Abstract
Guided by computational analysis, herein we report the design, synthesis and evaluation of four novel diazine-based histone deacetylase inhibitors (HDACis). The targets of interest (TOI) are analogues of panobinostat, one of the most potent and versatile HDACi reported. By simply replacing the phenyl core of panobinostat with that of a diazine derivative, docking studies against HDAC2 and HDAC8 revealed that the four analogues exhibit inhibition activities comparable to that of panobinostat. Multistep syntheses afforded the visualized targets TOI1, TOI2, TOI3-rev and TOI4 whose biological evaluation confirmed the strength of HDAC8 inhibition with TOI4 displaying the greatest efficacy at varying concentrations. The results of this study lay the foundation for future design strategies toward more potent HDACis for HDAC8 isozymes and further therapeutic applications for neuroblastoma.
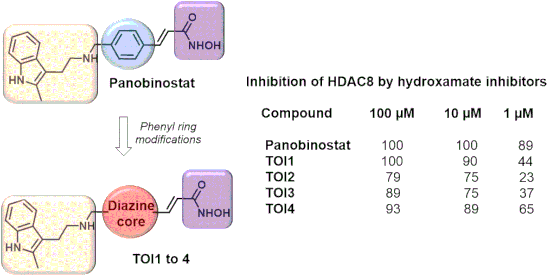
Graphical Abstract
Introduction
One of the most important posttranslational modifications involve acetylation/deacetylation of histone proteins by histone deacetylases (HDACs) [1]. HDACs belong to an important family of enzymes consisting of 18 isozymes. They control protein acetylation, which is a change that occurs after translation. In addition, they regulate gene transcription, cell differentiation, cell cycle progression and apoptosis by targeting both histone and non-histone proteins. The balance between acetylation and deacetylation is pivotal for typical cell function. Abnormal or increased HDAC expression has been reported in several human tumors and cancer cell lines [2]. As such, the development of novel HDAC inhibitors (HDACis) has become a rapidly evolving area where targeted inhibition has emerged in clinical research as a potential therapeutic approach for the treatment of various cancers as well as neurodegenerative disorders and immune related diseases [3-5]. Of specific interests are Class I HDAC isozymes, HDAC2 and HDAC8, which are important targets in cancer models as both are associated with high risk diseases such as prostate cancer and neuroblastoma [6-8]. Compounds such as vorinostat, givinostat and panobinostat have been successfully applied as HDAC inhibitors [3]. Among these drugs, panobinostat (Farydak, Novartis) an FDA approved drug, has been recognized as a pan-deacetylase inhibitor [9,10]. As a hydroxamic acid pan-HDACi, it is zinc-dependent, capable of binding in a bidentate fashion to the zinc-containing catalytic domain of the HDACs, and classified as highly potent amongst traditional HDACis [11]. According to previous reports, panobinostat not only induces apoptosis in cells, but also stimulates cell growth inhibition, and cell-cycle arrest in a time- and dose-dependent manner. Thus, panobinostat has demonstrated high therapeutic potential in anticancer efforts. Although panobinostat offers a versatile approach for the inhibition of cancer cell growth and survival, a lack of selectivity and bioavailability can cause negative molecular and clinical effects, specifically in combination therapies.
Despite advances in Class I HDAC inhibition, there remains an obvious need to develop compounds having better therapeutic properties as a single-agent therapeutic drug. Our recent research based on computational studies indicated heterocyclic cores as suitable surrogates for the central core of the hydroxamate derivative, panobinostat [12]. It should be noted that TOI3-rev in this article is different from TOI3 in the previous reporting [12]. Here, the 1,2-diazole ring has been replaced with that of a pyrimidine core. Given the abundance of literature regarding analogues having modifications of the indole amine unit and vinylogous hydroxamic moieties [13-15], the non-availability of the central core modification stimulated our interest toward altering the central core to evaluate efficacy. We found particular interest in the replacement of the phenyl ring with diazine cores (pyridazines, pyrimidines and pyrazines) as their docking values were on par with that of the parent molecule, panobinostat (Figure 1). Given that the diazine-containing compounds are considered to be one of the most important classes of heterocycles, their presence in a plethora of pharmacology and drug molecules motivated us to synthesize these analogues and subject them towards biological evaluation [16-19]. In addition, our envisioned dinitrogen heterocycle cores may have increased interactions in the binding pockets, and thus leading to a better therapeutic activity.
![[1860-5397-16-59-1]](/bjoc/content/figures/1860-5397-16-59-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of the target diazine-based surrogates for the central core of panobinostat.
Figure 1: Chemical structures of the target diazine-based surrogates for the central core of panobinostat.
Aiming to provide a strategy to address our global objective of developing single agent therapeutic hydroxamate derivatives, we began the synthesis of four leading HDAC8 diazine-based HDACis: TOI1, TOI2, TOI3-rev and TOI4 (Figure 1). Herein, we provide a summary of the design process followed by an outline of the multistep synthesis and preliminary biological evaluation of each target. HDAC8 was selected for testing due to its unique structure and multifaceted functional activities [4,5,20,21]. HDAC8 is also upregulated in neuroblastoma, a childhood pediatric cancer, hence considered a drug target for this cancer subtype [22,23]. Despite a multifaceted array of alternative treatment options for neuroblastoma, some patient cohorts who are considered high risk at the time of diagnosis face poor prognosis [24,25]. Recent studies indicated that HDAC8 inhibition induces differentiated phenotypes and reduces neuroblastoma growth in vitro and in vivo with few adverse effects [22]. However, there are very few effective therapeutic options in neuroblastoma that inhibit HDAC8 [26]. Thus, the design of novel HDAC8 inhibitors as potential neuroblastoma therapeutics could be valuable to expand the treatment options for this patient population [27]. Therefore, we sought to focus on the biological evaluation of our proposed inhibitors in HDAC8. This study aimed at offering additional therapeutic options to be used in conjunction with or in place of panobinostat while providing a rationale design guideline towards HDACis.
Results and Discussion
Molecular design
Docking analysis is a well-established technique that is utilized to evaluate interactions in important biological receptors such as that of HDACs [28-30]. Previously, we implored docking to predict the interactions between the active sites of HDAC2 and HDAC8 molecular frameworks with similar structures to that of panobinostat [12]. In developing such analogues, the hydroxamate tails and indole capping moiety were maintained as both are essential to binding at the active site of HDAC2 and HDAC8. Results suggested that TOI1, TOI2, and TOI4 [12] would be inhibitors exhibiting similar potency as that of panobinostat. The reported −log(Kd) values were 8.93, 8.64 and 8.25, respectively, with panobinostat possessing a docking score of 8.47. Considering the effects of structural diversity, TOI3-rev was included in the library and computationally evaluated to determine its potential as an HDAC8 inhibitor. TOI3-rev possesses a −log(Kd) value of 8.36 suggesting that it too would be on a par with panobinostat (8.47).
