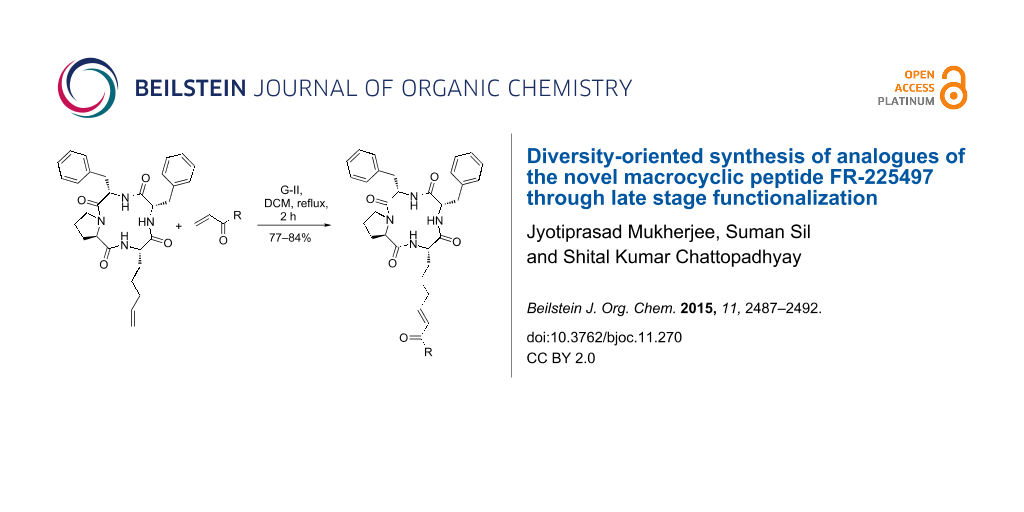
Abstract
A concise synthetic approach to a class of biologically interesting cyclic tetrapeptides is reported which involves a late-stage functionalization of a macrocyclic scaffold through cross metathesis in an attempt to create diversity. The utility of this protocol is demonstrated through the preparation of three structural analogues of the important naturally occurring histone deacetylase inhibitor FR-225497.

Graphical Abstract
Introduction
Diversity-oriented synthesis (DOS) has been established as an important paradigm in drug discovery [1-7]. Although the major focus is on the synthesis of small molecular libraries [8], macrocyclic compounds are currently expanding the medicinal chemistry space [9,10]. Macrocyclic natural products [11] with significant levels of biological activity, and their analogues [12] are therefore receiving attention as targets for diversity-oriented synthesis. A class of cyclic tetra peptides of the general structure 1 (Figure 1) displays important histone deacetylase inhibition property relevant to drug design against a number of diseases ranging from antifungal, antimicrobial to arrest of proliferation of several cell types of epithelial and hematological origin [13]. The compounds possess three distinct regions such as a surface recognition head group, a spacer group and a metal-binding end group which fold to provide suitable conformation necessary for their biological activity [14]. For example, compounds 2 and 4 possess identical head groups and the same spacer region; but differ in their end groups. Although there are excellent synthetic approaches to nearly all of such known targets [15], the development of a simple diversity-oriented approach suitable for modification of the compounds remains desirable.
![[1860-5397-11-270-1]](/bjoc/content/figures/1860-5397-11-270-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of FR-225497 (4) and related cyclic tetrapeptides.
Figure 1: Structure of FR-225497 (4) and related cyclic tetrapeptides.
Results and Discussion
We thought that a cross metathesis reaction [16,17] on a macrocyclic template would constitute a direct approach for grafting the pendant alkyl chain of 1, as demonstrated retrosynthetically in Figure 2. Our initial focus was on the preparation of analogues of the macrocyclic compound 4 through variation of the end groups as it has been shown [18] to have potent activities in the antiproliferation assays with human Jurkat and HT-29 tumor cell lines. Moreover; it also showed excellent immunosuppressive effects suggesting its use as prophylactic agent in organ transplantation. It contains 2-amino-8-oxo-decanoic acid (Aoda) component whereas the trapoxins 2 and 3 contain a 2-amino-8-oxo-9,10-epoxydecanoic acid (Aoe) as the unusual amino acid. The cyclic tetrapeptide scaffold represented by structure III is nearly identical to the compounds 2–4, the difference being in the D-proline/D-pipecolic acid segment. The difference may be responsible for the increased activity of compound 4 compared to 2 or 3 [18].
