Abstract
A novel and practical asymmetric synthesis of dapoxetine hydrochloride by using the chiral auxiliary (S)-tert-butanesulfinamide was explored. The synthesis was concise, mild, and easy to perform. The overall yield and stereoselectivity were excellent.

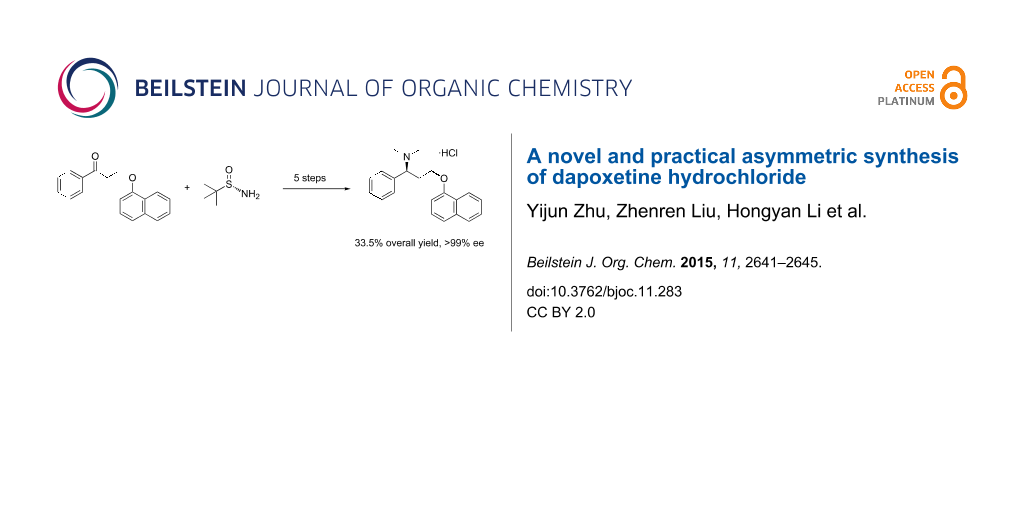
Graphical Abstract
Introduction
Premature ejaculation (PE) is the most frequent form of ejaculatory dysfunction with a distribution of 39% of the general male population [1,2]. Dapoxetine hydrochloride (1, (S)-(+)-N,N-dimethyl-[3-(naphthalen-1-yloxy)-1-phenylpropyl]amine hydrochloride, Figure 1) was approved by EMA in 2009 for the special treatment of PE [3,4]. By virtue of its fast acting property and rapid elimination from the body, it is one of the more effective and safe drugs for treating PE.
![[1860-5397-11-283-1]](/bjoc/content/figures/1860-5397-11-283-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
For this reason, the synthesis of this interesting drug has attracted great attention, especially asymmetric synthesis approaches. However, only a few methods have been reported for the synthesis of enantiopure dapoxetine hydrochloride. The earlier methods included chiral/enzymatic resolution [5], whereas the newer approaches encompass asymmetric dihydroxylation of trans-methyl cinnamate or cinnamyl alcohol [6], chiral azetidin-2,3-dione [7], asymmetric C–H amination reactions of a prochiral sulfamate [8], oxazaborolidine reduction of 3-chloropropiophenone or ketone [9], and an imidazolidin-2-one chiral auxiliary mediated acetate aldol reaction [10]. However, these methods are undermined by poor yield, low enantioselectivity, and complex synthetic procedure.
Chiral tert-butanesulfinamide, developed by García Ruano and Ellman, has been proven to be a broadly useful reagent for the preparation of chiral amines via the chiral N-tert-butanesulfinylimine intermediates [11,12]. Due to its high diastereoselectivity and convenient cleavage of the N-tert-butanesulfinyl group, it has become an excellent chiral auxiliary in the synthesis of chiral amine compounds [13]. This work was devoted to develop an efficient synthetic route for the synthesis of (S)-dapoxetine (1) through this chiral auxiliary.
Results and Discussion
Herein, a novel and practical synthesis of 1 (Scheme 1) based on (S)-tert-butanesulfinamide (2) was developed.
