unfortunately, analytical conditions to separate the enantiomers of 2d, so we could measure the enantiomeric excess of this starting material, could not be found. At this stage it is not possible to provide a full mechanistic rationalization of these reactions. It is also not immediately obvious how to improve yields and enantiomeric excesses. The kinetic studies suggest that the two reactions work through very different mechanisms and it strikes us as remarkable how both systems give roughly the same enantioselectivity and poor yield, yet have significantly different pathways.
Conclusion
The Cu-catalyzed AAA of alkylzirconium reagents to racemic heterocyclic electrophiles was explored. After extensive examination, two different methods for obtaining 3,6-dihydro-2H-pyran derivatives with respectable levels of ee (≈83% ee) were developed. Unfortunately, the yields were poor in both cases. Kinetic studies were performed to help to understand the difficulties associated with these reactions. While we were not able to resolve the issues of yield in these studies, this work reveals remarkable mechanistic diversity in Cu-catalysed asymmetric alkylation reactions to racemic starting materials.
Supporting Information
| Supporting Information File 1: Additional material. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Faber, K. Chem. – Eur. J. 2001, 7, 5004. doi:10.1002/1521-3765(20011203)7:23<5004::AID-CHEM5004>3.0.CO;2-X
Return to citation in text: [1] -
Vedejs, E.; Jure, M. Angew. Chem., Int. Ed. 2005, 44, 3974. doi:10.1002/anie.200460842
Return to citation in text: [1] -
Mohr, J. T.; Ebner, D. C.; Stoltz, B. M. Org. Biomol. Chem. 2007, 5, 3571. doi:10.1039/b711159m
Return to citation in text: [1] -
Miller, L. C.; Sarpong, R. Chem. Soc. Rev. 2011, 40, 4550. doi:10.1039/c1cs15069c
Return to citation in text: [1] -
Trost, B. M.; Bunt, R. C. J. Am. Chem. Soc. 1994, 116, 4089. doi:10.1021/ja00088a059
Return to citation in text: [1] -
Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395. doi:10.1021/cr9409804
Return to citation in text: [1] -
Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258. doi:10.1002/anie.200605113
Return to citation in text: [1] -
Huerta, F. F.; Minidis, A. B. E.; Bäckvall, J.-E. Chem. Soc. Rev. 2001, 30, 321. doi:10.1039/b105464n
Return to citation in text: [1] -
Trost, B. M.; Fandrick, D. R. Aldrichimica Acta 2007, 40, 59.
Return to citation in text: [1] -
Norinder, J.; Bäckvall, J.-E. Chem. – Eur. J. 2007, 13, 4094. doi:10.1002/chem.200601684
Return to citation in text: [1] -
Langlois, J.-B.; Alexakis, A. Chem. Commun. 2009, 3868. doi:10.1039/b907722g
Return to citation in text: [1] -
Langlois, J.-B.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 447. doi:10.1002/adsc.200900790
Return to citation in text: [1] -
Katcher, M. H.; Doyle, A. G. J. Am. Chem. Soc. 2010, 132, 17402. doi:10.1021/ja109120n
Return to citation in text: [1] -
Hamilton, J. Y.; Hauser, N.; Sarlah, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2014, 53, 10759. doi:10.1002/anie.201406077
Return to citation in text: [1] -
Misale, A.; Niyomchon, S.; Luparia, M.; Maulide, N. Angew. Chem., Int. Ed. 2014, 53, 7068. doi:10.1002/anie.201309074
