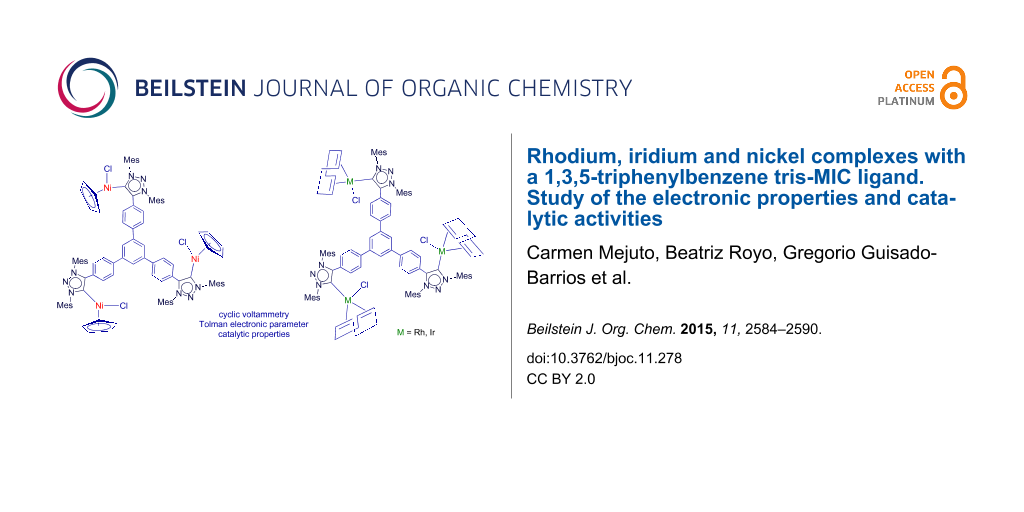
Abstract
The coordination versatility of a 1,3,5-triphenylbenzene-tris-mesoionic carbene ligand is illustrated by the preparation of complexes with three different metals: rhodium, iridium and nickel. The rhodium and iridium complexes contained the [MCl(COD)] fragments, while the nickel compound contained [NiCpCl]. The preparation of the tris-MIC (MIC = mesoionic carbene) complex with three [IrCl(CO)2] fragments, allowed the estimation of the Tolman electronic parameter (TEP) for the ligand, which was compared with the TEP value for a related 1,3,5-triphenylbenzene-tris-NHC ligand. The electronic properties of the tris-MIC ligand were studied by cyclic voltammetry measurements. In all cases, the tris-MIC ligand showed a stronger electron-donating character than the corresponding NHC-based ligands. The catalytic activity of the tri-rhodium complex was tested in the addition reaction of arylboronic acids to α,β-unsaturated ketones.

Graphical Abstract
Introduction
Highly symmetrical poly-NHCs are a very interesting type of ligands, because they allow the preparation of a variety of supramolecular assemblies that include molecular squares and triangles [1-6], cylinder-like structures [7-13], organometallic polymers [14-22] and even organometallic mesoporous materials [21,22]. Another interesting feature of this special type of poly-NHCs is its ability to form multimetallic catalysts whose catalytic performances can be compared with analogous monometallic NHC complexes [23-25]. On several occasions their activity has proven higher than the activities shown by their monometallic counterparts [23,26]. In the last few years we became interested in the design of several types of C3v-symmetric tris-NHCs, both for the preparation of self-assembly molecular cages [12,13,27] and for the design of discrete trimetallic molecules whose catalytic performances were explored [23,25,28,29]. Among these ligands, we found those featuring a nanoscale distance between the metals especially interesting [13,27] because for these systems a catalytic cooperation between the active metal sites should not be expected. As a consequence all catalytic improvements should be assigned to reasons dealing with supramolecular interactions [30] or with the higher nanolocal concentration of metal sites in the multimetallic catalyst [31]. In this context, we obtained the 1,3,5-triphenylbenzene-based C3-symmetrical tris-NHC ligand A (Scheme 1), which was coordinated to rhodium and iridium [25]. The catalytic activity of the trirhodium complex was tested in the addition reaction of arylboronic acids to 2-cyclohexen-1-one, where it showed good activity. The same ligand was also used for the preparation of nanometer-sized cylinder-like structures of Cu, Ag and Au [13].
![[1860-5397-11-278-i1]](/bjoc/content/inline/1860-5397-11-278-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Schematic representation of ligands A and B.
Scheme 1: Schematic representation of ligands A and B.
As mesoionic carbenes (MICs) are known to be stronger electron donors compared to NHCs [32-36], and because poly-MIC ligands are less explored [37-46], we very recently synthesized the tris-diarylated-(1,2,3-triazol-5-ylidene)-1,3,5-triphenylbenzene-based ligand (B). This ligand afforded trisilver and trigold cages with very interesting rearranging properties when mixed with the related 1,3,5-triphenylbenzene-based tris-NHC Ag cages [27]. Shortly afterwards, Sarkar and co-workers used a dialkylated (1,2,3-triazol-5-ylidene)-1,3,5-triphenylbenzene-based ligand for the preparation of the corresponding tris-Ir(III) and Pd(II) complexes and tested them for their catalytic activities [47].
Based on these previous findings, we herein report the synthesis of the tri-metallic complexes of Rh(I), Ir(I) and Ni(II) with the tris-MIC ligand B. The preparation of these complexes gives us an excellent opportunity to compare the electronic properties of the tris-MIC ligand B with those of its tris-NHC analogue, A. The catalytic activity of the tris-MIC-trirhodium complex was tested in the addition reaction of arylboronic acids to 2-cyclohexen-1-one and compared to the results obtained with the tris-NHC analogue.