![[1860-5397-11-283-i1]](/bjoc/content/inline/1860-5397-11-283-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
3-(Naphthalen-1-yloxy)-1-phenylpropan-1-one (3), which was commercially available from J&K Chemical Ltd., was chosen as a key building block to be condensed with 2 to form the imine. The reaction in the presence of Ti(OEt)4 gave compound 4 in 78% yield [14] (Scheme 1).
The diastereoselective reduction of imine 4 (Scheme 2) was the key step in this route. Accordingly, various conditions were screened and the results are presented in Table 1.
![[1860-5397-11-283-i2]](/bjoc/content/inline/1860-5397-11-283-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 1: Conditions for the reduction of sulfinylimine 4.
| entry | reductant | solvent | T (°C) | time (h) | crude product (5:5’:5”) | dea (%) |
|---|---|---|---|---|---|---|
| 1 | NaBH4 (0.8 equiv) | THF | 25 | 1 | 28%:9%:62% | 51 |
| 2 | NaBH4 (0.8 equiv) | THF | −30 | 3 | 56%:11%:33% | 67 |
| 3 | NaBH4 (0.8 equiv) | THF | −70 | 4 | 60%:12%:28% | 67 |
| 4b | NaBH4 (0.8 equiv) | THF | 25 | 2 | 67%:22%:11% | 61 |
| 5b | NaBH4 (0.8 equiv) | THF | −30 | 2.5 | 70%:21%:2% | 54 |
| 6 | NaBH3CN (1 equiv) | THF | −20 | 2 | NDc | |
| 7 | BH3 (1 equiv) | THF | −20 | 2 | 82%:14%:4% | 71 |
| 8 | BH3 (0.8 equiv) | THF | −20 | 2 | 81%:13%:6% | 72 |
| 9 | BH3 (0.6 equiv) | THF | −20 | 6 | 76%:14%:6% | 69 |
| 10 | BH3 (0.8 equiv) | THF | 0 | 1 | 81%:17%:2% | 66 |
| 11 | BH3 (0.8 equiv) | THF | 25 | 0.5 | 81%:15%:4% | 69 |
| 12 | BH3 (0.8 equiv) | MTBE | 25 | 1 | 80%:11%:9% | 76 |
| 13 | BH3 (0.8 equiv) | 2-MeTHF | 25 | 1 | 80%:16%:4% | 67 |
| 14 | BH3 (0.8 equiv) | IPEd | 25 | 1 | 85%:10%:4% | 78 |
| 15 | BH3 (0.8 equiv) | IPEd | 0 | 1.5 | 87%:5%:1% | 89 |
| 16 | BH3 (0.8 equiv) | IPEd | −25 | 3 | e | 85 |
aDiastereoisomeric excess of 5 and 5’; badded AcOH (0.1 equiv) in the reaction; cno products were detected; ddiisopropyl ether; e5:5’:4 55%:4%:40%.
Following a procedure reported in the literature [14], the reduction of 4 was carried out with NaBH4 in THF at 25 °C for 1 h (Table 1, entry 1). However, the main product was proven to be the denaphthalenyloxy compound 5’’ by 1H NMR and MS while the desired sulfinamide 5 was obtained only in a yield of 28%. The amount of denaphthalenyloxy was greatly reduced when the reaction temperature was decreased to −30 °C (Table 1, entry 2), but no significant improvement was achieved by further decreasing the temperature (Table 1, entry 3). It was assumed that after reduction, the basicity resulting from NaBH4 might lead to the denaphthalenyloxylation. Therefore, AcOH was used as an additive in the reaction. The results showed that the denaphthalenyloxylation was almost negligible, but the diastereoselectivity was not good enough (Table 1, entry 5). Although mild reductant, NaBH3CN, was then applied in the reaction, no reaction took place (Table 1, entry 6). When BH3 was used to react with 4 at −20 °C for 2 h, the result was promising in terms of both yield and de (Table 1, entry 7). The data of entries 7–11 indicated that 0.8 equiv of the reductant BH3 was sufficient and the optimized temperature was 0–25 °C when the reaction was carried out in THF. When other solvents were tested (Table 1, entries 12–14), it was found that diisopropyl ether gave the best result. Finally, the reaction was performed with 0.8 equiv BH3 in isopropyl ether at 0 °C for 1.5 h (Table 1, entry 15) and the de of the crude product was 89%. Compound 5 was isolated in pure form from the crude reaction mixture by recrystallization from 10% ethyl acetate/n-heptane in 79.2% yield with 99.0% de.