Our previous study showed that the hydroxamate tail of TOI1, TOI2, and TOI4 formed a bidentate interaction with the Zn2+ ion at the base of the HDAC8 receptor, which is consistent with the crystal structure of this class of inhibitors in HDACs [31-34]. Again, our work here extends the investigation focusing on HDAC8 due to the need for novel neuroblastoma therapeutics. Each compound was also shown to have two parallel-displaced π–π interactions, one with Phe-152 and the other with Phe-208. The mode of binding for each compound was similar to panobinostat in HDAC8 (Figure 2). Two differences were however observed in the mode of binding of TOI4. It was shown that the pyridazine ring did not lay as planar in the gorge as the pyrazine and pyrimidine rings of TOI1 and TOI2, respectively. The indole ring of TOI4 was also contorted upward, forming a T-shaped π–π interaction with Phe-207 while both the indole rings of TOI1 and TOI2 were shown to be flipped downward forming a T-shaped π–π interaction with Phe-208.
![[1860-5397-16-59-2]](/bjoc/content/figures/1860-5397-16-59-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Docking pose for panobinostat and panobinostat derivatives in the HDAC8 receptor. (a) Overlay of all compounds investigated in this study in the HDAC8 active site: panobinostat (green), TOI1 (purple), TOI2 (yellow) and TOI4 (grey); (b) TOI3-rev (pink) docking pose in active site; (c) TOI4 (grey) docking pose in the active site.
Figure 2: Docking pose for panobinostat and panobinostat derivatives in the HDAC8 receptor. (a) Overlay of al...
TOI4 is the only diazine compound having two nitrogens directly next to each other in the core ring structure. The nitrogen atoms in the pyridazine ring are positioned so that they do not sit in the center of the phenyl rings of Phe-152 or Phe-208. This results in the pyridazine ring of TOI4 shifted closer to the side of the gorge having Phe-152 compared to the other derivative (Figure 2). This shift allows for the indole ring of TOI4 to fit in a small hydrophobic pocket at the surface, which is created by the phenyl ring of Phe-207. Like the previously studied compounds (TOI1, TOI2, and TOI4), TOI3-rev is also shown to bind to the HDAC8 receptor by forming a bidentate interaction with the Zn2+ ion. TOI3-rev, like that of TOI1, TOI2, and TOI4 produces two parallel-displaced π–π interactions with Phe-152 and Phe-208 (Figure 2). The pyrimidine ring of TOI3-rev also lays planar in the gorge similar to the pyrazine and pyrimidine rings of TOI1 and TOI2. The indole ring of TOI3-rev is tilted downward forming a T-shaped π–π interaction with Phe-208.
In review of the computational results, the inclusion of the nitrogen atoms in the core ring structure of panobinostat produced compounds with predicted binding affinities similar to panobinostat. Thus, aiming to develop improved compounds to effectively target HDAC8, the synthesis of TOI inhibitors and their evaluation was undertaken.
Synthesis
Equipped with molecular targets achieved via theory, the synthesis of the hypothesized compounds commenced with commercially available starting materials (Figure 3). The central core building blocks for TOI1, TOI2, TOI3-rev and TOI4 were selected based on their ability to tether the indole amine and the zinc binding group in a para-relationship to each other.
![[1860-5397-16-59-3]](/bjoc/content/figures/1860-5397-16-59-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: General building blocks for the visualized targets.
Figure 3: General building blocks for the visualized targets.
Initial efforts focused on synthesizing analogue TOI1 (Scheme 1). Acid 1 was converted to the corresponding methyl ester 5 via esterification reaction using methanol mediated by sulfuric acid under heating conditions to provide the compound in 81% yield. The methyl ester 5 was reduced to aldehyde 6 using DIBAL-H at −78 °C. While TLC analysis revealed complete conversion of the ester to aldehyde, the isolated yield was poor (20%). The obstacle was overcome by using a modified Fieser work-up procedure to yield the aldehyde 6 in high yield, 78%. Then, aldehyde 6 was converted to the α,β-unsaturated trans-ester 7 through a Wittig reaction with the phosphorane synthon 8, which was derived from ethyl bromoacetate at 60 °C for 8 h in THF in 72% yield. The exclusive formation of the trans-isomer was confirmed by 1H NMR studies, namely the presence of the olefin at δ 7.00 and 7.71 with a J value of 15 Hz (Supporting Information File 1, Figure S3).
![[1860-5397-16-59-i1]](/bjoc/content/inline/1860-5397-16-59-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction conditions: a) MeOH, H2SO4 (5 drops), MS 4 Å (2 pieces), 68 °C, 8 h, 81%; b) DIBAL-H (1.2 equiv), 6 h, −78 °C, THF, 78%; c) phosphorane 8 (2.0 equiv), THF, 8 h, 60 °C, 72%; d) SeO2, dioxane, 110 °C, 8 h, 61%; e) indolamine 10 (1.1 equiv) DCE, sodium triacetoxyborohydride (STAB, 1.1 equiv), TEA (2 equiv), rt, 63%; f) NaOH at −10 °C, NH2OH·H2O at −10 °C, MeOH , rt, 12 h, 55%.
Scheme 1: Reaction conditions: a) MeOH, H2SO4 (5 drops), MS 4 Å (2 pieces), 68 °C, 8 h, 81%; b) DIBAL-H (1.2 ...
Next, oxidation of the methyl group of 7 under SeO2 conditions at 110 °C provided the ethyl acrylate aldehyde 9 in 61% yield. The next step involved the crucial reductive amination reaction between aldehyde 9 with indolamine 10, which had been obtained via Fischer indole synthesis – the reaction of phenylhydrazine with 5-chloro-2-pentanone [35]. Initial reduction attempts using sodium triacetoxyborohydride (STAB) as the reducing agent provided predominantly starting material and negligible potential product as monitored by TLC. However, addition of 2 equivalents of triethylamine to the reaction mixture facilitated the formation of the product, compound 11 in 63% yield. The product was confirmed by 1H NMR and 13C NMR as shown in Supporting Information File 1.
Finally, the ethyl ester 11 was converted to the hydroxamic acid derivative, TOI1 using the bidentate nucleophile hydroxylamine either under neutral or basic conditions [36,37]. We first explored neutral conditions where aqueous hydroxylamine was added to compound 11 in methanol, and a predominant polar spot was observed by TLC. However, the isolated product was not the expected TOI1, as 1H NMR revealed two new peaks at δ 4.53 and 2.66 ppm (Supporting Information File 1, Figure S36) presumably indicating that a favorable Michael addition followed by intramolecular cyclization or vice versa provided compound 12, which was validated by 13C and DEPT NMR studies (Supporting Information File 1 Figures S37and S38).