Results and Discussion
Complex 2 was obtained by the in situ deprotonation of the tris-triazolium salt 1 with potassium bis(trimethyl)silyl amide (KHMDS) in the presence of [RhCl(COD)]2 in THF at −78 °C (Scheme 2). It was isolated in 84% yield after purification by column chromatography. For the preparation of the related iridium(I) complex, we found it more convenient to use a preparative method inspired by a recent work by Plenio and co-workers [48]. In this way refluxing a mixture of 1 with [IrCl(COD)]2 in the presence of K2CO3 in acetone for 24 h gave the triiridium(I) complex 3 in 60% yield after purification. The complexes 2 and 3 were characterized by NMR and mass spectrometry. Both, the 1H and the 13C NMR spectra of the complexes were consistent with the expected threefold symmetry of the molecules, as exemplified by the appearance of one only signal for the carbene carbons, at 173.4 (1JRh–C = 41.5 Hz) and 172.1 ppm, for 2 and 3, respectively. The 1H NMR spectra of complexes 2 and 3 exhibited relatively broad signals, which may indicate a fluxional behavior. This may be likely caused by the combined rotation around the C–C sigma bonds of the tris-MIC ligand and the Ccarbene–M bond.
![[1860-5397-11-278-i2]](/bjoc/content/inline/1860-5397-11-278-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of rhodium(I), iridium(I), and nickel(II) complexes of ligand B.
Scheme 2: Synthesis of rhodium(I), iridium(I), and nickel(II) complexes of ligand B.
In order to widen the coordination scope of the tris-MIC ligand B, the corresponding Ni(II) complex was also synthesized. The reaction of 1 with [NiCp2] in the presence of NEt4Cl in refluxing dioxane, afforded the tris-MIC complex of Ni(II) 4, as a red solid in 40% yield after purification. The three-fold symmetry of this complex was also confirmed by its NMR spectra. In the 1H NMR a singlet assigned to the equivalent 15 protons of the three cyclopentadienyl rings was observed. As already indicated for complexes 2 and 3 the 1H NMR spectrum of complex 4 showed broad signals as a consequence of the fluxionality due to the rotation about the C–C sigma bonds of the ligand, and the Ccarbene–Ni bond. The 13C NMR spectrum showed the distinctive signal due to the metalation of the carbene carbon at 151.4 ppm, which is in the same region of the previously reported [NiCpCl(MIC)] complex (148 ppm) [49]. The trimetallic nature of the complex was further confirmed by mass spectrometry, which revealed a peak at m/z 810.7, assigned to [M − 2Cl]2+. Compound 4 is very interesting, because despite the fact that many [NiCpX(NHC)] complexes have already been reported [49-53], to our knowledge, this is the first tris-MIC-trinickel complex described so far.
To further evaluate the electron-donating character of the tris-MIC ligand B, the iridium hexacarbonyl complex 5 was obtained by bubbling carbon monoxide into a solution of 3 in CH2Cl2. The resulting yellow solid was obtained in 93% yield. The IR spectrum of a CH2Cl2 solution of 5 showed the characteristic CO stretching bands at 2057 and 1972 cm−1 from which a Tolman electronic parameter (TEP) of 2042 cm−1 could be estimated by using the well-accepted correlations [54-56]. This obtained TEP value is slightly lower than the one shown by the tris-NHC analogue A, for which the reported TEP was 2045 cm−1, therefore suggesting that the tris-MIC ligand B is a stronger electron donor than ligand A. However, this comparison must be taken with care, because the tris-carbene ligands A and B, not only differ in the nature of their carbenes (MIC vs NHC), but also in their substituents at the carbene rings, which may also affect the electronic nature of the ligands. It worth mentioning, that the monometallic complex [IrCl(MIC)(CO)2] (MIC = 1,3-bis(2,6-diisopropylphenyl)-4-phenyl-1,2,3-triazolylidene), which may be considered as the monometallic analogue of complex 5, displays an average CO stretching frequency at 2018 cm−1 [32]. This frequency is 4 cm−1 higher than the average frequency observed for 5 (2014 cm−1), indicating a stronger electron-donating character of the tris-MIC ligand. In a similar way, the Tolman electronic parameter of 1-ethyl-3-phenylimidazolyidene (which may be considered as the monocarbene analogue of A) is 2053 cm−1 [57], therefore 8 cm−1 higher than that reported for A. Scheme 3 displays the comparison of the TEP values of the triscarbene ligands A and B, and their related monocarbenes. The results clearly indicate that the tritopic nature of ligands A and B is significantly improving the electron-donating ability of the ligands which by no means should be regarded as the simple combination of the three monocarbenes that constitute the branches of these tris-carbenes.
![[1860-5397-11-278-i3]](/bjoc/content/inline/1860-5397-11-278-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Tolman electronic parameters (TEP) for A, B and their related monocarbenes.
Scheme 3: Tolman electronic parameters (TEP) for A, B and their related monocarbenes.
To gain further insight into the electronic properties of the tris-MIC ligand B, cyclic voltammetry studies of 2 and 3 (Figure 1) and 4 (Figure 2) were performed. The rhodium complex 2 showed an irreversible wave at E1/2 = 0.56 mV, while the iridium complex 3 displayed a pseudo-reversible wave at a half-wave potential of E1/2 = 0.63 mV. Compared to the cyclic voltammetry data obtained for the analogous Rh and Ir complexes with the tris-NHC ligand A (E1/2 = 0.67 mV, for both complexes) [25], the observed lower E1/2 values for complexes 2 and 3 are consistent with a stronger electron-donating character of the tris-MIC ligand B. Further the observation of only one redox wave for both complexes 2 and 3 is consistent with the electronic disconnection of the three metals in both trimetallic complexes.
