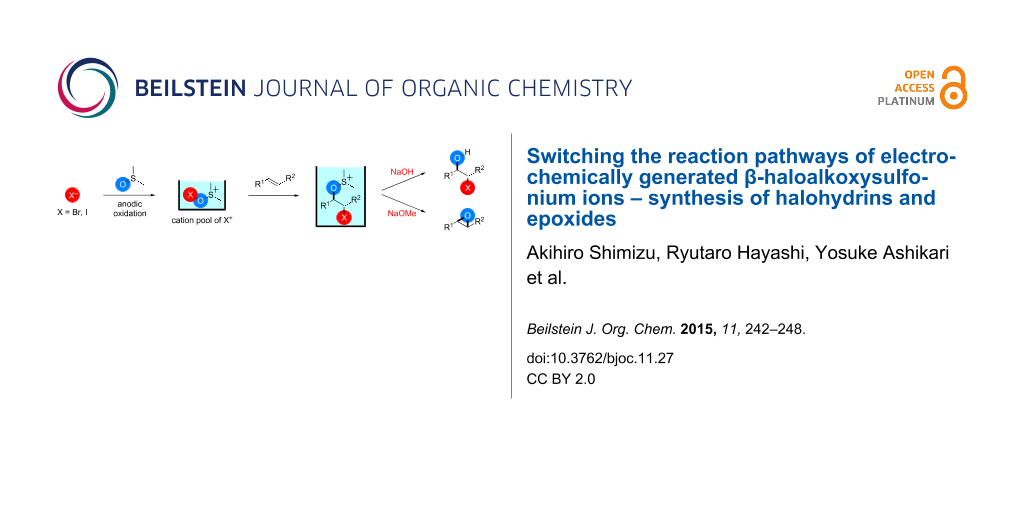
Abstract
β-Haloalkoxysulfonium ions generated by the reaction of electrogenerated Br+ and I+ ions stabilized by dimethyl sulfoxide (DMSO) reacted with sodium hydroxide and sodium methoxide to give the corresponding halohydrins and epoxides, respectively, whereas the treatment with triethylamine gave α-halocarbonyl compounds.

Graphical Abstract
Introduction
Alkene difunctionalization through three-membered ring halonium ion intermediates [1] is an important transformation in organic synthesis. Usually the halonium ions such as bromonium or iodonium ions are generated by the reaction of alkenes with Br2 and I2 [2]. However, the most straightforward method is the reaction of alkenes with halogen cations such as Br+ and I+. The I+ cation pool exists as reported by Filimonov et al. [3], although the used solvent is concentrated sulfuric acid which is therefore not compatible with most organic compounds.
Electrochemical oxidation [4-11] is a potent technique to generate and accumulate highly reactive cationic species in solution (the “cation pool” method) [12-17]. Although halogen cations are too unstable to accumulate in solution as “cation pools”, halogen cations stabilized by an appropriate stabilizing agent that coordinates the cations can be accumulated in the solution. For example, “I+” cations stabilized by acetonitrile (CH3CN) [18-20] or by trimethyl orthoformate (TMOF) [21] were reported in the literature. Recently, we reported that dimethyl sulfoxide (DMSO) can also be used to effectively stabilize halogen cations (Scheme 1) [22].
![[1860-5397-11-27-i1]](/bjoc/content/inline/1860-5397-11-27-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of halohydrins and epoxides through β-haloalkoxysulfonium ions generated by the reaction of alkenes with DMSO-stabilized halogen cations.
Scheme 1: Synthesis of halohydrins and epoxides through β-haloalkoxysulfonium ions generated by the reaction ...