Marred with these observations, compound 11 was treated with aqueous hydroxylamine in the presence of strong base (i.e., 10 equivalents of methanolic sodium hydroxide or aqueous sodium hydroxide) at 0 °C. The reaction was monitored by TLC and it revealed that methanolic sodium hydroxide provided a cleaner reaction than aqueous sodium hydroxide conditions. The reaction mixture was quenched with a saturated ammonium chloride solution at 0 °C after 12 h, the solvent was evaporated, and the compound was subjected to reversed-phase column chromatography using C-18 silica gel. After initial unsuccessful purification protocols with water/ACN or water/THF solvent systems, we identified an optimized water/methanol mixture to provide the pure product TOI1 in 55% yield. The isolated compound was thoroughly characterized by spectroscopic techniques.
Having successfully establish reaction conditions for the synthesis of TOI1, we then focused our efforts on the generation of regioisomers TOI2 and TOI3-rev, respectively. Initial attempts to oxidize the methyl group at the benzylic position in starting materials 2 and 3 to provide the corresponding aldehyde compounds 13 and 14 failed, despite using rigorous reaction conditions of SeO2 or alternative strong oxidizing agents (e.g., MnO2 and oxone). Thus, we considered the critical role of the electronic effects of the nitrogen atoms on this cyclic substrate, and then we revised our synthetic strategy by a) tethering an alkene functional group on the aromatic ring and b) then conducting the oxidation of the benzylic group to afford the aldehyde product. Towards this end, we performed a Suzuki coupling reaction between boronic acid 15 with chloro compound 2 (Scheme 2). To the best of our knowledge, there is no report of a Suzuki coupling reaction using boronic acid 15 in the literature. However, we generated this required boronic acid from the corresponding methyl propiolate [38].
![[1860-5397-16-59-i2]](/bjoc/content/inline/1860-5397-16-59-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction conditions: a) boronic acid 15 (1.3 equiv), PdCl2(PPh3)2 (0.1 equiv), dioxane/H2O (3:1), Na2HPO4 (2.0 equiv), TEA (4.0 equiv), 90 °C, 15 h, 55% for 16 and 71% for 18; b) SeO2 , different conditions, 0%; c) SeO2 (2.0 equiv), dioxane, 110 °C, 12 h, 54%.
Scheme 2: Reaction conditions: a) boronic acid 15 (1.3 equiv), PdCl2(PPh3)2 (0.1 equiv), dioxane/H2O (3:1), Na...
Next we investigated reaction conditions for the reaction of compound 2 with boronic acid 15 using different variables (Supporting Information File 1, Table S1). Gratifyingly, after surveying several reaction conditions, we successfully obtained the desired product 16 in 35% yield using PdCl2(PPh3)2 and Na2HPO4 as the base in a dioxane/water system under heating conditions of 90 °C.
To improve the overall chemical yields, we evaluated several organic bases and found that TEA (4.0 equiv) improved the yield to 55%. This success was attributed to the improved increased solubility of TEA in the reaction mixture under heating conditions. In parallel, similar reaction conditions were used for compound 3 and the final coupled product 18 was obtained in 71% yield. The product was confirmed by the expected chemical shift at δ 6.50 and 7.54 ppm with a J value of 15 Hz for compound 16 (Supporting Information File 1, Figure S7) and δ 6.57 and 7.63 ppm with a J value of 15 Hz for compound 18 (Supporting Information File 1, Figure S9) as inferred by 1H NMR analysis.
The resulting Suzuki-coupled products 16 and 18, were subjected to benzylic oxidation expecting the olefin functionality would facilitate the corresponding aldehydes 17 and 19, respectively. Surprisingly, the methyl group in compound 16 did not undergo oxidation under SeO2 conditions as observed for TOI1 whereas under the same reaction conditions, compound 18 readily underwent oxidation to yield compound 19 in 54% yields.
Confounded by this observation, extensive experimentation varying solvent and temperature were evaluated, but none were fruitful. Alternatively to synthesize compound 17, we adapted the strategy as depicted in Scheme 3. Compound 20 was converted into compound 17 via intermediate 21 using a previously reported literature procedure [39]. The intermediate 21 was then subjected to the Suzuki reaction using conditions already developed to provide the ester aldehyde 17 in overall yield of 16% for two steps. Having identified synthetically suitable conditions for compound 17, we scaled up the reaction to complete the final two steps, the reductive amination reaction and the hydroxamic acid preparation. Using the same reaction conditions developed for TOI1, we proceeded with precursors 22 and 23, which were obtained in 61% and 68% yield, respectively. The desired hydroxamic acid TOI2 and TOI3-rev were obtained in 49% and 51% yield, respectively, after C18 silica gel purification procedure using a methanol/water (1:1) solvent system.
![[1860-5397-16-59-i3]](/bjoc/content/inline/1860-5397-16-59-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction conditions: a) 5-bromo-2-chloropyrimidine (1 equiv), ethyl formate (1.5 equiv), THF (20 mL), n-butyllithium (0.6 equiv, 2.5 M) in hexane, −100 °C, 2 h, 42%; b) boronic acid (1.3 equiv), PdCl2(PPh3)2 (0.1 equiv), dioxane/water (8:2), Na2HPO4 (2.0 equiv), TEA (4.0 equiv), 95 °C, 15 h, 40%; c) indolamine 10 (1.1 equiv) DCE, STAB (1.0 equiv), TEA (2 equiv), rt, 61% for 22, 68% for 23; d) NaOH at −10 °C, NH2OH·H2O at −10 °C, MeOH, rt, 12 h, 49% for TIO2 and 51% for TIO3-rev.
Scheme 3: Reaction conditions: a) 5-bromo-2-chloropyrimidine (1 equiv), ethyl formate (1.5 equiv), THF (20 mL...
Finally, we focused our efforts towards the synthesis of the last analogue TOI4. From the above observations, we hypothesize that successful synthesis of TOI4 would rely on generating the key intermediate aldehyde 25 (Scheme 4).
![[1860-5397-16-59-i4]](/bjoc/content/inline/1860-5397-16-59-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction conditions: a) boronic acid 15 (1.3 equiv), PdCl2(PPh3)2 (0.1 equiv), dioxane/H2O (8:2, Na2HPO4 (2.0 equiv), TEA (4.0 equiv), 95 °C, 15 h, 41%; b) SeO2 (2.0 equiv), dioxane , 110 °C, 16 h, 52%; c) indolamine 10 (1.1 equiv), DCE, STAB (1.0 equiv), TEA (2 equiv), rt, 38%; d) NaOH at −10 °C, NH2OH·H2O at −10 °C, MeOH, rt, 12 h, 44%.