![[1860-5397-11-278-1]](/bjoc/content/figures/1860-5397-11-278-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: CV plots of complexes 2 (a), and 3 (b). Experiments were carried out using 1 mM solutions of the complexes in dry CH2Cl2 with 0.1 M [NBu4][PF6] as the supporting electrolyte, 100 mVs−1 scan rate, Fc+/Fc used as internal standard with E1/2 (Fc/Fc+) = 0.44 V vs SCE.
Figure 1: CV plots of complexes 2 (a), and 3 (b). Experiments were carried out using 1 mM solutions of the co...
![[1860-5397-11-278-2]](/bjoc/content/figures/1860-5397-11-278-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: CV plot (a) and relevant DPV section (b) of complex 4. Experiments were carried out using 1 mM solution of the complex in dry CH2Cl2 with 0.1 M [NBu4][PF6] as the supporting electrolyte, 100 mVs−1 scan rate, Fc+/ Fc used as internal standard with E1/2 (Fc/Fc+)= 0.44V vs SCE.
Figure 2: CV plot (a) and relevant DPV section (b) of complex 4. Experiments were carried out using 1 mM solu...
The cyclic voltammetry diagram of the tri-Ni(II) complex 4 is shown in Figure 2, together with the differential pulse voltammetry (DPV) plot. The complex shows a quasi-reversible wave at a half-wave potential of E1/2 = 0.64 mV, which is significantly lower than the half-wave potential (E1/2 = 0.72 mV) exhibited by the monometallic NHC-based complex [NiCpCl(IMes)] [50] (IMes = 1,3-mesitylimidazolylidene). This is in agreement with the stronger electron-donating character for the ligand in 4 compared to IMes. From the differential pulse voltammetry (DPV) analysis generated for 4 it can be seen that there is only one redox event taking place, thus evidencing that the trimetallic complex 4 contains three nickel fragments that are essentially decoupled.
Since we previously evaluated the catalytic properties of the tris-NHC Rh(I) complex 6 (Scheme 4) in the rhodium-catalyzed addition of arylboronic acids to α,β-unsaturated ketones [25] we decided to study complex 2 in the same reaction. From these comparative data it was assumed to gaining more information about the effects of the nature of the carbene ligand while maintaining a similar structural environment on the catalyst. The catalytic addition of arylboronic acids to α,β-unsaturated ketones [58-62] is a process for which several Rh(I)-NHC complexes have afforded excellent activities and chemoselectivities [58,63,64].
![[1860-5397-11-278-i4]](/bjoc/content/inline/1860-5397-11-278-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Schematic representation of complex 6.
Scheme 4: Schematic representation of complex 6.
For the catalytic experiments, the arylation of 2-cyclohexen-1-one with several arylboronic acids was studied. The results obtained with 0.066 mol % catalyst 2 were compared with those previously obtained with catalyst 6. As can be seen from the data collected in Table 1, the activity of complex 2 is lower than that shown by complex 6, both in terms of conversion and selectivity. For all reactions carried out in the presence of catalyst 2, deborylation of the boronic acids took place. This side reaction explains the differences found between conversions and yields for all reactions performed. This observation is more relevant for the case of the use of 4-methoxyphenylboronic acid, for which the formation of anisole is the dominant process (Table 1, entries 5 and 6).
Table 1: 1,4-Addition of arylboronic acids to 2-cyclohex-1-one.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-278-i5.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | R | Conversion (yield, %) |
|---|---|---|---|
| 1 | 6 | H | 100 (91)b |
| 2 | 2 | H | 66 (43)c |
| 3 | 6 | Me | 85 (69)b |
| 4 | 2 | Me | 63 (34)c |
| 5 | 6 | OMe | 48 (16)c |
| 6 | 2 | OMe | 52 (15)c |
aReaction conditions: Catalyst (0.066 mol %), 2-cyclohexen-1-one (0.5 mmol), KOH (0.09 mmol), ArB(OH)2 (0.6 mmol), dry toluene (3 mL). Conversions were determined by gas chromatography (GC), using banisol or c2,4,6-trimethylphenol as internal standards. Yields are given in parentheses. The results given for the use of complex 6 were taken from reference [25].
Conclusion
In summary, this work illustrated the high coordination versatility of a nanosized tris-MIC ligand, by obtaining a series of Rh(I), Ir(I) and Ni(II) complexes. Interestingly, the tris-MIC complex of Ni is the first trimetallic Ni complex with a tris-carbene ligand. The electron-donating properties of the ligand were assessed by cyclic voltammetry and by IR spectroscopy of the corresponding carbonylated tris-Ir(I) complex. Both techniques indicate that the ligand is a stronger electron donor than its related tris-NHC analogue. It is even more important to mention that the tris-MIC ligand is a stronger electron donor than its more closely related mono-MIC ligand – a situation that is also true for the tris-NHC ligand A, compared to its related mono-NHC counterpart. This indicates that the triphenylbenzene core is significantly increasing the electron-donating character of the ligand, compared to the related monocarbenes, thus proving that the tris-carbene ligands should not be regarded as a simple combination of three independent monocarbenes.
Supporting Information
| Supporting Information File 1: Experimental details and copies of spectra. | ||
| Format: PDF | Size: 584.5 KB | Download |
Acknowledgements
We gratefully acknowledge financial support from MINECO of Spain (CTQ2014-51999-P), UJI (P11B2014-02), Generalitat Valenciana (GV/2015/097) and FCT-Fundação para a Ciência e a Tecnologia (UID/Multi/04551/2013 and PTDC/QEQ- QIN/0565/2012). C. M. is thankful to the FPI program for a fellowship, B. R. thanks FCT for IF/00346/2013 and G. G.-B. thanks the MINECO for a postdoctoral grant (FPDI-2013-16525). The authors are grateful to the Serveis Centrals d’Instrumentació Científica (SCIC) of the Universitat Jaume I for providing with spectroscopic and X-ray facilities, and to FCT (RECI/BBB-BQB/0230/2012).