![[1860-5397-11-270-2]](/bjoc/content/figures/1860-5397-11-270-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Synthetic planning for the design of analogues of 4.
Figure 2: Synthetic planning for the design of analogues of 4.
Accommodating some of these common features, we prepared the macrocyclic template 13 (Scheme 1) through the acyclic tetrapeptide Boc-L-Bhag-L-Phe-L-Phe-D-Pro-OMe (11) (Bhag = bishomoallylglycine) [19]. The latter was accessed by solution phase peptide coupling between L-Phe-D-Pro-OMe (6) (prepared by hydrogenolysis of the corresponding carbamate 5) and L-Cbz-Phe-OH (7) followed by elaboration of the resulting tripeptide 8 via N-terminus deprotection leading to 9 followed by a second peptide coupling of the latter with the known Boc-L-Bhag 10 [20]. The doubly protected tetrapeptide 11 was obtained in an overall yield of 38% in a linear sequence of four steps from 5. The two termini of 11 were then sequentially deprotected involving ester hydrolysis of 11→12 followed by N-deprotection of the latter leading to a crude TFA salt in readiness for a subsequent macrocyclization. The macrocyclization proceeded well under our previously developed conditions [21] to deliver the template 13 in an acceptable yield of 73% over two steps.
![[1860-5397-11-270-i1]](/bjoc/content/inline/1860-5397-11-270-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of the macrocyclic template 13.
Scheme 1: Preparation of the macrocyclic template 13.
We next focused our attention on the crucial cross metathesis reaction of 13. Attempted CM of 13 (Scheme 2) with ethyl vinyl ketone in the presence of Grubbs’ 1st generation catalyst (G-I) proved to be unsuccessful in our hands under a range of conditions. However, changing to the 2nd-generation catalyst [(1,3-bis-(2,4,6-trimethylphenyl)-2-imidazolidinylidene)dichloro(phenylmethylene)(trichlorohexylphosphine)ruthenium, G-II] smoothly effected the desired transformation within a short period of time. Compound 14 was obtained as a single (E)-isomer, as expected. Saturation of the double bond in the latter furnished nor-FR225497 (15) in an overall yield of ≈70% over two steps from template 13.
![[1860-5397-11-270-i2]](/bjoc/content/inline/1860-5397-11-270-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of nor-FR225497 (15).
Scheme 2: Synthesis of nor-FR225497 (15).
We opted for the CM studies of 13 with olefins 17 and 20 (Scheme 3) for possible incorporation of additional oxygenation pattern in the 9-10 positions of the Aoda fragment in the macrocyclic target in view of the occurrence of such functionalities in compounds 2 and 3 and other related ones [22]. The known olefin 17 [23] was prepared by a straightforward oxidation–vinylation–oxidation sequence on 3-(benzyloxy)propanol (16). On the other hand, a vinylation–oxidation sequence on the known aldehyde 18 [24] led to the conjugated olefin 20 through the intermediate 19 (not purified) in good yields.
![[1860-5397-11-270-i3]](/bjoc/content/inline/1860-5397-11-270-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of oxygenated ethyl vinyl ketones.
Scheme 3: Synthesis of oxygenated ethyl vinyl ketones.