Scheme 4: Reaction conditions: a) boronic acid 15 (1.3 equiv), PdCl2(PPh3)2 (0.1 equiv), dioxane/H2O (8:2, Na2...
Initially, we explored the Suzuki coupling reaction for substrate 4. Gratifyingly, the reaction product 24 was produced in 41% yield from our developed synthetic strategy. Then, compound 24 was subjected to an SeO2 oxidation reaction. The oxidation reaction was performed at 110 °C for 16 h to furnish the aldehyde 25 in 52% yield. Next, successful coupling of the aldehyde 25 with indolamine 10 yielded the expected product 26 in 38% yield. Compound 26 was converted to hydroxamic acid TIO4 in 44% yield under NaOH conditions in methanol and purified using C18 column chromatography. All the final compounds were thoroughly characterized by NMR and mass spectrometry (see Supporting Information File 1).
Having successfully synthesized the targets TOI1 to TOI4, our next aim was to evaluate them biologically.
Inhibition assay
The biochemical evaluation of the proposed inhibitors TOI1, TOI2, TOI3-rev, and TOI4 was performed to experimentally determine the potency of HDAC8 inhibition. The obtained results, as shown in Table 1, aligned with the predicted computational studies. In a homogeneous fluorogenic assay, the HDAC activity is quenched with a fluorescent dye that is tethered to an acetyllysine-containing peptide. If the acetyl moiety of the fluorophore is enzymatically hydrolyzed by HDAC8, it will produce a strongly fluorescent signal at 360 nm. Table 1 shows the percentage of HDAC8 inhibition at 100, 10, and 1 µM concentration of the designed inhibitors and panobinostat. All compounds displayed HDAC8 inhibition as predicted by our computational studies [12]. Complete inhibition of HDAC8 was observed for panobinostat at 100 µM or 10 µM concentration, while only 89% inhibition was recorded at 1 µM concentration. The in vitro IC50 values for panobinostat against HDAC8 have been shown to be 277 nM [11]. In vivo studies measuring the IC50 values of panobinostat have also been performed, however, since panobinostat is a pan-DAC inhibitor it has been difficult for researchers to specifically correlate its IC50 value for HDAC8 in a physiological system [40]. Theoretical studies predicted TOI1 as the most potent inhibitor over TOI2, TOI3-rev, and TOI4. TOI1 was shown to be the most potent inhibitor designed in this study producing 100% inhibition at a 100 µM, 90% inhibition at a 10 µM, and 44% at a 1 µM concentration. At a 100 µM concentration, TOI2, TOI3-rev and TOI4 inhibited 79%, 89%, and 93% of HDAC8 activity, respectively. Strong inhibition was seen at a 10 µM concentration for all inhibitors. At a concentration of 1 µM all inhibitors showed less inhibition of HDAC8 than panobinostat. It was noted that TOI2 and TOI3-rev produced similar inhibition results against HDAC8. Both of these inhibitors have pyrimidine rings in the core; thus, the data suggests that while this ring structure did produce inhibition, it was not as effective as the inclusion of a pyrazine ring. This finding is consistent with our computational data which demonstrated that TOI1 would slightly outperform TOI2 and TOI3-rev.
Table 1: Percent inhibition of HDAC8 by hydroxamate inhibitors.
| compound | concentration | ||
| 100 µM | 10 µM | 1 µM | |
| panobinostat (n = 4)a | 100 ± 0.0 | 100 ± 0.0 | 89 ± 13.4 |
| TOI1 (n = 4) | 100 ± 0.0 | 90 ± 21.1 | 44 ± 11.5 |
| TOI2 (n = 4) | 79 ± 8.1 | 75 ± 13.4 | 23 ± 26.9 |
| TOI3-rev (n = 4) | 89 ± 7.0 | 75 ± 26.9 | 37 ± 18.1 |
|
TOI4 (n = 4 @ 100 and 10 µM,
n = 3 @ 1 µM) |
93 ± 8.1 | 89 ± 13.4 | 65 ± 21.4 |
| TCAb | 93 ± 14.0 | ||
an = number of replicates for the assay; btrichostatin A (TCA) as an inhibitor control.
It is interesting to note that TOI1, which was shown to be better than TOI4 at the 100 µM level, was less potent than TOI4 at low concentration (1 µM). TOI4, which was predicted to be slightly less potent than TOI1, TOI2, and TO3-rev computationally was revealed via experiment to be a better inhibitor than TOI2 and TOI3-rev. The result was observed presumably due to a variation in the binding mode for TOI4 that differs from that of TOI1, TOI2, and TOI3-rev. TOI4 was shown to observe a slightly different orientation of the core heterocyclic ring as well as a change in the orientation of the terminal indole ring. It is not clear if this difference in binding mode affected the ability of TOI4 to maintain a higher potency at lower concentrations. However, this observation does raise an interesting question: “Are certain modes of binding more effective at different concentrations and if so how can this be accurately modelled computationally?” Taken together, this data indicates that while TOI1 is the best inhibitor of the four compounds presented herein, TOI4 remains effective against HDAC8 even at lower concentrations.
Conclusion
Our investigation provides a successful synthetic strategy towards four new analogues of panobinostat having diazines as the central core and details the results of their biochemical evaluation. Computational data corroborated that the substitution of benzene in the molecular framework of panobinostat for a nitrogen-containing heterocycle in the core ring structure would enhance the pharmacological properties while maintaining the level of HDAC8 inhibition. The targets TOI1–4 were synthesized from commercially available starting materials in moderate yields. The synthesized compounds displayed potent activity against HDAC8; thus, emphasizing the advantages of drug design on a theoretical basis. These efforts led to the design of potential analogues that warrant further studies to develop therapeutic agents for neuroblastoma. Future research will be aimed at investigating the HDAC class specificity of these designed analogues and evaluating their overall potential to inhibit neuroblastoma cell growth.
Experimental
General
Reagents and solvents were purchased from commercial sources and used without further purification unless otherwise specified. Tetrahydrofuran (THF), ether, dichloromethane (DCM), and dimethylformamide (DMF) were degassed in 20 L drums and passed through two sequential purification columns (activated alumina; molecular sieves for DMF) under a positive argon atmosphere. Thin-layer chromatography (TLC) was performed on SiO2-60 F254 aluminum plates with visualization by ultraviolet (UV) detection at 254 nm or staining. Flash column chromatography was performed using Purasil SiO2-60, 230–400 mesh from Whatman. NMR spectra were recorded on a BRUKER AV500 spectrometer (operating at 500 MHz for 1H and 125 MHz for 13C acquisitions). Chemical shifts were reported as ppm relative to the solvent residual peak (CHCl3: 7.26 ppm for 1H, 77.2 ppm for 13C). Data are reported as follows: chemical shifts, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet, br = broad), coupling constant J (Hz), and integration. High-resolution mass spectra were recorded using an ESI–TOF mass spectrometer (Agilent 6220 Time-of-Flight), gas temperature – 350 °C, drying gas (N2) – 8.0 L/min, mobile phase(s): methanol with 0.1% formic acid, flow rate: 0.2 mL/min, sample preparation: The sample was dissolved in 1 drop of chloroform and diluted with 1 mL methanol.