References
-
Hahn, F. E.; Radloff, C.; Pape, T.; Hepp, A. Organometallics 2008, 27, 6408–6410. doi:10.1021/om801007u
Return to citation in text: [1] -
Radloff, C.; Hahn, F. E.; Pape, T.; Fröhlich, R. Dalton Trans. 2009, 7215–7222. doi:10.1039/b907896g
Return to citation in text: [1] -
Radloff, C.; Weigand, J. J.; Hahn, F. E. Dalton Trans. 2009, 9392–9394. doi:10.1039/b916651c
Return to citation in text: [1] -
Conrady, F. M.; Fröhlich, R.; Schulte to Brinke, C.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2011, 133, 11496–11499. doi:10.1021/ja205021p
Return to citation in text: [1] -
Schmidtendorf, M.; Pape, T.; Hahn, F. E. Angew. Chem., Int. Ed. 2012, 51, 2195–2198. doi:10.1002/anie.201107227
Return to citation in text: [1] -
Viciano, M.; Sanaú, M.; Peris, E. Organometallics 2007, 26, 6050–6054. doi:10.1021/om7007919
Return to citation in text: [1] -
Hahn, F. E.; Radloff, C.; Pape, T.; Hepp, A. Chem. – Eur. J. 2008, 14, 10900–10904. doi:10.1002/chem.200801877
Return to citation in text: [1] -
Radloff, C.; Gong, H.-Y.; Schulte to Brinke, C.; Pape, T.; Lynch, V. M.; Sessler, J. L.; Hahn, F. E. Chem. – Eur. J. 2010, 16, 13077–13081. doi:10.1002/chem.201002276
Return to citation in text: [1] -
Rit, A.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2010, 132, 4572–4573. doi:10.1021/ja101490d
Return to citation in text: [1] -
Rit, A.; Pape, T.; Hepp, A.; Hahn, F. E. Organometallics 2011, 30, 334–347. doi:10.1021/om101102j
Return to citation in text: [1] -
Wang, D.; Zhang, B.; He, C.; Wu, P.; Duan, C. Chem. Commun. 2010, 46, 4728–4730. doi:10.1039/c000793e
Return to citation in text: [1] -
Segarra, C.; Guisado-Barrios, G.; Hahn, F. E.; Peris, E. Organometallics 2014, 33, 5077–5080. doi:10.1021/om500729b
Return to citation in text: [1] [2] -
Sinha, N.; Roelfes, F.; Hepp, A.; Mejuto, C.; Peris, E.; Hahn, F. E. Organometallics 2014, 33, 6898–6904. doi:10.1021/om500973b
Return to citation in text: [1] [2] [3] [4] -
Guerret, O.; Solé, S.; Gornitzka, H.; Teichert, M.; Trinquier, G.; Bertrand, G. J. Am. Chem. Soc. 1997, 119, 6668–6669. doi:10.1021/ja964191a
Return to citation in text: [1] -
Karimi, B.; Akhavan, P. F. Chem. Commun. 2011, 47, 7686–7688. doi:10.1039/c1cc00017a
Return to citation in text: [1] -
Karimi, B.; Akhavan, P. F. Inorg. Chem. 2011, 50, 6063–6072. doi:10.1021/ic2000766
Return to citation in text: [1] -
Karimi, B.; Akhavan, P. F. Chem. Commun. 2009, 3750–3752. doi:10.1039/b902096a
Return to citation in text: [1] -
Boydston, A. J.; Bielawski, C. W. Dalton Trans. 2006, 4073–4077. doi:10.1039/b607696n
Return to citation in text: [1] -
Mercs, L.; Neels, A.; Albrecht, M. Dalton Trans. 2008, 5570–5576. doi:10.1039/b809721f
Return to citation in text: [1] -
Mercs, L.; Neels, A.; Stoeckli-Evans, H.; Albrecht, M. Dalton Trans. 2009, 7168–7178. doi:10.1039/b907018d
Return to citation in text: [1] -
Zhang, C.; Wang, J.-J.; Liu, Y.; Ma, H.; Yang, X.-L.; Xu, H.-B. Chem. – Eur. J. 2013, 19, 5004–5008. doi:10.1002/chem.201203975
Return to citation in text: [1] [2] -
Choi, J.; Yang, H. Y.; Kim, H. J.; Son, S. U. Angew. Chem., Int. Ed. 2010, 49, 7718–7722. doi:10.1002/anie.201003101
Return to citation in text: [1] [2] -
Gonell, S.; Poyatos, M.; Peris, E. Angew. Chem., Int. Ed. 2013, 52, 7009–7013. doi:10.1002/anie.201302686
Return to citation in text: [1] [2] [3] -
Guisado-Barrios, G.; Hiller, J.; Peris, E. Chem. – Eur. J. 2013, 19, 10405–10411. doi:10.1002/chem.201300486
Return to citation in text: [1] -
Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Mata, J. A.; Hahn, F. E.; Peris, E. Chem. Sci. 2014, 5, 1723–1732. doi:10.1039/c3sc53126k
Return to citation in text: [1] -
Mejuto, C.; Guisado-Barrios, G.; Gusev, D.; Peris, E. Chem. Commun. 2015, 51, 13914–13917. doi:10.1039/C5CC05114B
Return to citation in text: [1] [2] [3] -
Segarra, C.; Linke, J.; Mas-Marzá, E.; Kuck, D.; Peris, E. Chem. Commun. 2013, 49, 10572–10574. doi:10.1039/c3cc46155f
Return to citation in text: [1] -