Then purified 5 was hydrolyzed in methanol with HCl/EtOH solution at room temperature and dissociated with NaHCO3 to give the primary amine 6 in 90.0% yield. The reductive amination of 6 under Eschweiler–Clarke conditions furnished (S)-dapoxetine 7 with excellent enantiopurity (99.3% ee) in 74.7% yield. After salt formation and recrystallization, the target compound 1 was obtained. The optical rotation value of compound 1 was consistent with that previously reported [15], which confirmed that the S-enantiomer of dapoxetine hydrochloride was synthesized successfully by using this route.
Conclusion
In summary, a novel and stereoselective synthesis of dapoxetine hydrochloride starting from commercially available 3-(naphthalen-1-yloxy)-1-phenylpropan-1-one in five linear steps (33.5% overall yield) via introduction of the chiral auxiliary (S)-tert-butanesulfinamide was developed. This method was easy to perform and both the purity and yield of the product were excellent.
Experimental
All solvents and reagents were of reagent grade and used without further purification. 1H and 13C NMR spectra were recorded using a Bruker 400 MHz spectrometer with TMS as an internal standard. HPLC analyses were recorded with on a Dionex Ultimate 3000 chromatograph and chiral HPLC analyses were recorded with an Agilent 1100 Series spectrometer.
Preparation of (S)-2-methyl-N-(3-(naphthalen-1-yloxy)-1-phenylpropylidene)propane-2-sulfinamide (4): To a solution of 3 (30 g, 0.11 mol) and (S)-tert-butanesulfinamide (14.7 g, 0.12 mol) in THF (300 mL), Ti(OEt)4 (61.8 g, 0.22 mol) was added under N2 atmosphere and the mixture was refluxed at 65 °C for about 8 h. Upon completion (as determined by TLC), the reaction mixture was first cooled to rt and then quenched with ethyl acetate (300 mL) and brine (300 mL), then stirred for 1 h, filtered, and the filtrate was washed with brine and dried over Na2SO4 and concentrated. The residue was purified via flash chromatography with petrol ether/ethyl acetate (20:1) to give 4 as a pale yellow solid. Yield 32.3 g (78.3%); mp 49–51 °C; ![[Graphic 1]](/bjoc/content/inline/1860-5397-11-283-i3.svg?max-width=637&scale=1.18182) −10 (c 0.8, CDCl3); 1H NMR (400 MHz, CDCl3/TMS) δ 8.10 (d, J = 8 Hz, 1H), 7.79 (d, J = 8 Hz, 1H), 7.51–7.28 (m, 8H), 6.81 (d, J = 7.6 Hz, 1H), 4.62–4.49 (m, 2H), 4.02–3.79 (m, 2H), 1.39 (s, 9H); 13C NMR (100 MHz, CDCl3/TMS) δ 197.7, 154.4, 137.0, 134.6, 133.3, 128.7, 128.2, 127.4, 126.3, 125.8, 125.7, 125.1, 122.0, 120.5, 105.0, 77.3, 77.0, 76.6, 63.9, 38.2; HRMS (ES+) m/z: [M + Na]+ calcd for C23H25NO2NaS, 402.1504; found, 402.1493.
−10 (c 0.8, CDCl3); 1H NMR (400 MHz, CDCl3/TMS) δ 8.10 (d, J = 8 Hz, 1H), 7.79 (d, J = 8 Hz, 1H), 7.51–7.28 (m, 8H), 6.81 (d, J = 7.6 Hz, 1H), 4.62–4.49 (m, 2H), 4.02–3.79 (m, 2H), 1.39 (s, 9H); 13C NMR (100 MHz, CDCl3/TMS) δ 197.7, 154.4, 137.0, 134.6, 133.3, 128.7, 128.2, 127.4, 126.3, 125.8, 125.7, 125.1, 122.0, 120.5, 105.0, 77.3, 77.0, 76.6, 63.9, 38.2; HRMS (ES+) m/z: [M + Na]+ calcd for C23H25NO2NaS, 402.1504; found, 402.1493.