Return to citation in text: [1] -
Ito, H.; Kunii, S.; Sawamura, M. Nat. Chem. 2010, 2, 972. doi:10.1038/nchem.801
Return to citation in text: [1] -
Langlois, J.-B.; Emery, D.; Mareda, J.; Alexakis, A. Chem. Sci. 2012, 3, 1062. doi:10.1039/C2SC00868H
Return to citation in text: [1] -
Delvos, L. B.; Oestreich, M. Synthesis 2015, 47, 924. doi:10.1055/s-0034-1380157
Return to citation in text: [1] -
Maksymowicz, R. M.; Roth, P. M. C.; Fletcher, S. P. Nat. Chem. 2012, 4, 649. doi:10.1038/nchem.1394
Return to citation in text: [1] -
Sidera, M.; Roth, P. M. C.; Maksymowicz, R. M.; Fletcher, S. P. Angew. Chem., Int. Ed. 2013, 52, 7995. doi:10.1002/anie.201303202
Return to citation in text: [1] -
Maciver, E. E.; Maksymowicz, R. M.; Wilkinson, N.; Roth, P. M. C.; Fletcher, S. P. Org. Lett. 2014, 16, 3288. doi:10.1021/ol501292x
Return to citation in text: [1] -
Roth, P. M. C.; Fletcher, S. P. Org. Lett. 2015, 17, 912. doi:10.1021/acs.orglett.5b00021
Return to citation in text: [1] -
Rideau, E.; Mäsing, F.; Fletcher, S. P. Synthesis 2015, 47, 2217. doi:10.1055/s-0034-1379928
Return to citation in text: [1] -
Caprioglio, D.; Fletcher, S. P. Chem. Commun. 2015, 51, 14866. doi:10.1039/C5CC05805H
Return to citation in text: [1] -
Maksymowicz, R. M.; Bissette, A. J.; Fletcher, S. P. Chem. – Eur. J. 2015, 21, 5668. doi:10.1002/chem.201405855
Return to citation in text: [1] -
You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi:10.1038/nature14089
Return to citation in text: [1] [2] [3] [4] [5] -
Sidera, M.; Fletcher, S. P. Chem. Commun. 2015, 51, 5044. doi:10.1039/C5CC00421G
Return to citation in text: [1] -
Morken, J. P.; Didiuk, M. T.; Visser, M. S.; Hoveyda, A. H. J. Am. Chem. Soc. 1994, 116, 3123. doi:10.1021/ja00086a052
Return to citation in text: [1] -
Fu, G. C.; Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc. 1993, 115, 9856. doi:10.1021/ja00074a085
Return to citation in text: [1] -
Clark, J. S. Chem. Commun. 2006, 3571. doi:10.1039/b601839d
Return to citation in text: [1] -
Aggarwal, V. K.; Staubitz, A. C.; Owen, M. Org. Process Res. Dev. 2006, 10, 64. doi:10.1021/op058013q
Return to citation in text: [1] -
Weissman, S. A.; Anderson, N. G. Org. Process Res. Dev. 2015, 19, 1605. doi:10.1021/op500169m
Return to citation in text: [1] -
Owen, M. R.; Luscombe, C.; Lai-Wah; Godbert, S.; Crookes, D. L.; Emiabata-Smith, D. Org. Process Res. Dev. 2001, 5, 308. doi:10.1021/op000024q
Return to citation in text: [1] -
Chen, J. J.; Nugent, T. C.; Lu, C. V.; Kondapally, S.; Giannousis, P.; Wang, Y.; Wilmot, J. T. Org. Process Res. Dev. 2003, 7, 313. doi:10.1021/op034018g
Return to citation in text: [1] -
Denmark, S. E.; Butler, C. R. J. Am. Chem. Soc. 2008, 130, 3690. doi:10.1021/ja7100888
Return to citation in text: [1] -
Arnold, K.; Batsanov, A. S.; Davies, B.; Whiting, A. Green Chem. 2008, 10, 124. doi:10.1039/B712008G
Return to citation in text: [1] -
Peñafiel, I.; Pastor, I. M.; Yus, M. Eur. J. Org. Chem. 2013, 1479. doi:10.1002/ejoc.201201066
Return to citation in text: [1] -
Harper, K. C.; Sigman, M. S. J. Org. Chem. 2013, 78, 2813. doi:10.1021/jo4002239