Gonell, S.; Alabau, R. G.; Poyatos, M.; Peris, E. Chem. Commun. 2013, 49, 7126–7128. doi:10.1039/c3cc44109a
Return to citation in text: [1] -
Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1660–1733. doi:10.1039/C3CS60027K
Return to citation in text: [1] -
Helms, B.; Fréchet, J. M. J. Adv. Synth. Catal. 2006, 348, 1125–1148. doi:10.1002/adsc.200606095
Return to citation in text: [1] -
Bouffard, J.; Keitz, B. K.; Tonner, R.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R. H.; Bertrand, G. Organometallics 2011, 30, 2617–2627. doi:10.1021/om200272m
Return to citation in text: [1] [2] -
Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759–4762. doi:10.1002/anie.201001864
Return to citation in text: [1] -
Keitz, B. K.; Bouffard, J.; Bertrand, G.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8498–8501. doi:10.1021/ja203070r
Return to citation in text: [1] -
Aldeco-Perez, E.; Rosenthal, A. J.; Donnadieu, B.; Parameswaran, P.; Frenking, G.; Bertrand, G. Science 2009, 326, 556–559. doi:10.1126/science.1178206
Return to citation in text: [1] -
Mathew, P.; Neels, A.; Albrecht, M. J. Am. Chem. Soc. 2008, 130, 13534–13535. doi:10.1021/ja805781s
Return to citation in text: [1] -
Zamora, M. T.; Ferguson, M. J.; Cowie, M. Organometallics 2012, 31, 5384–5395. doi:10.1021/om300423z
Return to citation in text: [1] -
Maity, R.; van der Meer, M.; Sarkar, B. Dalton Trans. 2015, 44, 46–49. doi:10.1039/C4DT03239J
Return to citation in text: [1] -
Hohloch, S.; Kaiser, S.; Duecker, F. L.; Bolje, A.; Maity, R.; Košmrlj, J.; Sarkar, B. Dalton Trans. 2015, 44, 686–693. doi:10.1039/C4DT02879A
Return to citation in text: [1] -
Maity, R.; Hohloch, S.; Su, C.-Y.; van der Meer, M.; Sarkar, B. Chem. – Eur. J. 2014, 20, 9952–9961. doi:10.1002/chem.201402838
Return to citation in text: [1] -
Keske, E. C.; Zenkina, O. V.; Wang, R.; Crudden, C. M. Organometallics 2012, 31, 456–461. doi:10.1021/om201104f
Return to citation in text: [1] -
Keske, E. C.; Zenkina, O. V.; Wang, R.; Crudden, C. M. Organometallics 2012, 31, 6215–6221. doi:10.1021/om3005228
Return to citation in text: [1] -
Cai, J.; Yang, X.; Arumugam, K.; Bielawski, C. W.; Sessler, J. L. Organometallics 2011, 30, 5033–5037. doi:10.1021/om200670f
Return to citation in text: [1] -
Maity, R.; Van der Meer, M.; Hohloch, S.; Sarkar, A. Organometallics 2015, 34, 3090–3096. doi:10.1021/acs.organomet.5b00365
Return to citation in text: [1] -
Bezuidenhout, D. I.; Kleinhans, G.; Guisado-Barrios, G.; Liles, D. C.; Ung, G.; Bertrand, G. Chem. Commun. 2014, 50, 2431–2433. doi:10.1039/c3cc49385g
Return to citation in text: [1] -
Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Organometallics 2011, 30, 6017–6021. doi:10.1021/om200844b
Return to citation in text: [1] -
Maity, R.; Mekic, A.; van der Meer, M.; Verma, A.; Sarkar, B. Chem. Commun. 2015, 51, 15106–15109. doi:10.1039/C5CC05506G
Return to citation in text: [1] -
Savka, R.; Plenio, H. Dalton Trans. 2015, 44, 891–893. doi:10.1039/C4DT03449J
Return to citation in text: [1] -
Wei, Y.; Petronilho, A.; Mueller-Bunz, H.; Albrecht, M. Organometallics 2014, 33, 5834–5844. doi:10.1021/om500593s
Return to citation in text: [1] [2] -
Luca, O. R.; Thompson, B. A.; Takase, M. K.; Crabtree, R. H. J. Organomet. Chem. 2013, 730, 79–83. doi:10.1016/j.jorganchem.2012.10.038
Return to citation in text: [1] [2] -
Abernethy, C. D.; Cowley, A. H.; Jones, R. A. J. Organomet. Chem. 2000, 596, 3–5. doi:10.1016/S0022-328X(99)00557-4
Return to citation in text: [1] -
Kelly, R. A., III; Scott, N. M.; Diez-González, S.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, 3442–3447. doi:10.1021/om0501879
Return to citation in text: [1] -
Valdés, H.; Poyatos, M.; Ujaque, G.; Peris, E. Chem. – Eur. J. 2015, 21, 1578–1588. doi:10.1002/chem.201404618
Return to citation in text: [1] -
Chianese, A. R.; Li, X.; Janzen, M. C.; Faller, J. W.; Crabtree, R. H. Organometallics 2003, 22, 1663–1667. doi:10.1021/om021029+