Additional chemical synthesis details can be found in Supporting Information File 1.
HDAC8 enzymatic activity assay
A fluorogenic assay (BPS Bioscience, catalog # 50008) was performed to evaluate the inhibition potential of the designed inhibitors. The assay was carried out using the supplier’s instructions. TOI1, TOI2, TOI3-rev, TOI4, and panobinostat were evaluated at concentrations of 100 µM, 10 µM, and 1 µM in a 96 well plate. Trichostatin A (TCA), which was used as an inhibitor control, was also evaluated at 100 µM. The blank consisted of HDAC assay buffer without the addition of inhibitor or HDAC8, while the positive control consisted of HDAC8 without inhibitor added. The substrate concentration was 5 µM, and HDAC8 concentration per well was 4 ng/µL. The reaction was initiated by addition of enzyme. The fluorogenic substrate was excited at 360 nm and the emission signal was detected at 460 nm using a Biotek Synergy HTX multimodal plate reader after incubation for 30 minutes.
Preparation of the HDAC8 receptor for docking
The HDAC8 crystal structure (protein database pdb: 1W22) [32] was utilized as the docking receptor for all compounds. This receptor is crystalized as a dimer thus only the A chain was prepared for docking using Sylbyl-X 2.1. A protocol defining the regions of hydrogen donor, acceptor and hydrophobic character was created using the SFXC protocol [41-43]. The Zn2+ ion which is known to be essential in the hydroxamate class of HDAC inhibitors was included in the preparation of the receptor.
Preparation of inhibitor compounds for docking
Compounds were drawn in ChemDraw and the converted for 2D to 3D using Marvin Sketch. The energy gradient optimization method was used to perform an initial minimization in Marvin Sketch. A full structural minimization was performed in UCSF Chimera [44] using conjugate gradient followed by steepest decent. In Sybyl-X multiple conformers of each potential inhibitor compound were created for docking analysis using the prep protocol Docking >1 parameter.
Molecular docking of inhibitors in the HDAC8 receptor
The inhibitor candidates were docked into the receptor using the Surflex-Dock Geom (SFXC) protocol [41-43] to evaluate the binding affinity of the ligand for the HDAC8 receptor. The C-scoring method was used to calculate these binding affinities and binding scores are given in −log10(Kd) values [45]. Docking simulations where ran considering conformers at pH 7 to simulate the physiological conditions where the pH is 7.4. Results were analyzed in both Sybyl-X and UCSF Chimera.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 2.9 MB | Download |
Funding
For the synthesis of the nitrogen containing heterocyclic cores, S.B., S.V., K.D. S.A. and D. L. W. appreciate financial support from the National Science Foundation under Grant Numbers CHE-1652094. X. A. M., L. V. G. and S. V. S. would like to thank the William Randolph Hearst Fellowship for the funding.
References
-
Strahl, B. D.; Allis, C. D. Nature 2000, 403, 41–45. doi:10.1038/47412
Return to citation in text: [1] -
Barneda-Zahonero, B.; Parra, M. Mol. Oncol. 2012, 6, 579–589. doi:10.1016/j.molonc.2012.07.003
Return to citation in text: [1] -
Manal, M.; Chandrasekar, M. J. N.; Gomathi Priya, J.; Nanjan, M. J. Bioorg. Chem. 2016, 67, 18–42. doi:10.1016/j.bioorg.2016.05.005
Return to citation in text: [1] [2] -
Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R. J.; Jung, M. Trends Pharmacol. Sci. 2015, 36, 481–492. doi:10.1016/j.tips.2015.04.013
Return to citation in text: [1] [2] -
Ingham, O. J.; Paranal, R. M.; Smith, W. B.; Escobar, R. A.; Yueh, H.; Snyder, T.; Porco, J. A., Jr.; Bradner, J. E.; Beeler, A. B. ACS Med. Chem. Lett. 2016, 7, 929–932. doi:10.1021/acsmedchemlett.6b00239
Return to citation in text: [1] [2] -
West, A. C.; Johnstone, R. W. J. Clin. Invest. 2014, 124, 30–39. doi:10.1172/jci69738
Return to citation in text: [1] -
Ononye, S. N.; van Heyst, M.; Falcone, E. M.; Anderson, A. C.; Wright, D. L. Pharm. Pat. Anal. 2012, 1, 207–221. doi:10.4155/ppa.12.21
Return to citation in text: [1] -
Glaser, K. B. Biochem. Pharmacol. 2007, 74, 659–671. doi:10.1016/j.bcp.2007.04.007
Return to citation in text: [1] -
Prince, H. M.; Bishton, M. J.; Johnstone, R. W. Future Oncol. 2009, 5, 601–612. doi:10.2217/fon.09.36
Return to citation in text: [1] -
Garnock-Jones, K. P. Drugs 2015, 75, 695–704. doi:10.1007/s40265-015-0388-8
Return to citation in text: [1] -
Atadja, P. Cancer Lett. 2009, 280, 233–241. doi:10.1016/j.canlet.2009.02.019
Return to citation in text: [1] [2] -
Stoddard, S. V.; May, X. A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D. L. Mol. Inf. 2019, 38, 1800080. doi:10.1002/minf.201800080
Return to citation in text: [1] [2] [3] [4] [5] -
Zhang, Y.; Feng, J.; Jia, Y.; Xu, Y.; Liu, C.; Fang, H.; Xu, W. Eur. J. Med. Chem. 2011, 46, 5387–5397. doi:10.1016/j.ejmech.2011.08.045
Return to citation in text: [1] -
Lee, H.-Y.; Lee, J.-F.; Kumar, S.; Wu, Y.-W.; HuangFu, W.-C.; Lai, M.-J.; Li, Y.-H.; Huang, H.-L.; Kuo, F.-C.; Hsiao, C.-J.; Cheng, C.-C.; Yang, C.-R.; Liou, J.-P. Eur. J. Med. Chem. 2017, 125, 1268–1278. doi:10.1016/j.ejmech.2016.11.033
Return to citation in text: [1] -
Chen, Y.; Wang, X.; Xiang, W.; He, L.; Tang, M.; Wang, F.; Wang, T.; Yang, Z.; Yi, Y.; Wang, H.; Niu, T.; Zheng, L.; Lei, L.; Li, X.; Song, H.; Chen, L. J. Med. Chem. 2016, 59, 5488–5504. doi:10.1021/acs.jmedchem.6b00579
Return to citation in text: [1] -
Guo, Z. Acta Pharm. Sin. B 2017, 7, 119–136. doi:10.1016/j.apsb.2016.06.003
Return to citation in text: [1] -
Flefel, E.; Tantawy, W.; El-Sofany, W.; El-Shahat, M.; El-Sayed, A.; Abd-Elshafy, D. Molecules 2017, 22, 148. doi:10.3390/molecules22010148
Return to citation in text: [1] -
Martins, P.; Jesus, J.; Santos, S.; Raposo, L.; Roma-Rodrigues, C.; Baptista, P.; Fernandes, A. Molecules 2015, 20, 16852–16891. doi:10.3390/molecules200916852
Return to citation in text: [1] -
Robarge, K. D.; Lee, W.; Eigenbrot, C.; Ultsch, M.; Wiesmann, C.; Heald, R.; Price, S.; Hewitt, J.; Jackson, P.; Savy, P.; Burton, B.; Choo, E. F.; Pang, J.; Boggs, J.; Yang, A.; Yang, X.; Baumgardner, M. Bioorg. Med. Chem. Lett. 2014, 24, 4714–4723. doi:10.1016/j.bmcl.2014.08.008
Return to citation in text: [1] -
Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039
Return to citation in text: [1] -
Wolfson, N. A.; Pitcairn, C. A.; Fierke, C. A. Biopolymers 2013, 99, 112–126. doi:10.1002/bip.22135
Return to citation in text: [1] -
Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W. C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; Deubzer, H. E.; Milde, T.; Witt, O.; Oehme, I. Cell Death Dis. 2015, 6, e1657. doi:10.1038/cddis.2015.24
Return to citation in text: [1] [2] -
Oehme, I.; Deubzer, H. E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S. M.; von Deimling, A.; Fischer, M.; Witt, O. Clin. Cancer Res. 2009, 15, 91–99. doi:10.1158/1078-0432.ccr-08-0684
Return to citation in text: [1] -
Kushner, B. H. J. Nucl. Med. 2004, 45, 1172–1188.