The pools of stabilized halogen cations enable alkene difunctionalization. We previously reported that the reaction of alkenes with DMSO-stabilized halogen cations such as Br+ and I+ gave β-haloalkoxysulfonium ions and their subsequent treatment with triethylamine gave α-halocarbonyl compounds through Swern–Moffatt-type oxidation [23-27]. Recently reaction integration [28-31] has received significant research interest because it enhances the power and speed of organic syntheses and this is an example of integration of an electrochemical reaction and a chemical reaction using a reactive intermediate. Herein, we report that the reaction pathways of β-haloalkoxysulfonium ions can be switched to give different products by changing the base, thus expanding the utility of the present method. The treatment of β-haloalkoxysulfonium ions 3-X with sodium hydroxide gave the corresponding halohydrins 5-X, while the treatment with sodium methoxide gave epoxides 6 (Scheme 1).
Results and Discussion
Reactions of β-bromoalkoxysulfonium ions generated from (Z)-5-decene
We first examined the reactions of β-bromoalkoxysulfonium ion 3a-Br generated by the reaction of (Z)-5-decene (2a) with Br+/DMSO (1-Br) [21] (Scheme 1, X = Br). Bu4NBr in DMSO/CH2Cl2 (1:9 v/v) was electrochemically oxidized at −78 °C in a divided cell using Bu4NBF4 as a supporting electrolyte until 2.1 F/mol of electricity was applied. After addition of 2a to the solution, the mixture was stirred at 0 °C to give 3a-Br, which was characterized by NMR spectroscopy [22]. The treatment of 3a-Br with triethylamine gave α-bromoketone 4a-Br in 83% yield [22]. However, the treatment of 3a-Br with NaOH gave bromohydrin 5a-Br in 89% yield as shown in Table 1. These phenomena can be explained as follows: Due to the steric repulsion, triethylamine cannot attack the sulfur atom in 3a-Br and acts as base to abstract a proton attached to the carbon adjacent to the sulfur. The formed carbanion part of the resulting sulfur ylide abstracts a proton attached to the carbon adjacent to the oxygen to give α-bromoketone 4a-Br by the Swern–Moffatt-type oxidation mechanism [23-27]. On the other hand, the hydroxide ion attacks the sulfur atom in 3a-Br and cleaves the S–O bond to give the alkoxide ion, which is protonated by water to give bromohydrin 5a-Br (Scheme 2). The stereochemistry determined by NMR (5a-Br was synthesized using NBS according to the literature; see Supporting Information File 1) indicated that the addition of Br+ and DMSO across the C–C double bond was anti-selective, which is consistent with the results reported previously [22].
Table 1: Reaction of 3a-X (X = Br, I) with different bases.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-27-i4.svg?max-width=637&scale=1.0)
|
|||||||
| % Yield of productb | |||||||
|---|---|---|---|---|---|---|---|
| X = Br | X = I | ||||||
| Base | 4a-Br | 5a-Br | 6a | 4a-Ir | 5a-I | 6a | |
| Et3N/CH2Cl2 | 83 | ND | ND | 85 | ND | 1 | |
| NaOH/H2O | ND | 89 | 2 | ND | 84 | 1 | |
| NaOMe/MeOH | ND | ND | 95 | ND | ND | 96 | |
aThe electrolysis was carried out using 1.3 equiv of Bu4NBr or Bu4NI (based on the alkene which was added after electrolysis) with 2.1 F/mol of electricity based on Bu4NBr or Bu4NI. bYields were determined by GC.
![[1860-5397-11-27-i2]](/bjoc/content/inline/1860-5397-11-27-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed reaction mechanisms for the syntheses of bromohydrin 5a-Br and epoxide 6a.
Scheme 2: Proposed reaction mechanisms for the syntheses of bromohydrin 5a-Br and epoxide 6a.
Treatment of 3a-Br with NaOMe resulted in a different product, namely epoxide 6a in 95% yield. In this case, the methoxide ion attacks the sulfur atom and cleaves the S–O bond under formation of an alkoxide ion. The latter intramolecularly attacks the carbon atom bearing the bromine substituent to give epoxide 6a (Scheme 2). Presumably, the protonation of the alkoxide ion with MeOH is slower than the intramolecular nucleophilic attack. We could not exclude the possibility that a protonated DMSO molecule presumably generated by the reaction of 3a-Br with the hydroxide ion protonates the alkoxide ion to give 5a-Br, while a methylated DMSO molecule presumably generated by the reaction of 3a-Br with the methoxide ion cannot protonate the alkoxide ion, which converts to 6a. The stereochemistry determined by NMR [32] is consistent with a mechanism involving the back-side attack of the alkoxide ion to form epoxide 6a.