Return to citation in text: [1] -
Kelly, R. A., III; Clavier, H.; Giudice, S.; Scott, N. M.; Stevens, E. D.; Bordner, J.; Samardjiev, I.; Hoff, C. D.; Cavallo, L.; Nolan, S. P. Organometallics 2008, 27, 202–210. doi:10.1021/om701001g
Return to citation in text: [1] -
Nelson, D. J.; Nolan, S. P. Chem. Soc. Rev. 2013, 42, 6723–6753. doi:10.1039/c3cs60146c
Return to citation in text: [1] -
Gusev, D. G. Organometallics 2009, 28, 6458–6461. doi:10.1021/om900654g
Return to citation in text: [1] -
Truscott, B. J.; Fortman, G. C.; Slawin, A. M. Z.; Nolan, S. P. Org. Biomol. Chem. 2011, 9, 7038–7041. doi:10.1039/c1ob06112g
Return to citation in text: [1] [2] -
Pucheault, M.; Darses, S.; Genet, J.-P. Tetrahedron Lett. 2002, 43, 6155–6157. doi:10.1016/S0040-4039(02)01288-1
Return to citation in text: [1] -
Pucheault, M.; Darses, S.; Genet, J.-P. Eur. J. Org. Chem. 2002, 3552–3557. doi:10.1002/1099-0690(200211)2002:21<3552::AID-EJOC3552>3.0.CO;2-4
Return to citation in text: [1] -
Sakai, M.; Hayashi, H.; Miyaura, N. Organometallics 1997, 16, 4229–4231. doi:10.1021/om9705113
Return to citation in text: [1] -
Hayashi, T.; Takahashi, M.; Takaya, Y.; Ogasawara, M. J. Am. Chem. Soc. 2002, 124, 5052–5058. doi:10.1021/ja012711i
Return to citation in text: [1] -
Peñafiel, I.; Pastor, I. M.; Yus, M.; Esteruelas, M. A.; Oliván, M. Organometallics 2012, 31, 6154–6161. doi:10.1021/om300498e
Return to citation in text: [1] -
Bratko, I.; Guisado-Barrios, G.; Favier, I.; Mallet-Ladeira, S.; Teuma, E.; Peris, E.; Gómez, M. Eur. J. Org. Chem. 2014, 2160–2167. doi:10.1002/ejoc.201301220
Return to citation in text: [1]
| 58. | Truscott, B. J.; Fortman, G. C.; Slawin, A. M. Z.; Nolan, S. P. Org. Biomol. Chem. 2011, 9, 7038–7041. doi:10.1039/c1ob06112g |
| 63. | Peñafiel, I.; Pastor, I. M.; Yus, M.; Esteruelas, M. A.; Oliván, M. Organometallics 2012, 31, 6154–6161. doi:10.1021/om300498e |
| 64. | Bratko, I.; Guisado-Barrios, G.; Favier, I.; Mallet-Ladeira, S.; Teuma, E.; Peris, E.; Gómez, M. Eur. J. Org. Chem. 2014, 2160–2167. doi:10.1002/ejoc.201301220 |
| 25. | Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g |
| 1. | Hahn, F. E.; Radloff, C.; Pape, T.; Hepp, A. Organometallics 2008, 27, 6408–6410. doi:10.1021/om801007u |
| 2. | Radloff, C.; Hahn, F. E.; Pape, T.; Fröhlich, R. Dalton Trans. 2009, 7215–7222. doi:10.1039/b907896g |
| 3. | Radloff, C.; Weigand, J. J.; Hahn, F. E. Dalton Trans. 2009, 9392–9394. doi:10.1039/b916651c |
| 4. | Conrady, F. M.; Fröhlich, R.; Schulte to Brinke, C.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2011, 133, 11496–11499. doi:10.1021/ja205021p |
| 5. | Schmidtendorf, M.; Pape, T.; Hahn, F. E. Angew. Chem., Int. Ed. 2012, 51, 2195–2198. doi:10.1002/anie.201107227 |
| 6. | Viciano, M.; Sanaú, M.; Peris, E. Organometallics 2007, 26, 6050–6054. doi:10.1021/om7007919 |
| 23. | Gonell, S.; Poyatos, M.; Peris, E. Angew. Chem., Int. Ed. 2013, 52, 7009–7013. doi:10.1002/anie.201302686 |
| 24. | Guisado-Barrios, G.; Hiller, J.; Peris, E. Chem. – Eur. J. 2013, 19, 10405–10411. doi:10.1002/chem.201300486 |
| 25. | Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g |
| 37. | Zamora, M. T.; Ferguson, M. J.; Cowie, M. Organometallics 2012, 31, 5384–5395. doi:10.1021/om300423z |
| 38. | Maity, R.; van der Meer, M.; Sarkar, B. Dalton Trans. 2015, 44, 46–49. doi:10.1039/C4DT03239J |
| 39. | Hohloch, S.; Kaiser, S.; Duecker, F. L.; Bolje, A.; Maity, R.; Košmrlj, J.; Sarkar, B. Dalton Trans. 2015, 44, 686–693. doi:10.1039/C4DT02879A |
| 40. | Maity, R.; Hohloch, S.; Su, C.-Y.; van der Meer, M.; Sarkar, B. Chem. – Eur. J. 2014, 20, 9952–9961. doi:10.1002/chem.201402838 |
| 41. | Keske, E. C.; Zenkina, O. V.; Wang, R.; Crudden, C. M. Organometallics 2012, 31, 456–461. doi:10.1021/om201104f |
| 42. | Keske, E. C.; Zenkina, O. V.; Wang, R.; Crudden, C. M. Organometallics 2012, 31, 6215–6221. doi:10.1021/om3005228 |