Cross metathesis of template 13 with olefin 17 proceeded rapidly under the developed conditions to provide product 21 (Scheme 4) in good yield. Saturation of the double bond in the later with hydrogen also proceeded without events to provide the 10-hydroxylated product 22. Similarly, CM with olefin 20 proceeded well analogously to provide the macrocyclic product 23. Compound 23 was further transformed to the C9,C10-dihydroxylated macrocyclic product 25 via two conventional steps viz. saturation of the double bond leading to 24 followed by an acid-mediated deprotection of the dioxolane ring in the latter. The macrocyclic products 15, 22 and 25 have the nearly same head group and spacer as in the trapoxins and FR-225497 but differ in the end groups. A α-hydroxyketone or a α,ß-dihydroxyketone moiety in the end-region may prove to be beneficial for SAR studies in view of the importance of these functionalities in metal binding.
![[1860-5397-11-270-i4]](/bjoc/content/inline/1860-5397-11-270-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of analogues of FR-225497.
Scheme 4: Synthesis of analogues of FR-225497.
Conclusion
In conclusion, we have been able to demonstrate that cross metathesis reaction on a cyclic tetrapeptide template is a viable strategy for the synthesis of a class of macrocyclic natural product-based analogues. The developed protocol may provide scope for judicious manipulation of the spacer region as well as the metal-binding domain attached to a particular surface recognition part present in the class of these compounds since histone deacetylase activity has been correlated to zinc-binding ability of the 8-oxo moiety in some of such compounds [25]. Moreover, the opportunity of incorporation of other functional groups compatible with CM reaction appears possible. The protocol developed is simple and high yielding. This apparently overlooked approach may therefore find application in the assembly of focused library of simplified agents for possible SAR studies.
Supporting Information
| Supporting Information File 1: Experimental details and analytical data of all new compounds as well as copies of their 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 3.9 MB | Download |
References
-
Burke, M. D.; Schreiber, S. L. Angew. Chem., Int. Ed. 2004, 43, 46. doi:10.1002/anie.200300626
Return to citation in text: [1] -
Wetzel, S.; Bon, R. S.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2011, 50, 10800. doi:10.1002/anie.201007004
Return to citation in text: [1] -
Nadin, A.; Hattotuwagama, C.; Churcher, I. Angew. Chem., Int. Ed. 2012, 51, 1114. doi:10.1002/anie.201105840
Return to citation in text: [1] -
O'Connor, C. J.; Beckmann, H. S. G.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 4444. doi:10.1039/c2cs35023h
Return to citation in text: [1] -
Dandapani, S.; Comer, E.; Duvall, J. R.; Munoz, B. Future Med. Chem. 2012, 4, 2279. doi:10.4155/fmc.12.178
Return to citation in text: [1] -
Barker, A.; Kettle, J. G.; Nowak, T.; Pease, J. E. Drug Discovery Today 2013, 18, 298. doi:10.1016/j.drudis.2012.10.008
Return to citation in text: [1] -
Doveston, R.; Marsden, S.; Nelson, A. Drug Discovery Today 2014, 19, 813. doi:10.1016/j.drudis.2013.11.006
Return to citation in text: [1] -
Eckert, H. Molecules 2012, 17, 1074. doi:10.3390/molecules17011074
Return to citation in text: [1] -
Cordier, C.; Morton, D.; Murrison, S.; Nelson, A.; O'Leary-Steele, C. Nat. Prod. Rep. 2008, 25, 719. doi:10.1039/b706296f
Return to citation in text: [1] -
Ishido-Llobet, A.; Georgiou, K. H.; Galloway, W. R. J. D.; Giacomini, E.; Hansen, M. R.; Méndez-Abt, G.; Tan, Y. S.; Carro, L.; Sore, H. F.; Spring, D. R. Org. Biomol. Chem. 2015, 13, 4570. doi:10.1039/C5OB00371G
Return to citation in text: [1] -
Madsen, C. M.; Clausen, M. H. Eur. J. Org. Chem. 2011, 3107. doi:10.1002/ejoc.201001715
Return to citation in text: [1] -
Maier, M. E. Org. Biomol. Chem. 2015, 13, 5302. doi:10.1039/C5OB00169B
Return to citation in text: [1] -
Zhang, J.; Zhong, Q. Cell. Mol. Life Sci. 2014, 71, 3885. doi:10.1007/s00018-014-1656-6
See for a review.