Preparation of (S)-2-methyl-N-((S)-3-(naphthalen-1-yloxy)-1-phenylpropyl)propane-2-sulfinamide (5): To a suspension of 4 (20 g, 53 mmol) in diisopropyl ether (300 mL), BH3/THF (10 mL, 42.2 mmol) was added dropwise at −5 to 0 °C. After this addition, the reaction mixture was stirred for 1.5 h. The color of the reaction changed from yellow to white and TLC showed the complete consumption of 4. Then, ethyl acetate (200 mL) and water (100 mL) were added and the mixture was stirred for 5 min and then separated. The organic phase was washed with brine, dried over Na2SO4, and filtered and concentrated to obtain the crude product. The crude product was crystallized from an n-heptane/ethyl acetate mixture (9:1) to get pure 5 as an off-white solid. Yield 15.9 g (79.2%); mp 60–61 °C; ![[Graphic 2]](/bjoc/content/inline/1860-5397-11-283-i4.svg?max-width=637&scale=1.18182) 65.8 (c 1, CDCl3); de 99.0%; 1H NMR (400 MHz, CDCl3/TMS) δ 8.20 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.51–7.28 (m, 8H), 6.67 (d, J = 7.6 Hz, 1H), 4.80 (m, 1H), 4.15 (m, 1H), 4.02 (m, 1H), 3.58 (d, NH), 2.66 (m, 1H), 2.35 (m, 1H), 1.22 (s, 9H); 13C NMR (100 MHz, CDCl3/TMS) δ 154.5, 141.9, 134.6, 128.9, 128.1, 127.4, 127.2, 126.4, 125.7, 125.3, 122.0, 120.4, 104.6, 94.5, 77.3, 77.2, 77.0, 76.7, 64.7, 56.8, 55.9, 36.5, 22.6; HRMS (ES+) m/z: [M + H]+ calcd for C23H28NO2S, 382.1841; found, 382.1842.
65.8 (c 1, CDCl3); de 99.0%; 1H NMR (400 MHz, CDCl3/TMS) δ 8.20 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.51–7.28 (m, 8H), 6.67 (d, J = 7.6 Hz, 1H), 4.80 (m, 1H), 4.15 (m, 1H), 4.02 (m, 1H), 3.58 (d, NH), 2.66 (m, 1H), 2.35 (m, 1H), 1.22 (s, 9H); 13C NMR (100 MHz, CDCl3/TMS) δ 154.5, 141.9, 134.6, 128.9, 128.1, 127.4, 127.2, 126.4, 125.7, 125.3, 122.0, 120.4, 104.6, 94.5, 77.3, 77.2, 77.0, 76.7, 64.7, 56.8, 55.9, 36.5, 22.6; HRMS (ES+) m/z: [M + H]+ calcd for C23H28NO2S, 382.1841; found, 382.1842.
Preparation of (S)-3-(naphthalen-1-yloxy)-1-phenylpropan-1-amine (6): To a solution of 5 (12 g, 31.5 mmol) dissolved in methanol (60 mL), 28% HCl/EtOH (9 mL, 63 mmol) was added at 10–20 °C. The mixture was stirred for 1 h at room temperature. Then, the mixture was concentrated and the obtained crude residue was resuspended with MTBE (70 mL) to give pure hydrochloride 6. The solid was suspended in DCM (50 mL), and saturated aqueous NaHCO3 solution (15 mL) was added and stirred until the mixture was no longer turbid. The organic phase was washed with brine, dried over Na2SO4, and filtered and concentrated to give 6 as a pale yellow oil. Yield 7.4 g (90.0%); ![[Graphic 3]](/bjoc/content/inline/1860-5397-11-283-i5.svg?max-width=637&scale=1.18182) 66.1 (c 0.3, CDCl3); 1H NMR (400 MHz, CDCl3/TMS) δ 8.26 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.51–7.28 (m, 8H), 6.75 (d, 1H), 4.35 (m, 1H), 4.23 (m, 1H), 4.11 (m, 1H), 2.34 (m, 2H); 13C NMR (100 MHz, CDCl3/TMS) δ 154.6, 145.0, 134.6, 128.7, 127.4, 127.4, 126.4, 126.3, 125.8, 125.1, 122.0, 120.3, 104.8, 77.3, 77.0, 76.7, 65.5, 53.7, 38.5; MS (ES+) m/z: 278 [M + H]+.