Reactions of β-iodoalkoxysulfonium ions generated from (Z)-5-decene
We next examined the reactions of β-iodoalkoxysulfonium ion 3a-I generated by the reaction of (Z)-5-decene (2a) with I+/DMSO (1-I) cation pool [22] (Scheme 1, X = I). Bu4NI in DMSO/CH2Cl2 (1:9 v/v) was electrochemically oxidized at −78 °C in a divided cell using Bu4NBF4 as a supporting electrolyte until 2.1 F/mol of electricity was applied. After addition of 2a to the solution, the mixture was stirred at 0 °C to give 3a-I, which was characterized by NMR spectroscopy [22]. The treatment of 3a-I with triethylamine gave α-iodoketone 4a-I in 85% yield as we reported previously [22]. However, the treatment of 3a-I with NaOH and NaOMe gave iodohydrin 5a-I in 84% yield and epoxide 6a in 96% yield, respectively (Table 1). The stereochemistry as determined by NMR (5a-I was synthesized using I2 and H2O2; see Supporting Information File 1) indicated that the addition of I+ and DMSO across the C–C double bond was anti-selective as anticipated.
Synthesis of halohydrins and epoxides from various alkenes
The present method was successfully applied to the synthesis of halohydrins and epoxides from various alkenes. The reactions of alkenes with 1-X followed by the treatment with NaOH gave the corresponding halohydrins as shown in Table 2. The reactions of E and Z isomers of 1-phenyl-1-propene (2d) with 1-Br gave 5d-Br and 5d’-Br, respectively (Table 2, entries 7 and 9), indicating the anti-addition of Br+ and DMSO across the C–C double bond. The reaction with 1-I also gave the anti-addition products (Table 2, entries 8 and 10). Therefore, the reaction is stereospecific, and the stereochemistry is consistent with the proposed reaction mechanism (Scheme 2). The addition of Br+ or I+ and DMSO to unsymmetrically substituted olefins 2c and 2d regioselectively gave bromohydrins as single regioisomers (Table 2, entries 5–10). The regioselectivity of the products can be explained by the stability of carbocations (benzyl > secondary > primary). In the case of terminal alkene 2c, Br and I were introduced to a primary carbon atom, whereas OH was introduced to a secondary carbon atom. In the case of styrene derivative 2d, Br and I were introduced to a secondary carbon, whereas OH was introduced to the benzyl carbon. DMSO seems to attack the more positively charged carbon of the three-membered ring bromonium ion or iodonium ion.
Table 2: Synthesis of halohydrins by the reaction of 1-X with alkenes followed by the treatment with NaOH.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-27-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Alkene | Product | Yield (%)b |
|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-27-i6.svg?max-width=637&scale=1.0)
2a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-27-i7.svg?max-width=637&scale=1.0)
5a-Br, 5a-I |
5a-Br: 87 |
| 2 | 5a-I: 84c | ||
| 3 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-27-i8.svg?max-width=637&scale=1.0)
2b (Z:E = 72:28) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-27-i9.svg?max-width=637&scale=1.0)
5b-Br, 5b-I |
5b-Br: 74
(trans:cis = 79:21) |
| 4 |
5b-I: 94
(trans:cis = 71:29) |
||
| 5 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-27-i10.svg?max-width=637&scale=1.0)
2c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-27-i11.svg?max-width=637&scale=1.0)
5c-Br, 5c-I |
5c-Br: 57 |
| 6 | 5c-I: 53 | ||
| 7 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-27-i12.svg?max-width=637&scale=1.0)
(E)-2d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-27-i13.svg?max-width=637&scale=1.0)
5d-Br, 5d-I |
5d-Br: 73 |
| 8 | 5d-I: 35 | ||
| 9 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-27-i14.svg?max-width=637&scale=1.0)
(Z)-2d |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-27-i15.svg?max-width=637&scale=1.0)
5d’-Br, 5d’-I |
5d’-Br: 75 |
| 10 | 5d’-I: 51 | ||
aThe electrolysis of Bu4NBr and Bu4NI was carried out using 1.3 equiv of Bu4NX (based on the alkene which was added after the electrolysis) with 2.1 F/mol of electricity based on Bu4NX. bIsolated yield. cYield was determined by GC.