Return to citation in text: [1] -
Brandl, A.; Heinzel, T.; Krämer, O. H. Biol. Cell 2009, 101, 193. doi:10.1042/BC20080158
Return to citation in text: [1] -
Newkirk, T. L.; Bowers, A. A.; Williams, R. M. Nat. Prod. Rep. 2009, 26, 1293. doi:10.1039/b817886k
Return to citation in text: [1] -
O'Leary, D. J.; O'Neil, G. W. Cross-Metathesis. In Handbook of Metathesis; Grubbs, R. H.; Wenzel, A. G.; O'Leary, D. J.; Khosravi, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp 171–294. doi:10.1002/9783527674107.ch16
Return to citation in text: [1] -
Żukowska, K.; Grela, K. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier; Vol. 5, pp 1257–1301. doi:10.1016/B978-0-08-097742-3.00527-9
Return to citation in text: [1] -
Abe, F.; Hino, M.; Mori, H.; Takase, S.; Yoshimura, S. Inhibitor of Histone deacetylase. WO Patent WO2000/008048 A2, Feb 17, 2000.
Return to citation in text: [1] [2] -
Traoré, M.; Mietton, F.; Maubon, D.; Peuchmaur, M.; Hilário, F. F.; Pereira de Freitas, R.; Bougdour, A.; Curt, A.; Maynadier, M.; Vial, H.; Pelloux, H.; Hakimi, M.-A.; Wong, Y.-S. J. Org. Chem. 2013, 78, 3655. doi:10.1021/jo4001492
See for a recent scaffold-based approach.
Return to citation in text: [1] -
Alcón, M.; Moyano, A.; Pericàs, M. A.; Riera, A. Tetrahedron: Asymmetry 1999, 10, 4639. doi:10.1016/S0957-4166(99)00525-X
Return to citation in text: [1] -
Chattopadhyay, S. K.; Singha, S. K. Synlett 2010, 555. doi:10.1055/s-0029-1219180
Return to citation in text: [1] -
Bolden, J. E.; Peart, M. J.; Johnstone, R. W. Nat. Rev. Drug Discovery 2006, 5, 769. doi:10.1038/nrd2133
Return to citation in text: [1] -
Yamauchi, N.; Natsubori, Y.; Murae, T. Bull. Chem. Soc. Jpn. 2000, 73, 2513. doi:10.1246/bcsj.73.2513
Return to citation in text: [1] -
Chattopadhyay, A.; Mamdapur, V. R. J. Org. Chem. 1995, 60, 585. doi:10.1021/jo00108a020
Return to citation in text: [1] -
Madsen, A. S.; Kirstensen, H. M. E.; Lanz, G.; Olsen, C. A. ChemMedChem 2014, 9, 614. doi:10.1002/cmdc.201300433
Return to citation in text: [1]
| 24. | Chattopadhyay, A.; Mamdapur, V. R. J. Org. Chem. 1995, 60, 585. doi:10.1021/jo00108a020 |
| 25. | Madsen, A. S.; Kirstensen, H. M. E.; Lanz, G.; Olsen, C. A. ChemMedChem 2014, 9, 614. doi:10.1002/cmdc.201300433 |
| 1. | Burke, M. D.; Schreiber, S. L. Angew. Chem., Int. Ed. 2004, 43, 46. doi:10.1002/anie.200300626 |
| 2. | Wetzel, S.; Bon, R. S.; Kumar, K.; Waldmann, H. Angew. Chem., Int. Ed. 2011, 50, 10800. doi:10.1002/anie.201007004 |
| 3. | Nadin, A.; Hattotuwagama, C.; Churcher, I. Angew. Chem., Int. Ed. 2012, 51, 1114. doi:10.1002/anie.201105840 |
| 4. | O'Connor, C. J.; Beckmann, H. S. G.; Spring, D. R. Chem. Soc. Rev. 2012, 41, 4444. doi:10.1039/c2cs35023h |