66.1 (c 0.3, CDCl3); 1H NMR (400 MHz, CDCl3/TMS) δ 8.26 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.51–7.28 (m, 8H), 6.75 (d, 1H), 4.35 (m, 1H), 4.23 (m, 1H), 4.11 (m, 1H), 2.34 (m, 2H); 13C NMR (100 MHz, CDCl3/TMS) δ 154.6, 145.0, 134.6, 128.7, 127.4, 127.4, 126.4, 126.3, 125.8, 125.1, 122.0, 120.3, 104.8, 77.3, 77.0, 76.7, 65.5, 53.7, 38.5; MS (ES+) m/z: 278 [M + H]+.
Preparation of dapoxetine ((S)-N,N-dimethyl-3-(naphthalen-1-yloxy)-1-phenylpropan-1-amine, 7): To a 50 mL round-bottomed flask, 6 (6 g, 21.6 mmol), 98% HCOOH (3.9 mL, 54.1 mmol) and an aqueous solution of 30% formaldehyde (9.7 mL, 108 mmol) were added at room temperature. The reaction mixture was heated to 85 °C for 8 h and quenched with saturated aqueous NaHCO3 solution (pH ≈8). The aqueous layer was extracted with EtOAc (20 mL, twice). The organic phase was washed with water, brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography to afford dapoxetine as a colorless oil. Yield 4.95 g (74.7%); chiral purity (HPLC): 99.63%; 1H NMR (400 MHz, CDCl3/TMS) δ 8.26 (d, J = 9.2 Hz, 1H), 7.79 (d, J = 9.2 Hz, 1H), 7.47 (m, 2H), 7.35–7.28 (m, 7H), 6.67 (d, J = 8 Hz, 1H), 4.13–4.08 (m, 1H), 3.98–3.94 (m, 1H), 3.64–3.60 (m, 1H), 2.67–2.62 (m, 1H), 2.33–2.31 (m, 1H), 2.29 (s, 6H); 13C NMR (100 MHz, CDCl3/TMS) δ 154.7, 134.5, 128.6, 128.2, 127.4, 126.3, 125.8, 125.0, 122.0, 120.1, 104.7, 77.3, 77.0, 76.6, 67.8, 65.7, 42.7, 33.0; MS (ES+) m/z: 306 [M + H]+.
Preparation of dapoxetine hydrochloride (1): To a solution of 7 (3 g, 9.8 mmol) dissolved in diisopropyl ether (30 mL), 28% HCl/EtOH (1.3 mL, 1.2 equiv) was added dropwise at room temperature. A white solid was precipitated and filtered to obtain the crude 1 (2.8 g). The solid was recrystallized from isopropyl alcohol/n-hexane (9 mL:8 mL) to give the product. Yield 2.7 g (80.4%); mp 178–180 °C; ![[Graphic 4]](/bjoc/content/inline/1860-5397-11-283-i6.svg?max-width=637&scale=1.18182) 126.4 (c 1, methanol) [lit. [15] mp 180–184 °C;
126.4 (c 1, methanol) [lit. [15] mp 180–184 °C; ![[Graphic 5]](/bjoc/content/inline/1860-5397-11-283-i7.svg?max-width=637&scale=1.18182) 131.7 (c 1, methanol)]; chiral purity (HPLC): >99% ee; 1H NMR (400 MHz, DMSO-d6/TMS) δ 11.21 (brs, 1H, HCl), 8.06–6.73 (m, 12H), 4.71 (m, 1H), 4.11 (m, 1H), 3.75 (m, 1H), 2.95 (m, 1H), 2.92 (s, 3H), 2.73 (m, 1H), 2.58 (s, 3H); 13C NMR (100 MHz, DMSO-d6/TMS) δ 153.7, 134.0, 132.5, 129.9, 129.7, 129.0, 127.4, 126.5, 126.1, 125.2, 124.9, 121.8, 120.2, 105.1, 67.3, 64.6, 41.4, 40.3, 40.1, 39.9, 39.7, 39.5, 39.3, 39.0, 29.6; MS (ES+) m/z: 306 [M + H]+.