The reaction of 1-X with alkenes followed by the treatment with NaOMe gave the corresponding epoxides as shown in Table 3. Alkenes having an alkoxycarbonyl group gave the corresponding epoxides in moderate yields (Table 3, entries 11–14). Diene 2f reacted with 1-Br and 1-I to give monoepoxide 6f in moderate yields (Table 3, entries 13 and 14). Interestingly, 2g reacted with 1-Br to give 6g but not with 1-I (Table 3, entries 15 and 16), although the reason is not clear at present. The facial selectivity of the reaction is the opposite to that of the epoxidation using conventional reagents such as m-chloroperoxybenzoic acid (mCPBA) which epoxidizes alkenes from the less hindered face [33,34]. In this reaction, Br+ adds to the C–C double bond of 2g from the less hindered face to form the corresponding three-membered ring bromonium ion intermediate. Subsequently, DMSO attacks the bromonium ion from the more hindered face to form the corresponding β-haloalkoxysulfonium ion. The treatment of the β-haloalkoxysulfonium ion with NaOMe cleaves the O–S bond to generate the alkoxide ion, which attacks the carbon atom bearing bromine to give epoxide 6g. Therefore, the installation of the oxygen atom takes place from the more hindered face.
Table 3: Synthesis of epoxides by the reaction of 1-X with alkenes followed by the treatment with NaOMe.a
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-27-i16.svg?max-width=637&scale=1.0)
|
||||
| Entry | Alkene | Product | X | Yield (%)b |
|---|---|---|---|---|
| 1 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-27-i17.svg?max-width=637&scale=1.0)
2a |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-27-i18.svg?max-width=637&scale=1.0)
6a |
Br | 95c |
| 2 | I | 96c | ||
| 3 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-27-i19.svg?max-width=637&scale=1.0)
2b (Z:E = 72:28) |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-27-i20.svg?max-width=637&scale=1.0)
6b |
Br |
68
(cis:trans = 74:26) |
| 4 | I |
89
(cis:trans = 74:26) |
||
| 5 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-27-i21.svg?max-width=637&scale=1.0)
2c |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-27-i22.svg?max-width=637&scale=1.0)
6c |
Br | 73c |
| 6 | I | 86c | ||
| 7 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-27-i23.svg?max-width=637&scale=1.0)
(E)-2d |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-27-i24.svg?max-width=637&scale=1.0)
6d |
Br | 53 |
| 8 | I | 38d | ||
| 9 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-27-i25.svg?max-width=637&scale=1.0)
(Z)-2d |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-27-i26.svg?max-width=637&scale=1.0)
6d’ |
Br | 60 |
| 10 | I | 67d | ||
| 11 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-27-i27.svg?max-width=637&scale=1.0)
2e |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-27-i28.svg?max-width=637&scale=1.0)
6e |
Br | 52e |
| 12 | I | 57e | ||
| 13 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-27-i29.svg?max-width=637&scale=1.0)
2f |
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-27-i30.svg?max-width=637&scale=1.0)
6f |
Br | 49e |
| 14 | I | 47e | ||
| 15 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-27-i31.svg?max-width=637&scale=1.0)
2g |
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-27-i32.svg?max-width=637&scale=1.0)
6g |
Br | 69 |
| 16 | I | 0 | ||
aThe electrolysis was carried out using 1.3 equiv of Bu4NBr or Bu4NI (based on the alkene which was added after electrolysis) with 2.1 F/mol of electricity based on Bu4NBr or Bu4NI. bIsolated yield. cYield was determined by GC. d2.0 Equiv of Bu4NI was used. eReacted with 2.5 equiv of NaOMe for 2 h.