| 5. | Dandapani, S.; Comer, E.; Duvall, J. R.; Munoz, B. Future Med. Chem. 2012, 4, 2279. doi:10.4155/fmc.12.178 |
| 6. | Barker, A.; Kettle, J. G.; Nowak, T.; Pease, J. E. Drug Discovery Today 2013, 18, 298. doi:10.1016/j.drudis.2012.10.008 |
| 7. | Doveston, R.; Marsden, S.; Nelson, A. Drug Discovery Today 2014, 19, 813. doi:10.1016/j.drudis.2013.11.006 |
| 22. | Bolden, J. E.; Peart, M. J.; Johnstone, R. W. Nat. Rev. Drug Discovery 2006, 5, 769. doi:10.1038/nrd2133 |
| 11. | Madsen, C. M.; Clausen, M. H. Eur. J. Org. Chem. 2011, 3107. doi:10.1002/ejoc.201001715 |
| 23. | Yamauchi, N.; Natsubori, Y.; Murae, T. Bull. Chem. Soc. Jpn. 2000, 73, 2513. doi:10.1246/bcsj.73.2513 |
| 9. | Cordier, C.; Morton, D.; Murrison, S.; Nelson, A.; O'Leary-Steele, C. Nat. Prod. Rep. 2008, 25, 719. doi:10.1039/b706296f |
| 10. | Ishido-Llobet, A.; Georgiou, K. H.; Galloway, W. R. J. D.; Giacomini, E.; Hansen, M. R.; Méndez-Abt, G.; Tan, Y. S.; Carro, L.; Sore, H. F.; Spring, D. R. Org. Biomol. Chem. 2015, 13, 4570. doi:10.1039/C5OB00371G |
| 20. | Alcón, M.; Moyano, A.; Pericàs, M. A.; Riera, A. Tetrahedron: Asymmetry 1999, 10, 4639. doi:10.1016/S0957-4166(99)00525-X |
| 21. | Chattopadhyay, S. K.; Singha, S. K. Synlett 2010, 555. doi:10.1055/s-0029-1219180 |
| 16. | O'Leary, D. J.; O'Neil, G. W. Cross-Metathesis. In Handbook of Metathesis; Grubbs, R. H.; Wenzel, A. G.; O'Leary, D. J.; Khosravi, E., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp 171–294. doi:10.1002/9783527674107.ch16 |
| 17. | Żukowska, K.; Grela, K. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier; Vol. 5, pp 1257–1301. doi:10.1016/B978-0-08-097742-3.00527-9 |
| 18. | Abe, F.; Hino, M.; Mori, H.; Takase, S.; Yoshimura, S. Inhibitor of Histone deacetylase. WO Patent WO2000/008048 A2, Feb 17, 2000. |
| 15. | Newkirk, T. L.; Bowers, A. A.; Williams, R. M. Nat. Prod. Rep. 2009, 26, 1293. doi:10.1039/b817886k |
| 19. |
Traoré, M.; Mietton, F.; Maubon, D.; Peuchmaur, M.; Hilário, F. F.; Pereira de Freitas, R.; Bougdour, A.; Curt, A.; Maynadier, M.; Vial, H.; Pelloux, H.; Hakimi, M.-A.; Wong, Y.-S. J. Org. Chem. 2013, 78, 3655. doi:10.1021/jo4001492
See for a recent scaffold-based approach. |
| 14. | Brandl, A.; Heinzel, T.; Krämer, O. H. Biol. Cell 2009, 101, 193. doi:10.1042/BC20080158 |
| 13. |
Zhang, J.; Zhong, Q. Cell. Mol. Life Sci. 2014, 71, 3885. doi:10.1007/s00018-014-1656-6
See for a review. |
| 18. | Abe, F.; Hino, M.; Mori, H.; Takase, S.; Yoshimura, S. Inhibitor of Histone deacetylase. WO Patent WO2000/008048 A2, Feb 17, 2000. |
© 2015 Mukherjee et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)