131.7 (c 1, methanol)]; chiral purity (HPLC): >99% ee; 1H NMR (400 MHz, DMSO-d6/TMS) δ 11.21 (brs, 1H, HCl), 8.06–6.73 (m, 12H), 4.71 (m, 1H), 4.11 (m, 1H), 3.75 (m, 1H), 2.95 (m, 1H), 2.92 (s, 3H), 2.73 (m, 1H), 2.58 (s, 3H); 13C NMR (100 MHz, DMSO-d6/TMS) δ 153.7, 134.0, 132.5, 129.9, 129.7, 129.0, 127.4, 126.5, 126.1, 125.2, 124.9, 121.8, 120.2, 105.1, 67.3, 64.6, 41.4, 40.3, 40.1, 39.9, 39.7, 39.5, 39.3, 39.0, 29.6; MS (ES+) m/z: 306 [M + H]+.
Supporting Information
| Supporting Information File 1: 1H NMR, 13C NMR and ESIMS spectra of compounds 1, 4, 5, 5”, 6 and 7 and chiral HPLC diagrams of 1 and 7. | ||
| Format: PDF | Size: 854.9 KB | Download |
References
-
Althof, S. E. J. Urol. 2006, 175, 842–848. doi:10.1016/S0022-5347(05)00341-1
Return to citation in text: [1] -
Zhou, C.; Jiang, X.; Xu, Z.; Guo, L.; Wang, H.; Zhang, D.; Shi, B. J. Sex. Med. 2010, 7, 3750–3757. doi:10.1111/j.1743-6109.2009.01646.x
Return to citation in text: [1] -
Sorbera, L. A.; Castaner, J.; Castaner, R. M. Drugs Future 2004, 29, 1201–1205. doi:10.1358/dof.2004.029.12.865936
Return to citation in text: [1] -
Darcsi, A.; Tóth, G.; Kökösi, J.; Béni, S. J. Pharm. Biomed. Anal. 2014, 96, 272–277. doi:10.1016/j.jpba.2014.04.002
Return to citation in text: [1] -
Torre, O.; Gotor-Fernández, V.; Gotor, V. Tetrahedron: Asymmetry 2006, 17, 860–866. doi:10.1016/j.tetasy.2006.02.022
Return to citation in text: [1] -
Siddiqui, S. A.; Srinivasan, K. V. Tetrahedron: Asymmetry 2007, 18, 2099–2103. doi:10.1016/j.tetasy.2007.07.028
Return to citation in text: [1] -
Chincholkar, P. M.; Kale, A. S.; Gumaste, V. K.; Deshmukh, A. R. A. S. Tetrahedron 2009, 65, 2605–2609. doi:10.1016/j.tet.2008.11.042
Return to citation in text: [1] -
Kang, S.; Lee, H.-K. J. Org. Chem. 2010, 75, 237–240. doi:10.1021/jo902176s
Return to citation in text: [1] -
Kim, S. J.; Jeon, T. H.; Min, I. S.; Kim, I. S.; Jung, Y. H. Tetrahedron Lett. 2012, 53, 3680–3682. doi:10.1016/j.tetlet.2012.05.037
Return to citation in text: [1] -
Khatik, G. L.; Sharma, R.; Kumar, V.; Chouhan, M.; Nair, V. A. Tetrahedron Lett. 2013, 54, 5991–5993. doi:10.1016/j.tetlet.2013.08.059
Return to citation in text: [1] -
Cogan, D. A.; Liu, G.; Kim, K.; Backes, B. J.; Ellman, J. A. J. Am. Chem. Soc. 1998, 120, 8011–8019. doi:10.1021/ja9809206
Return to citation in text: [1] -
Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t
Return to citation in text: [1] -
Arava, V. R.; Gorentla, L.; Dubey, P. K. Beilstein J. Org. Chem. 2012, 8, 1366–1373. doi:10.3762/bjoc.8.158
Return to citation in text: [1] -
Borg, G.; Cogan, D. A.; Ellman, J. A. Tetrahedron Lett. 1999, 40, 6709–6712. doi:10.1016/S0040-4039(99)01351-9
Return to citation in text: [1] [2] -
Dave, A. M.; Patel, D. J.; Kumar, R.; Dwivedi, S. D. A process for preparing (+)-dapoxetine and its salts. WO Pat. Appl. WO2008035358A2, June 5, 2007.