Reaction mechanism
To confirm the mechanism shown in Scheme 2, the experiment was repeated using 18O-labeled DMSO (96% 18O)/CH2Cl2 (1:50 v/v). As outlined in Scheme 3, epoxide 6c containing 18O (94% 18O) was obtained in 81% yield, indicating that the oxygen atom in the product originated from DMSO. Since 18O-labeled DMSO can be easily obtained from H218O [35], the present transformation serves as a convenient method for synthesizing 18O-labeled epoxides, that can be used for various mechanistic and biological studies.
![[1860-5397-11-27-i3]](/bjoc/content/inline/1860-5397-11-27-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanistic study using 18O-DMSO.
Scheme 3: Mechanistic study using 18O-DMSO.
Conclusion
In conclusion, we found that the reaction pathways of β-haloalkoxysulfonium ions generated by the reaction of electrogenerated Br+ and I+ stabilized by dimethyl sulfoxide (DMSO) can be switched by changing the nature of the base. The present transformation serves as stereospecific route to halohydrins and epoxides from alkenes. The method is also useful for synthesizing 18O-labeled epoxides.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 5.5 MB | Download |
References
-
Olah, G. A.; Laali, K. K.; Wang, Q.; Prakash, G. K. S. Onium ions; Wiley: New York, 1998; pp 246–268.
Return to citation in text: [1] -
Rodriguez, J.; Dulcère, J.-P. Synthesis 1993, 1177–1205. doi:10.1055/s-1993-26022
Return to citation in text: [1] -
Chaikovski, V. K.; Kharlova, T. S.; Filimonov, V. D.; Saryucheva, T. A. Synthesis 1999, 748–750. doi:10.1055/s-1999-3475
Return to citation in text: [1] -
Moeller, K. D. Tetrahedron 2000, 56, 9527–9554. doi:10.1016/S0040-4020(00)00840-1
Return to citation in text: [1] -
Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/b512308a
Return to citation in text: [1] -
Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843
Return to citation in text: [1] -
Kirste, A.; Elsler, B.; Schnakenburg, G.; Waldvogel, S. R. J. Am. Chem. Soc. 2012, 134, 3571–3576. doi:10.1021/ja211005g
Return to citation in text: [1] -
Finney, E. E.; Ogawa, K. A.; Boydston, A. J. J. Am. Chem. Soc. 2012, 134, 12374–12377. doi:10.1021/ja304716r
Return to citation in text: [1] -
Sumi, K.; Saitoh, T.; Natsui, K.; Yamamoto, T.; Atobe, M.; Einaga, Y.; Nishiyama, S. Angew. Chem., Int. Ed. 2012, 51, 5443–5446. doi:10.1002/anie.201200878
Return to citation in text: [1] -
Morofuji, T.; Shimizu, A.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 5000–5003. doi:10.1021/ja402083e
Return to citation in text: [1] -
Yamaguchi, Y.; Okada, Y.; Chiba, K. J. Org. Chem. 2013, 78, 2626–2638. doi:10.1021/jo3028246
Return to citation in text: [1] -
Yoshida, J.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546–9549. doi:10.1021/ja9920112
Return to citation in text: [1] -
Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J. J. Am. Chem. Soc. 2000, 122, 10244–10245. doi:10.1021/ja002123p
Return to citation in text: [1] -
Yoshida, J.; Suga, S. Chem. – Eur. J. 2002, 8, 2650–2658. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S
Return to citation in text: [1] -
Suzuki, S.; Matsumoto, K.; Kawamura, K.; Suga, S.; Yoshida, J. Org. Lett. 2004, 6, 3755–3758. doi:10.1021/ol048524h
Return to citation in text: [1] -
Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J. J. Am. Chem. Soc. 2006, 128, 7710–7711. doi:10.1021/ja0625778
Return to citation in text: [1] -
Matsumoto, K.; Sanada, T.; Shimazaki, H.; Shimada, K.; Hagiwara, S.; Fujie, S.; Ashikari, Y.; Suga, S.; Kashimura, S.; Yoshida, J. Asian J. Org. Chem. 2013, 2, 325–329. doi:10.1002/ajoc.201300017
Return to citation in text: [1] -
Miller, L. L.; Kujawa, E. P.; Campbell, C. B. J. Am. Chem. Soc. 1970, 92, 2821–2825. doi:10.1021/ja00712a036
Return to citation in text: [1] -
Miller, L. L.; Watkins, B. F. J. Am. Chem. Soc. 1976, 98, 1515–1519. doi:10.1021/ja00422a039
Return to citation in text: [1] -
Midorikawa, K.; Suga, S.; Yoshida, J. Chem. Commun. 2006, 3794–3796. doi:10.1039/b607284d
Return to citation in text: [1] -
Shono, T.; Matsumura, Y.; Katoh, S.; Ikeda, K.; Kamada, T. Tetrahedron Lett. 1989, 30, 1649–1650. doi:10.1016/S0040-4039(00)99543-1
Return to citation in text: [1] [2] -
Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Kornblum, N.; Powers, J. W.; Anderson, G. J.; Jones, W. J.; Larson, H. O.; Levand, O.; Weaver, W. M. J. Am. Chem. Soc. 1957, 79, 6562. doi:10.1021/ja01581a057
Return to citation in text: [1] [2] -
Mancuso, A. J.; Swern, D. Synthesis 1981, 165–185. doi:10.1055/s-1981-29377
Return to citation in text: [1] [2] -
Phan, T. B.; Nolte, C.; Kobayashi, S.; Ofial, A. R.; Mayr, H. J. Am. Chem. Soc. 2009, 131, 11392–11401. doi:10.1021/ja903207b
Return to citation in text: [1] [2] -
Ashikari, Y.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2011, 133, 11840–11843. doi:10.1021/ja202880n
Return to citation in text: [1] [2] -
Ashikari, Y.; Nokami, T.; Yoshida, J. Org. Biomol. Chem. 2013, 11, 3322–3331. doi:10.1039/c3ob40315g
Return to citation in text: [1] [2] -
Schmidt, B. Pure Appl. Chem. 2009, 78, 469–476. doi:10.1351/pac200678020469
Return to citation in text: [1] -
Enders, D.; Hüttl, M. R. M.; Grondal, C.; Raabe, G. Nature 2006, 441, 861–863. doi:10.1038/nature04820
Return to citation in text: [1] -
Yoshida, J.; Saito, K.; Nokami, T.; Nagaki, A. Synlett 2011, 1189–1194. doi:10.1055/s-0030-1259946
Return to citation in text: [1] -
Suga, S.; Yamada, D.; Yoshida, J. Chem. Lett. 2010, 39, 404–406. doi:10.1246/cl.2010.404
Return to citation in text: [1] -
Brimeyer, M. O.; Mehrota, A.; Quici, S.; Nigam, A.; Regen, S. L. J. Org. Chem. 1980, 45, 4254–4255. doi:10.1021/jo01309a047