Return to citation in text: [1] -
Yoshikai, N.; Nakamura, E. Chem. Rev. 2012, 112, 2339. doi:10.1021/cr200241f
Return to citation in text: [1] -
Geurts, K.; Fletcher, S. P.; van Zijl, A. W.; Minnaard, A. J.; Feringa, B. L. Pure Appl. Chem. 2008, 80, 1025. doi:10.1351/pac200880051025
Return to citation in text: [1] -
Alexakis, A.; Bäckvall, J. E.; Krause, N.; Pàmies, O.; Diéguez, M. Chem. Rev. 2008, 108, 2796. doi:10.1021/cr0683515
Return to citation in text: [1] -
Harutyunyan, S. R.; den Hartog, T.; Geurts, K.; Minnaard, A. J.; Feringa, B. L. Chem. Rev. 2008, 108, 2824. doi:10.1021/cr068424k
Return to citation in text: [1] -
Alexakis, A.; Krause, N.; Woodward, S. Copper-Catalyzed Asymmetric Synthesis; Wiley-VCH: Weinheim, 2014. doi:10.1002/9783527664573
Return to citation in text: [1] -
Maougal, E.; Dalençon, S.; Pearson-Long, M. S. M.; Mathé-Allainmat, M.; Lebreton, J.; Legoupy, S. Synthesis 2014, 46, 3268. doi:10.1055/s-0034-1378663
Return to citation in text: [1]
| 1. | Faber, K. Chem. – Eur. J. 2001, 7, 5004. doi:10.1002/1521-3765(20011203)7:23<5004::AID-CHEM5004>3.0.CO;2-X |
| 2. | Vedejs, E.; Jure, M. Angew. Chem., Int. Ed. 2005, 44, 3974. doi:10.1002/anie.200460842 |
| 3. | Mohr, J. T.; Ebner, D. C.; Stoltz, B. M. Org. Biomol. Chem. 2007, 5, 3571. doi:10.1039/b711159m |
| 4. | Miller, L. C.; Sarpong, R. Chem. Soc. Rev. 2011, 40, 4550. doi:10.1039/c1cs15069c |
| 19. | Maksymowicz, R. M.; Roth, P. M. C.; Fletcher, S. P. Nat. Chem. 2012, 4, 649. doi:10.1038/nchem.1394 |
| 20. | Sidera, M.; Roth, P. M. C.; Maksymowicz, R. M.; Fletcher, S. P. Angew. Chem., Int. Ed. 2013, 52, 7995. doi:10.1002/anie.201303202 |
| 21. | Maciver, E. E.; Maksymowicz, R. M.; Wilkinson, N.; Roth, P. M. C.; Fletcher, S. P. Org. Lett. 2014, 16, 3288. doi:10.1021/ol501292x |
| 22. | Roth, P. M. C.; Fletcher, S. P. Org. Lett. 2015, 17, 912. doi:10.1021/acs.orglett.5b00021 |
| 23. | Rideau, E.; Mäsing, F.; Fletcher, S. P. Synthesis 2015, 47, 2217. doi:10.1055/s-0034-1379928 |
| 24. | Caprioglio, D.; Fletcher, S. P. Chem. Commun. 2015, 51, 14866. doi:10.1039/C5CC05805H |
| 25. | Maksymowicz, R. M.; Bissette, A. J.; Fletcher, S. P. Chem. – Eur. J. 2015, 21, 5668. doi:10.1002/chem.201405855 |
| 39. | Yoshikai, N.; Nakamura, E. Chem. Rev. 2012, 112, 2339. doi:10.1021/cr200241f |
| 40. | Geurts, K.; Fletcher, S. P.; van Zijl, A. W.; Minnaard, A. J.; Feringa, B. L. Pure Appl. Chem. 2008, 80, 1025. doi:10.1351/pac200880051025 |
| 41. | Alexakis, A.; Bäckvall, J. E.; Krause, N.; Pàmies, O.; Diéguez, M. Chem. Rev. 2008, 108, 2796. doi:10.1021/cr0683515 |
| 42. | Harutyunyan, S. R.; den Hartog, T.; Geurts, K.; Minnaard, A. J.; Feringa, B. L. Chem. Rev. 2008, 108, 2824. doi:10.1021/cr068424k |
| 43. | Alexakis, A.; Krause, N.; Woodward, S. Copper-Catalyzed Asymmetric Synthesis; Wiley-VCH: Weinheim, 2014. doi:10.1002/9783527664573 |
| 16. | Ito, H.; Kunii, S.; Sawamura, M. Nat. Chem. 2010, 2, 972. doi:10.1038/nchem.801 |
| 17. | Langlois, J.-B.; Emery, D.; Mareda, J.; Alexakis, A. Chem. Sci. 2012, 3, 1062. doi:10.1039/C2SC00868H |
| 18. | Delvos, L. B.; Oestreich, M. Synthesis 2015, 47, 924. doi:10.1055/s-0034-1380157 |