| 43. | Cai, J.; Yang, X.; Arumugam, K.; Bielawski, C. W.; Sessler, J. L. Organometallics 2011, 30, 5033–5037. doi:10.1021/om200670f |
| 44. | Maity, R.; Van der Meer, M.; Hohloch, S.; Sarkar, A. Organometallics 2015, 34, 3090–3096. doi:10.1021/acs.organomet.5b00365 |
| 45. | Bezuidenhout, D. I.; Kleinhans, G.; Guisado-Barrios, G.; Liles, D. C.; Ung, G.; Bertrand, G. Chem. Commun. 2014, 50, 2431–2433. doi:10.1039/c3cc49385g |
| 46. | Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Organometallics 2011, 30, 6017–6021. doi:10.1021/om200844b |
| 21. | Zhang, C.; Wang, J.-J.; Liu, Y.; Ma, H.; Yang, X.-L.; Xu, H.-B. Chem. – Eur. J. 2013, 19, 5004–5008. doi:10.1002/chem.201203975 |
| 22. | Choi, J.; Yang, H. Y.; Kim, H. J.; Son, S. U. Angew. Chem., Int. Ed. 2010, 49, 7718–7722. doi:10.1002/anie.201003101 |
| 27. | Mejuto, C.; Guisado-Barrios, G.; Gusev, D.; Peris, E. Chem. Commun. 2015, 51, 13914–13917. doi:10.1039/C5CC05114B |
| 14. | Guerret, O.; Solé, S.; Gornitzka, H.; Teichert, M.; Trinquier, G.; Bertrand, G. J. Am. Chem. Soc. 1997, 119, 6668–6669. doi:10.1021/ja964191a |
| 15. | Karimi, B.; Akhavan, P. F. Chem. Commun. 2011, 47, 7686–7688. doi:10.1039/c1cc00017a |
| 16. | Karimi, B.; Akhavan, P. F. Inorg. Chem. 2011, 50, 6063–6072. doi:10.1021/ic2000766 |
| 17. | Karimi, B.; Akhavan, P. F. Chem. Commun. 2009, 3750–3752. doi:10.1039/b902096a |
| 18. | Boydston, A. J.; Bielawski, C. W. Dalton Trans. 2006, 4073–4077. doi:10.1039/b607696n |
| 19. | Mercs, L.; Neels, A.; Albrecht, M. Dalton Trans. 2008, 5570–5576. doi:10.1039/b809721f |
| 20. | Mercs, L.; Neels, A.; Stoeckli-Evans, H.; Albrecht, M. Dalton Trans. 2009, 7168–7178. doi:10.1039/b907018d |
| 21. | Zhang, C.; Wang, J.-J.; Liu, Y.; Ma, H.; Yang, X.-L.; Xu, H.-B. Chem. – Eur. J. 2013, 19, 5004–5008. doi:10.1002/chem.201203975 |
| 22. | Choi, J.; Yang, H. Y.; Kim, H. J.; Son, S. U. Angew. Chem., Int. Ed. 2010, 49, 7718–7722. doi:10.1002/anie.201003101 |
| 13. | Sinha, N.; Roelfes, F.; Hepp, A.; Mejuto, C.; Peris, E.; Hahn, F. E. Organometallics 2014, 33, 6898–6904. doi:10.1021/om500973b |
| 7. | Hahn, F. E.; Radloff, C.; Pape, T.; Hepp, A. Chem. – Eur. J. 2008, 14, 10900–10904. doi:10.1002/chem.200801877 |
| 8. | Radloff, C.; Gong, H.-Y.; Schulte to Brinke, C.; Pape, T.; Lynch, V. M.; Sessler, J. L.; Hahn, F. E. Chem. – Eur. J. 2010, 16, 13077–13081. doi:10.1002/chem.201002276 |
| 9. | Rit, A.; Pape, T.; Hahn, F. E. J. Am. Chem. Soc. 2010, 132, 4572–4573. doi:10.1021/ja101490d |
| 10. | Rit, A.; Pape, T.; Hepp, A.; Hahn, F. E. Organometallics 2011, 30, 334–347. doi:10.1021/om101102j |
| 11. | Wang, D.; Zhang, B.; He, C.; Wu, P.; Duan, C. Chem. Commun. 2010, 46, 4728–4730. doi:10.1039/c000793e |
| 12. | Segarra, C.; Guisado-Barrios, G.; Hahn, F. E.; Peris, E. Organometallics 2014, 33, 5077–5080. doi:10.1021/om500729b |
| 13. | Sinha, N.; Roelfes, F.; Hepp, A.; Mejuto, C.; Peris, E.; Hahn, F. E. Organometallics 2014, 33, 6898–6904. doi:10.1021/om500973b |
| 32. | Bouffard, J.; Keitz, B. K.; Tonner, R.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R. H.; Bertrand, G. Organometallics 2011, 30, 2617–2627. doi:10.1021/om200272m |
| 33. | Guisado-Barrios, G.; Bouffard, J.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2010, 49, 4759–4762. doi:10.1002/anie.201001864 |
| 34. | Keitz, B. K.; Bouffard, J.; Bertrand, G.; Grubbs, R. H. J. Am. Chem. Soc. 2011, 133, 8498–8501. doi:10.1021/ja203070r |
| 35. | Aldeco-Perez, E.; Rosenthal, A. J.; Donnadieu, B.; Parameswaran, P.; Frenking, G.; Bertrand, G. Science 2009, 326, 556–559. doi:10.1126/science.1178206 |
| 36. | Mathew, P.; Neels, A.; Albrecht, M. J. Am. Chem. Soc. 2008, 130, 13534–13535. doi:10.1021/ja805781s |
| 13. | Sinha, N.; Roelfes, F.; Hepp, A.; Mejuto, C.; Peris, E.; Hahn, F. E. Organometallics 2014, 33, 6898–6904. doi:10.1021/om500973b |
| 27. | Mejuto, C.; Guisado-Barrios, G.; Gusev, D.; Peris, E. Chem. Commun. 2015, 51, 13914–13917. doi:10.1039/C5CC05114B |