Return to citation in text: [1] -
Gianini, M.; von Zelewsky, A. Synthesis 1996, 702–706. doi:10.1055/s-1996-4280
Return to citation in text: [1] -
Majetich, G.; Shimkus, J.; Li, Y. Tetrahedron Lett. 2010, 51, 6830–6834. doi:10.1016/j.tetlet.2010.10.068
Return to citation in text: [1] -
Fenselau, A. M.; Moffatt, J. G. J. Am. Chem. Soc. 1966, 88, 1762–1765. doi:10.1021/ja00960a033
Return to citation in text: [1]
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 1. | Olah, G. A.; Laali, K. K.; Wang, Q.; Prakash, G. K. S. Onium ions; Wiley: New York, 1998; pp 246–268. |
| 12. | Yoshida, J.; Suga, S.; Suzuki, S.; Kinomura, N.; Yamamoto, A.; Fujiwara, K. J. Am. Chem. Soc. 1999, 121, 9546–9549. doi:10.1021/ja9920112 |
| 13. | Suga, S.; Suzuki, S.; Yamamoto, A.; Yoshida, J. J. Am. Chem. Soc. 2000, 122, 10244–10245. doi:10.1021/ja002123p |
| 14. | Yoshida, J.; Suga, S. Chem. – Eur. J. 2002, 8, 2650–2658. doi:10.1002/1521-3765(20020617)8:12<2650::AID-CHEM2650>3.0.CO;2-S |
| 15. | Suzuki, S.; Matsumoto, K.; Kawamura, K.; Suga, S.; Yoshida, J. Org. Lett. 2004, 6, 3755–3758. doi:10.1021/ol048524h |
| 16. | Suga, S.; Matsumoto, K.; Ueoka, K.; Yoshida, J. J. Am. Chem. Soc. 2006, 128, 7710–7711. doi:10.1021/ja0625778 |
| 17. | Matsumoto, K.; Sanada, T.; Shimazaki, H.; Shimada, K.; Hagiwara, S.; Fujie, S.; Ashikari, Y.; Suga, S.; Kashimura, S.; Yoshida, J. Asian J. Org. Chem. 2013, 2, 325–329. doi:10.1002/ajoc.201300017 |
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 4. | Moeller, K. D. Tetrahedron 2000, 56, 9527–9554. doi:10.1016/S0040-4020(00)00840-1 |
| 5. | Sperry, J. B.; Wright, D. L. Chem. Soc. Rev. 2006, 35, 605–621. doi:10.1039/b512308a |
| 6. | Yoshida, J.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843 |
| 7. | Kirste, A.; Elsler, B.; Schnakenburg, G.; Waldvogel, S. R. J. Am. Chem. Soc. 2012, 134, 3571–3576. doi:10.1021/ja211005g |
| 8. | Finney, E. E.; Ogawa, K. A.; Boydston, A. J. J. Am. Chem. Soc. 2012, 134, 12374–12377. doi:10.1021/ja304716r |
| 9. | Sumi, K.; Saitoh, T.; Natsui, K.; Yamamoto, T.; Atobe, M.; Einaga, Y.; Nishiyama, S. Angew. Chem., Int. Ed. 2012, 51, 5443–5446. doi:10.1002/anie.201200878 |
| 10. | Morofuji, T.; Shimizu, A.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 5000–5003. doi:10.1021/ja402083e |
| 11. | Yamaguchi, Y.; Okada, Y.; Chiba, K. J. Org. Chem. 2013, 78, 2626–2638. doi:10.1021/jo3028246 |
| 32. | Brimeyer, M. O.; Mehrota, A.; Quici, S.; Nigam, A.; Regen, S. L. J. Org. Chem. 1980, 45, 4254–4255. doi:10.1021/jo01309a047 |
| 3. | Chaikovski, V. K.; Kharlova, T. S.; Filimonov, V. D.; Saryucheva, T. A. Synthesis 1999, 748–750. doi:10.1055/s-1999-3475 |
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 2. | Rodriguez, J.; Dulcère, J.-P. Synthesis 1993, 1177–1205. doi:10.1055/s-1993-26022 |