Return to citation in text: [1] [2]
| 1. | Althof, S. E. J. Urol. 2006, 175, 842–848. doi:10.1016/S0022-5347(05)00341-1 |
| 2. | Zhou, C.; Jiang, X.; Xu, Z.; Guo, L.; Wang, H.; Zhang, D.; Shi, B. J. Sex. Med. 2010, 7, 3750–3757. doi:10.1111/j.1743-6109.2009.01646.x |
| 7. | Chincholkar, P. M.; Kale, A. S.; Gumaste, V. K.; Deshmukh, A. R. A. S. Tetrahedron 2009, 65, 2605–2609. doi:10.1016/j.tet.2008.11.042 |
| 6. | Siddiqui, S. A.; Srinivasan, K. V. Tetrahedron: Asymmetry 2007, 18, 2099–2103. doi:10.1016/j.tetasy.2007.07.028 |
| 5. | Torre, O.; Gotor-Fernández, V.; Gotor, V. Tetrahedron: Asymmetry 2006, 17, 860–866. doi:10.1016/j.tetasy.2006.02.022 |
| 15. | Dave, A. M.; Patel, D. J.; Kumar, R.; Dwivedi, S. D. A process for preparing (+)-dapoxetine and its salts. WO Pat. Appl. WO2008035358A2, June 5, 2007. |
| 3. | Sorbera, L. A.; Castaner, J.; Castaner, R. M. Drugs Future 2004, 29, 1201–1205. doi:10.1358/dof.2004.029.12.865936 |
| 4. | Darcsi, A.; Tóth, G.; Kökösi, J.; Béni, S. J. Pharm. Biomed. Anal. 2014, 96, 272–277. doi:10.1016/j.jpba.2014.04.002 |
| 15. | Dave, A. M.; Patel, D. J.; Kumar, R.; Dwivedi, S. D. A process for preparing (+)-dapoxetine and its salts. WO Pat. Appl. WO2008035358A2, June 5, 2007. |
| 11. | Cogan, D. A.; Liu, G.; Kim, K.; Backes, B. J.; Ellman, J. A. J. Am. Chem. Soc. 1998, 120, 8011–8019. doi:10.1021/ja9809206 |
| 12. | Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t |
| 14. | Borg, G.; Cogan, D. A.; Ellman, J. A. Tetrahedron Lett. 1999, 40, 6709–6712. doi:10.1016/S0040-4039(99)01351-9 |
| 10. | Khatik, G. L.; Sharma, R.; Kumar, V.; Chouhan, M.; Nair, V. A. Tetrahedron Lett. 2013, 54, 5991–5993. doi:10.1016/j.tetlet.2013.08.059 |
| 14. | Borg, G.; Cogan, D. A.; Ellman, J. A. Tetrahedron Lett. 1999, 40, 6709–6712. doi:10.1016/S0040-4039(99)01351-9 |
| 9. | Kim, S. J.; Jeon, T. H.; Min, I. S.; Kim, I. S.; Jung, Y. H. Tetrahedron Lett. 2012, 53, 3680–3682. doi:10.1016/j.tetlet.2012.05.037 |
| 13. | Arava, V. R.; Gorentla, L.; Dubey, P. K. Beilstein J. Org. Chem. 2012, 8, 1366–1373. doi:10.3762/bjoc.8.158 |
© 2015 Zhu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)