Return to citation in text: [1] -
Brodeur, G. M. Nat. Rev. Cancer 2003, 3, 203–216. doi:10.1038/nrc1014
Return to citation in text: [1] -
Kolbinger, F. R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A. K.; Westermann, F.; Witt, O.; Oehme, I. Arch. Toxicol. 2018, 92, 2649–2664. doi:10.1007/s00204-018-2234-8
Return to citation in text: [1] -
Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039
Return to citation in text: [1] -
Di Micco, S.; Chini, M. G.; Terracciano, S.; Bruno, I.; Riccio, R.; Bifulco, G. Bioorg. Med. Chem. 2013, 21, 3795–3807. doi:10.1016/j.bmc.2013.04.036
Return to citation in text: [1] -
Leach, A. R.; Shoichet, B. K.; Peishoff, C. E. J. Med. Chem. 2006, 49, 5851–5855. doi:10.1021/jm060999m
Return to citation in text: [1] -
Ewing, T. J. A.; Makino, S.; Skillman, A. G.; Kuntz, I. D. J. Comput.-Aided Mol. Des. 2001, 15, 411–428. doi:10.1023/a:1011115820450
Return to citation in text: [1] -
Lauffer, B. E. L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; Ortwine, D. F.; Gunzner, J.; Modrusan, Z.; Neumann, L.; Koth, C. M.; Lupardus, P. J.; Kaminker, J. S.; Heise, C. E.; Steiner, P. J. Biol. Chem. 2013, 288, 26926–26943. doi:10.1074/jbc.m113.490706
Return to citation in text: [1] -
Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E. C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; Steinkuhler, C.; Di Marco, S. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15064–15069. doi:10.1073/pnas.0404603101
Return to citation in text: [1] [2] -
Finnin, M. S.; Donigian, J. R.; Cohen, A.; Richon, V. M.; Rifkind, R. A.; Marks, P. A.; Breslow, R.; Pavletich, N. P. Nature 1999, 401, 188–193. doi:10.1038/43710
Return to citation in text: [1] -
Rouanet-Mehouas, C.; Czarny, B.; Beau, F.; Cassar-Lajeunesse, E.; Stura, E. A.; Dive, V.; Devel, L. J. Med. Chem. 2017, 60, 403–414. doi:10.1021/acs.jmedchem.6b01420
Return to citation in text: [1] -
Grandberg, I. I. Chem. Heterocycl. Compd. 1974, 10, 501–510. doi:10.1007/bf00471313
Return to citation in text: [1] -
Wang, C.; Eessalu, T. E.; Barth, V. N.; Mitch, C. H.; Wagner, F. F.; Hong, Y.; Neelamegam, R.; Schroeder, F. A.; Holson, E. B.; Haggarty, S. J.; Hooker, J. M. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 29–38.