| 23. | Kornblum, N.; Powers, J. W.; Anderson, G. J.; Jones, W. J.; Larson, H. O.; Levand, O.; Weaver, W. M. J. Am. Chem. Soc. 1957, 79, 6562. doi:10.1021/ja01581a057 |
| 24. | Mancuso, A. J.; Swern, D. Synthesis 1981, 165–185. doi:10.1055/s-1981-29377 |
| 25. | Phan, T. B.; Nolte, C.; Kobayashi, S.; Ofial, A. R.; Mayr, H. J. Am. Chem. Soc. 2009, 131, 11392–11401. doi:10.1021/ja903207b |
| 26. | Ashikari, Y.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2011, 133, 11840–11843. doi:10.1021/ja202880n |
| 27. | Ashikari, Y.; Nokami, T.; Yoshida, J. Org. Biomol. Chem. 2013, 11, 3322–3331. doi:10.1039/c3ob40315g |
| 23. | Kornblum, N.; Powers, J. W.; Anderson, G. J.; Jones, W. J.; Larson, H. O.; Levand, O.; Weaver, W. M. J. Am. Chem. Soc. 1957, 79, 6562. doi:10.1021/ja01581a057 |
| 24. | Mancuso, A. J.; Swern, D. Synthesis 1981, 165–185. doi:10.1055/s-1981-29377 |
| 25. | Phan, T. B.; Nolte, C.; Kobayashi, S.; Ofial, A. R.; Mayr, H. J. Am. Chem. Soc. 2009, 131, 11392–11401. doi:10.1021/ja903207b |
| 26. | Ashikari, Y.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2011, 133, 11840–11843. doi:10.1021/ja202880n |
| 27. | Ashikari, Y.; Nokami, T.; Yoshida, J. Org. Biomol. Chem. 2013, 11, 3322–3331. doi:10.1039/c3ob40315g |
| 21. | Shono, T.; Matsumura, Y.; Katoh, S.; Ikeda, K.; Kamada, T. Tetrahedron Lett. 1989, 30, 1649–1650. doi:10.1016/S0040-4039(00)99543-1 |
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 22. | Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. J. Am. Chem. Soc. 2013, 135, 16070–16073. doi:10.1021/ja4092648 |
| 21. | Shono, T.; Matsumura, Y.; Katoh, S.; Ikeda, K.; Kamada, T. Tetrahedron Lett. 1989, 30, 1649–1650. doi:10.1016/S0040-4039(00)99543-1 |
| 33. | Gianini, M.; von Zelewsky, A. Synthesis 1996, 702–706. doi:10.1055/s-1996-4280 |
| 34. | Majetich, G.; Shimkus, J.; Li, Y. Tetrahedron Lett. 2010, 51, 6830–6834. doi:10.1016/j.tetlet.2010.10.068 |
| 18. | Miller, L. L.; Kujawa, E. P.; Campbell, C. B. J. Am. Chem. Soc. 1970, 92, 2821–2825. doi:10.1021/ja00712a036 |
| 19. | Miller, L. L.; Watkins, B. F. J. Am. Chem. Soc. 1976, 98, 1515–1519. doi:10.1021/ja00422a039 |
| 20. | Midorikawa, K.; Suga, S.; Yoshida, J. Chem. Commun. 2006, 3794–3796. doi:10.1039/b607284d |
| 28. | Schmidt, B. Pure Appl. Chem. 2009, 78, 469–476. doi:10.1351/pac200678020469 |
| 29. | Enders, D.; Hüttl, M. R. M.; Grondal, C.; Raabe, G. Nature 2006, 441, 861–863. doi:10.1038/nature04820 |
| 30. | Yoshida, J.; Saito, K.; Nokami, T.; Nagaki, A. Synlett 2011, 1189–1194. doi:10.1055/s-0030-1259946 |
| 31. | Suga, S.; Yamada, D.; Yoshida, J. Chem. Lett. 2010, 39, 404–406. doi:10.1246/cl.2010.404 |
| 35. | Fenselau, A. M.; Moffatt, J. G. J. Am. Chem. Soc. 1966, 88, 1762–1765. doi:10.1021/ja00960a033 |
© 2015 Shimizu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)