| 31. | Helms, B.; Fréchet, J. M. J. Adv. Synth. Catal. 2006, 348, 1125–1148. doi:10.1002/adsc.200606095 |
| 23. | Gonell, S.; Poyatos, M.; Peris, E. Angew. Chem., Int. Ed. 2013, 52, 7009–7013. doi:10.1002/anie.201302686 |
| 25. | Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g |
| 28. | Segarra, C.; Linke, J.; Mas-Marzá, E.; Kuck, D.; Peris, E. Chem. Commun. 2013, 49, 10572–10574. doi:10.1039/c3cc46155f |
| 29. | Gonell, S.; Alabau, R. G.; Poyatos, M.; Peris, E. Chem. Commun. 2013, 49, 7126–7128. doi:10.1039/c3cc44109a |
| 25. | Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g |
| 12. | Segarra, C.; Guisado-Barrios, G.; Hahn, F. E.; Peris, E. Organometallics 2014, 33, 5077–5080. doi:10.1021/om500729b |
| 13. | Sinha, N.; Roelfes, F.; Hepp, A.; Mejuto, C.; Peris, E.; Hahn, F. E. Organometallics 2014, 33, 6898–6904. doi:10.1021/om500973b |
| 27. | Mejuto, C.; Guisado-Barrios, G.; Gusev, D.; Peris, E. Chem. Commun. 2015, 51, 13914–13917. doi:10.1039/C5CC05114B |
| 23. | Gonell, S.; Poyatos, M.; Peris, E. Angew. Chem., Int. Ed. 2013, 52, 7009–7013. doi:10.1002/anie.201302686 |
| 26. | Mata, J. A.; Hahn, F. E.; Peris, E. Chem. Sci. 2014, 5, 1723–1732. doi:10.1039/c3sc53126k |
| 30. | Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1660–1733. doi:10.1039/C3CS60027K |
| 49. | Wei, Y.; Petronilho, A.; Mueller-Bunz, H.; Albrecht, M. Organometallics 2014, 33, 5834–5844. doi:10.1021/om500593s |
| 47. | Maity, R.; Mekic, A.; van der Meer, M.; Verma, A.; Sarkar, B. Chem. Commun. 2015, 51, 15106–15109. doi:10.1039/C5CC05506G |
| 48. | Savka, R.; Plenio, H. Dalton Trans. 2015, 44, 891–893. doi:10.1039/C4DT03449J |
| 25. | Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g |
| 58. | Truscott, B. J.; Fortman, G. C.; Slawin, A. M. Z.; Nolan, S. P. Org. Biomol. Chem. 2011, 9, 7038–7041. doi:10.1039/c1ob06112g |
| 59. | Pucheault, M.; Darses, S.; Genet, J.-P. Tetrahedron Lett. 2002, 43, 6155–6157. doi:10.1016/S0040-4039(02)01288-1 |
| 60. | Pucheault, M.; Darses, S.; Genet, J.-P. Eur. J. Org. Chem. 2002, 3552–3557. doi:10.1002/1099-0690(200211)2002:21<3552::AID-EJOC3552>3.0.CO;2-4 |
| 61. | Sakai, M.; Hayashi, H.; Miyaura, N. Organometallics 1997, 16, 4229–4231. doi:10.1021/om9705113 |
| 62. | Hayashi, T.; Takahashi, M.; Takaya, Y.; Ogasawara, M. J. Am. Chem. Soc. 2002, 124, 5052–5058. doi:10.1021/ja012711i |
| 25. | Mejuto, C.; Guisado-Barrios, G.; Peris, E. Organometallics 2014, 33, 3205–3211. doi:10.1021/om500547g |
| 50. | Luca, O. R.; Thompson, B. A.; Takase, M. K.; Crabtree, R. H. J. Organomet. Chem. 2013, 730, 79–83. doi:10.1016/j.jorganchem.2012.10.038 |
| 32. | Bouffard, J.; Keitz, B. K.; Tonner, R.; Guisado-Barrios, G.; Frenking, G.; Grubbs, R. H.; Bertrand, G. Organometallics 2011, 30, 2617–2627. doi:10.1021/om200272m |
| 49. | Wei, Y.; Petronilho, A.; Mueller-Bunz, H.; Albrecht, M. Organometallics 2014, 33, 5834–5844. doi:10.1021/om500593s |
| 50. | Luca, O. R.; Thompson, B. A.; Takase, M. K.; Crabtree, R. H. J. Organomet. Chem. 2013, 730, 79–83. doi:10.1016/j.jorganchem.2012.10.038 |
| 51. | Abernethy, C. D.; Cowley, A. H.; Jones, R. A. J. Organomet. Chem. 2000, 596, 3–5. doi:10.1016/S0022-328X(99)00557-4 |
| 52. | Kelly, R. A., III; Scott, N. M.; Diez-González, S.; Stevens, E. D.; Nolan, S. P. Organometallics 2005, 24, 3442–3447. doi:10.1021/om0501879 |
| 53. | Valdés, H.; Poyatos, M.; Ujaque, G.; Peris, E. Chem. – Eur. J. 2015, 21, 1578–1588. doi:10.1002/chem.201404618 |
| 54. | Chianese, A. R.; Li, X.; Janzen, M. C.; Faller, J. W.; Crabtree, R. H. Organometallics 2003, 22, 1663–1667. doi:10.1021/om021029+ |
| 55. | Kelly, R. A., III; Clavier, H.; Giudice, S.; Scott, N. M.; Stevens, E. D.; Bordner, J.; Samardjiev, I.; Hoff, C. D.; Cavallo, L.; Nolan, S. P. Organometallics 2008, 27, 202–210. doi:10.1021/om701001g |
| 56. | Nelson, D. J.; Nolan, S. P. Chem. Soc. Rev. 2013, 42, 6723–6753. doi:10.1039/c3cs60146c |
© 2015 Mejuto et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)