Return to citation in text: [1] -
Wang, H.; Yu, N.; Chen, D.; Lee, K. C. L.; Lye, P. L.; Chang, J. W. W.; Deng, W.; Ng, M. C. Y.; Lu, T.; Khoo, M. L.; Poulsen, A.; Sangthongpitag, K.; Wu, X.; Hu, C.; Goh, K. C.; Wang, X.; Fang, L.; Goh, K. L.; Khng, H. H.; Goh, S. K.; Yeo, P.; Liu, X.; Bonday, Z.; Wood, J. M.; Dymock, B. W.; Kantharaj, E.; Sun, E. T. J. Med. Chem. 2011, 54, 4694–4720. doi:10.1021/jm2003552
Return to citation in text: [1] -
Gravel, M.; Touré, B. B.; Hall, D. G. Org. Prep. Proced. Int. 2004, 36, 573–579. doi:10.1080/00304940409355974
Return to citation in text: [1] -
Kawasaki, T.; Nakaoda, M.; Takahashi, Y.; Kanto, Y.; Kuruhara, N.; Hosoi, K.; Sato, I.; Matsumoto, A.; Soai, K. Angew. Chem., Int. Ed. 2014, 53, 11199–11202. doi:10.1002/anie.201405441
Return to citation in text: [1] -
Ha, S.-D.; Reid, C.; Meshkibaf, S.; Kim, S. O. J. Biol. Chem. 2016, 291, 8745–8755. doi:10.1074/jbc.m115.695809
Return to citation in text: [1] -
Jain, A. N. J. Comput.-Aided Mol. Des. 2007, 21, 281–306. doi:10.1007/s10822-007-9114-2
Return to citation in text: [1] [2] -
Jain, A. N. J. Med. Chem. 2003, 46, 499–511. doi:10.1021/jm020406h
Return to citation in text: [1] [2] -
Jain, A. N. J. Comput.-Aided Mol. Des. 1996, 10, 427–440. doi:10.1007/bf00124474
Return to citation in text: [1] [2] -
Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. J. Comput. Chem. 2004, 25, 1605–1612. doi:10.1002/jcc.20084
Return to citation in text: [1] -
Warren, G. L.; Andrews, C. W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M. H.; Lindvall, M.; Nevins, N.; Semus, S. F.; Senger, S.; Tedesco, G.; Wall, I. D.; Woolven, J. M.; Peishoff, C. E.; Head, M. S. J. Med. Chem. 2006, 49, 5912–5931. doi:10.1021/jm050362n
Return to citation in text: [1]
| 40. | Ha, S.-D.; Reid, C.; Meshkibaf, S.; Kim, S. O. J. Biol. Chem. 2016, 291, 8745–8755. doi:10.1074/jbc.m115.695809 |
| 32. | Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E. C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; Steinkuhler, C.; Di Marco, S. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15064–15069. doi:10.1073/pnas.0404603101 |
| 41. | Jain, A. N. J. Comput.-Aided Mol. Des. 2007, 21, 281–306. doi:10.1007/s10822-007-9114-2 |
| 42. | Jain, A. N. J. Med. Chem. 2003, 46, 499–511. doi:10.1021/jm020406h |
| 43. | Jain, A. N. J. Comput.-Aided Mol. Des. 1996, 10, 427–440. doi:10.1007/bf00124474 |
| 3. | Manal, M.; Chandrasekar, M. J. N.; Gomathi Priya, J.; Nanjan, M. J. Bioorg. Chem. 2016, 67, 18–42. doi:10.1016/j.bioorg.2016.05.005 |
| 22. | Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W. C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; Deubzer, H. E.; Milde, T.; Witt, O.; Oehme, I. Cell Death Dis. 2015, 6, e1657. doi:10.1038/cddis.2015.24 |
| 6. | West, A. C.; Johnstone, R. W. J. Clin. Invest. 2014, 124, 30–39. doi:10.1172/jci69738 |
| 7. | Ononye, S. N.; van Heyst, M.; Falcone, E. M.; Anderson, A. C.; Wright, D. L. Pharm. Pat. Anal. 2012, 1, 207–221. doi:10.4155/ppa.12.21 |
| 8. | Glaser, K. B. Biochem. Pharmacol. 2007, 74, 659–671. doi:10.1016/j.bcp.2007.04.007 |
| 26. | Kolbinger, F. R.; Koeneke, E.; Ridinger, J.; Heimburg, T.; Müller, M.; Bayer, T.; Sippl, W.; Jung, M.; Gunkel, N.; Miller, A. K.; Westermann, F.; Witt, O.; Oehme, I. Arch. Toxicol. 2018, 92, 2649–2664. doi:10.1007/s00204-018-2234-8 |
| 3. | Manal, M.; Chandrasekar, M. J. N.; Gomathi Priya, J.; Nanjan, M. J. Bioorg. Chem. 2016, 67, 18–42. doi:10.1016/j.bioorg.2016.05.005 |
| 4. | Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R. J.; Jung, M. Trends Pharmacol. Sci. 2015, 36, 481–492. doi:10.1016/j.tips.2015.04.013 |
| 5. | Ingham, O. J.; Paranal, R. M.; Smith, W. B.; Escobar, R. A.; Yueh, H.; Snyder, T.; Porco, J. A., Jr.; Bradner, J. E.; Beeler, A. B. ACS Med. Chem. Lett. 2016, 7, 929–932. doi:10.1021/acsmedchemlett.6b00239 |
| 22. | Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W. C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; Deubzer, H. E.; Milde, T.; Witt, O.; Oehme, I. Cell Death Dis. 2015, 6, e1657. doi:10.1038/cddis.2015.24 |
| 23. | Oehme, I.; Deubzer, H. E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S. M.; von Deimling, A.; Fischer, M.; Witt, O. Clin. Cancer Res. 2009, 15, 91–99. doi:10.1158/1078-0432.ccr-08-0684 |
| 2. | Barneda-Zahonero, B.; Parra, M. Mol. Oncol. 2012, 6, 579–589. doi:10.1016/j.molonc.2012.07.003 |
| 24. | Kushner, B. H. J. Nucl. Med. 2004, 45, 1172–1188. |
| 25. | Brodeur, G. M. Nat. Rev. Cancer 2003, 3, 203–216. doi:10.1038/nrc1014 |
| 12. | Stoddard, S. V.; May, X. A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D. L. Mol. Inf. 2019, 38, 1800080. doi:10.1002/minf.201800080 |
| 16. | Guo, Z. Acta Pharm. Sin. B 2017, 7, 119–136. doi:10.1016/j.apsb.2016.06.003 |
| 17. | Flefel, E.; Tantawy, W.; El-Sofany, W.; El-Shahat, M.; El-Sayed, A.; Abd-Elshafy, D. Molecules 2017, 22, 148. doi:10.3390/molecules22010148 |
| 18. | Martins, P.; Jesus, J.; Santos, S.; Raposo, L.; Roma-Rodrigues, C.; Baptista, P.; Fernandes, A. Molecules 2015, 20, 16852–16891. doi:10.3390/molecules200916852 |
| 19. | Robarge, K. D.; Lee, W.; Eigenbrot, C.; Ultsch, M.; Wiesmann, C.; Heald, R.; Price, S.; Hewitt, J.; Jackson, P.; Savy, P.; Burton, B.; Choo, E. F.; Pang, J.; Boggs, J.; Yang, A.; Yang, X.; Baumgardner, M. Bioorg. Med. Chem. Lett. 2014, 24, 4714–4723. doi:10.1016/j.bmcl.2014.08.008 |