| 44. | Maougal, E.; Dalençon, S.; Pearson-Long, M. S. M.; Mathé-Allainmat, M.; Lebreton, J.; Legoupy, S. Synthesis 2014, 46, 3268. doi:10.1055/s-0034-1378663 |
| 8. | Huerta, F. F.; Minidis, A. B. E.; Bäckvall, J.-E. Chem. Soc. Rev. 2001, 30, 321. doi:10.1039/b105464n |
| 9. | Trost, B. M.; Fandrick, D. R. Aldrichimica Acta 2007, 40, 59. |
| 10. | Norinder, J.; Bäckvall, J.-E. Chem. – Eur. J. 2007, 13, 4094. doi:10.1002/chem.200601684 |
| 11. | Langlois, J.-B.; Alexakis, A. Chem. Commun. 2009, 3868. doi:10.1039/b907722g |
| 12. | Langlois, J.-B.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 447. doi:10.1002/adsc.200900790 |
| 13. | Katcher, M. H.; Doyle, A. G. J. Am. Chem. Soc. 2010, 132, 17402. doi:10.1021/ja109120n |
| 14. | Hamilton, J. Y.; Hauser, N.; Sarlah, D.; Carreira, E. M. Angew. Chem., Int. Ed. 2014, 53, 10759. doi:10.1002/anie.201406077 |
| 15. | Misale, A.; Niyomchon, S.; Luparia, M.; Maulide, N. Angew. Chem., Int. Ed. 2014, 53, 7068. doi:10.1002/anie.201309074 |
| 31. | Aggarwal, V. K.; Staubitz, A. C.; Owen, M. Org. Process Res. Dev. 2006, 10, 64. doi:10.1021/op058013q |
| 32. | Weissman, S. A.; Anderson, N. G. Org. Process Res. Dev. 2015, 19, 1605. doi:10.1021/op500169m |
| 33. | Owen, M. R.; Luscombe, C.; Lai-Wah; Godbert, S.; Crookes, D. L.; Emiabata-Smith, D. Org. Process Res. Dev. 2001, 5, 308. doi:10.1021/op000024q |
| 34. | Chen, J. J.; Nugent, T. C.; Lu, C. V.; Kondapally, S.; Giannousis, P.; Wang, Y.; Wilmot, J. T. Org. Process Res. Dev. 2003, 7, 313. doi:10.1021/op034018g |
| 35. | Denmark, S. E.; Butler, C. R. J. Am. Chem. Soc. 2008, 130, 3690. doi:10.1021/ja7100888 |
| 36. | Arnold, K.; Batsanov, A. S.; Davies, B.; Whiting, A. Green Chem. 2008, 10, 124. doi:10.1039/B712008G |
| 37. | Peñafiel, I.; Pastor, I. M.; Yus, M. Eur. J. Org. Chem. 2013, 1479. doi:10.1002/ejoc.201201066 |
| 5. | Trost, B. M.; Bunt, R. C. J. Am. Chem. Soc. 1994, 116, 4089. doi:10.1021/ja00088a059 |
| 6. | Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395. doi:10.1021/cr9409804 |
| 7. | Lu, Z.; Ma, S. Angew. Chem., Int. Ed. 2008, 47, 258. doi:10.1002/anie.200605113 |
| 38. | Harper, K. C.; Sigman, M. S. J. Org. Chem. 2013, 78, 2813. doi:10.1021/jo4002239 |
| 29. | Fu, G. C.; Nguyen, S. T.; Grubbs, R. H. J. Am. Chem. Soc. 1993, 115, 9856. doi:10.1021/ja00074a085 |
| 30. | Clark, J. S. Chem. Commun. 2006, 3571. doi:10.1039/b601839d |
| 26. | You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi:10.1038/nature14089 |
| 28. | Morken, J. P.; Didiuk, M. T.; Visser, M. S.; Hoveyda, A. H. J. Am. Chem. Soc. 1994, 116, 3123. doi:10.1021/ja00086a052 |
| 26. | You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi:10.1038/nature14089 |
| 26. | You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi:10.1038/nature14089 |
| 26. | You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi:10.1038/nature14089 |
| 27. | Sidera, M.; Fletcher, S. P. Chem. Commun. 2015, 51, 5044. doi:10.1039/C5CC00421G |
| 26. | You, H.; Rideau, E.; Sidera, M.; Fletcher, S. P. Nature 2015, 517, 351. doi:10.1038/nature14089 |
© 2015 Rideau and Fletcher; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)