| 45. | Warren, G. L.; Andrews, C. W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M. H.; Lindvall, M.; Nevins, N.; Semus, S. F.; Senger, S.; Tedesco, G.; Wall, I. D.; Woolven, J. M.; Peishoff, C. E.; Head, M. S. J. Med. Chem. 2006, 49, 5912–5931. doi:10.1021/jm050362n |
| 12. | Stoddard, S. V.; May, X. A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D. L. Mol. Inf. 2019, 38, 1800080. doi:10.1002/minf.201800080 |
| 4. | Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R. J.; Jung, M. Trends Pharmacol. Sci. 2015, 36, 481–492. doi:10.1016/j.tips.2015.04.013 |
| 5. | Ingham, O. J.; Paranal, R. M.; Smith, W. B.; Escobar, R. A.; Yueh, H.; Snyder, T.; Porco, J. A., Jr.; Bradner, J. E.; Beeler, A. B. ACS Med. Chem. Lett. 2016, 7, 929–932. doi:10.1021/acsmedchemlett.6b00239 |
| 20. | Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039 |
| 21. | Wolfson, N. A.; Pitcairn, C. A.; Fierke, C. A. Biopolymers 2013, 99, 112–126. doi:10.1002/bip.22135 |
| 44. | Pettersen, E. F.; Goddard, T. D.; Huang, C. C.; Couch, G. S.; Greenblatt, D. M.; Meng, E. C.; Ferrin, T. E. J. Comput. Chem. 2004, 25, 1605–1612. doi:10.1002/jcc.20084 |
| 9. | Prince, H. M.; Bishton, M. J.; Johnstone, R. W. Future Oncol. 2009, 5, 601–612. doi:10.2217/fon.09.36 |
| 10. | Garnock-Jones, K. P. Drugs 2015, 75, 695–704. doi:10.1007/s40265-015-0388-8 |
| 13. | Zhang, Y.; Feng, J.; Jia, Y.; Xu, Y.; Liu, C.; Fang, H.; Xu, W. Eur. J. Med. Chem. 2011, 46, 5387–5397. doi:10.1016/j.ejmech.2011.08.045 |
| 14. | Lee, H.-Y.; Lee, J.-F.; Kumar, S.; Wu, Y.-W.; HuangFu, W.-C.; Lai, M.-J.; Li, Y.-H.; Huang, H.-L.; Kuo, F.-C.; Hsiao, C.-J.; Cheng, C.-C.; Yang, C.-R.; Liou, J.-P. Eur. J. Med. Chem. 2017, 125, 1268–1278. doi:10.1016/j.ejmech.2016.11.033 |
| 15. | Chen, Y.; Wang, X.; Xiang, W.; He, L.; Tang, M.; Wang, F.; Wang, T.; Yang, Z.; Yi, Y.; Wang, H.; Niu, T.; Zheng, L.; Lei, L.; Li, X.; Song, H.; Chen, L. J. Med. Chem. 2016, 59, 5488–5504. doi:10.1021/acs.jmedchem.6b00579 |
| 41. | Jain, A. N. J. Comput.-Aided Mol. Des. 2007, 21, 281–306. doi:10.1007/s10822-007-9114-2 |
| 42. | Jain, A. N. J. Med. Chem. 2003, 46, 499–511. doi:10.1021/jm020406h |
| 43. | Jain, A. N. J. Comput.-Aided Mol. Des. 1996, 10, 427–440. doi:10.1007/bf00124474 |
| 12. | Stoddard, S. V.; May, X. A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D. L. Mol. Inf. 2019, 38, 1800080. doi:10.1002/minf.201800080 |
| 27. | Banerjee, S.; Adhikari, N.; Amin, S. A.; Jha, T. Eur. J. Med. Chem. 2019, 164, 214–240. doi:10.1016/j.ejmech.2018.12.039 |
| 28. | Di Micco, S.; Chini, M. G.; Terracciano, S.; Bruno, I.; Riccio, R.; Bifulco, G. Bioorg. Med. Chem. 2013, 21, 3795–3807. doi:10.1016/j.bmc.2013.04.036 |
| 29. | Leach, A. R.; Shoichet, B. K.; Peishoff, C. E. J. Med. Chem. 2006, 49, 5851–5855. doi:10.1021/jm060999m |
| 30. | Ewing, T. J. A.; Makino, S.; Skillman, A. G.; Kuntz, I. D. J. Comput.-Aided Mol. Des. 2001, 15, 411–428. doi:10.1023/a:1011115820450 |
| 12. | Stoddard, S. V.; May, X. A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D. L. Mol. Inf. 2019, 38, 1800080. doi:10.1002/minf.201800080 |
| 38. | Gravel, M.; Touré, B. B.; Hall, D. G. Org. Prep. Proced. Int. 2004, 36, 573–579. doi:10.1080/00304940409355974 |
| 39. | Kawasaki, T.; Nakaoda, M.; Takahashi, Y.; Kanto, Y.; Kuruhara, N.; Hosoi, K.; Sato, I.; Matsumoto, A.; Soai, K. Angew. Chem., Int. Ed. 2014, 53, 11199–11202. doi:10.1002/anie.201405441 |
| 35. | Grandberg, I. I. Chem. Heterocycl. Compd. 1974, 10, 501–510. doi:10.1007/bf00471313 |
| 36. | Wang, C.; Eessalu, T. E.; Barth, V. N.; Mitch, C. H.; Wagner, F. F.; Hong, Y.; Neelamegam, R.; Schroeder, F. A.; Holson, E. B.; Haggarty, S. J.; Hooker, J. M. Am. J. Nucl. Med. Mol. Imaging 2013, 4, 29–38. |
| 37. | Wang, H.; Yu, N.; Chen, D.; Lee, K. C. L.; Lye, P. L.; Chang, J. W. W.; Deng, W.; Ng, M. C. Y.; Lu, T.; Khoo, M. L.; Poulsen, A.; Sangthongpitag, K.; Wu, X.; Hu, C.; Goh, K. C.; Wang, X.; Fang, L.; Goh, K. L.; Khng, H. H.; Goh, S. K.; Yeo, P.; Liu, X.; Bonday, Z.; Wood, J. M.; Dymock, B. W.; Kantharaj, E.; Sun, E. T. J. Med. Chem. 2011, 54, 4694–4720. doi:10.1021/jm2003552 |
| 12. | Stoddard, S. V.; May, X. A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D. L. Mol. Inf. 2019, 38, 1800080. doi:10.1002/minf.201800080 |
| 31. | Lauffer, B. E. L.; Mintzer, R.; Fong, R.; Mukund, S.; Tam, C.; Zilberleyb, I.; Flicke, B.; Ritscher, A.; Fedorowicz, G.; Vallero, R.; Ortwine, D. F.; Gunzner, J.; Modrusan, Z.; Neumann, L.; Koth, C. M.; Lupardus, P. J.; Kaminker, J. S.; Heise, C. E.; Steiner, P. J. Biol. Chem. 2013, 288, 26926–26943. doi:10.1074/jbc.m113.490706 |
| 32. | Vannini, A.; Volpari, C.; Filocamo, G.; Casavola, E. C.; Brunetti, M.; Renzoni, D.; Chakravarty, P.; Paolini, C.; De Francesco, R.; Gallinari, P.; Steinkuhler, C.; Di Marco, S. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 15064–15069. doi:10.1073/pnas.0404603101 |
| 33. | Finnin, M. S.; Donigian, J. R.; Cohen, A.; Richon, V. M.; Rifkind, R. A.; Marks, P. A.; Breslow, R.; Pavletich, N. P. Nature 1999, 401, 188–193. doi:10.1038/43710 |
| 34. | Rouanet-Mehouas, C.; Czarny, B.; Beau, F.; Cassar-Lajeunesse, E.; Stura, E. A.; Dive, V.; Devel, L. J. Med. Chem. 2017, 60, 403–414. doi:10.1021/acs.jmedchem.6b01420 |
© 2020 Balasubramaniam et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)